
ABSTRACT
Background:
Clinical outcome of patients with chronic myeloid leukemia (CML) has improved dramatically since the introduction of tyrosine kinase inhibitors such as imatinib mesylate (IM). However, approximately 20-30% of patients experience IM resistance. SH-4-54, which targets the SH2 domains of both proteins STAT3 and STAT5, has been reported to exhibit anticancer activity in solid tumors. However, the roles of SH-4-54 in CML remain unclear. The aim was to explore whether SH-4-54 could overcome IM resistance and identify novel targets for CML.
Methods:
Cell viability was measured by CCK-8 assays after treatment of K562 and K562R cells with different concentrations of SH-4-54. Annexin V-FITC and PI were applied to assess the effects of SH-4-54 on cell apoptosis. Effects of SH-4-54 on the expression of proteins downstream of BCR::ABL1 were assessed by western blotting (WB). Effects of SH-4-54 on gene expression profile of CML cells were analyzed by Next generation sequence (NGS).
Results:
SH-4-54 inhibited the growth of CML cell lines with increasing concentration. SH-4-54 cytotoxic effects correlated with a significant induction of apoptosis. The results of WB analysis showed the downstream proteins of BCR::ABL1, such as STAT3 and STAT5, decreased after SH-4-54 treatment; moreover, the phosphorylation of both proteins were inhibited in dose-dependent manner. Using NGS, we obtained Mrna expression profiles in SH-4-54 treated K562 and K562R cells and identified differentially expressed mRNAs. Among these, STAT3 and STAT5 were markedly downregulated.
Conclusion:
SH-4-54 may overcome IM resistance and represent a promising novel approach to improve the outcome of CML.
Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disease characterized by the presence of the BCR::ABL1 fusion gene, which results from the Philadelphia chromosome, accounting for approximately 15% of all newly diagnosed cases of leukemia in adults. Clinical outcome of patients with CML has improved dramatically since the introduction of tyrosine kinase inhibitors (TKIs) such as imatinib mesylate (IM), which are now considered standard therapy for CML patients [Citation1]. Despite this success, several clinical challenges still exist. Approximately 20–30% of the patients eventually experience IM resistance. In addition, TKIs do not completely eradicate the leukemic stem cells [Citation2]. Therefore, new therapeutic strategies to cure CML are important and challenging tasks.
Signal Transducer and Activators of Transcription (STAT) are a seven-member family of cytoplasmic proteins that contribute to signal transcription and transmission that participate in normal cellular responses to growth factors, hormones, and cytokines. However, inappropriate or aberrant activation of certain STAT family members, particularly STAT3 and STAT5, is associated with a wide variety of human malignancies, including hematologic and solid cancers [Citation3–10]. STAT5 is constitutively activated in cell lines derived from CML patients. STAT3 and STAT5 are aberrantly tyrosine-phosphorylated in cancer cells and are considered as crucial effectors of BCR::ABL1 in CML [Citation10]. Mouse models of CML have demonstrated that STAT3 and STAT5 are both necessary for the initiation of the disease, furthermore, STAT5 is required for the maintenance of leukemia. In addition, reports have indicated that STAT3 and STAT5 mediate imatinib resistance of CML cells. Collectively, these data would undoubtedly define both proteins as important therapeutic targets in hematopoietic cancers. The development of selective pharmacological inhibitors has been the subject of intense investigations [Citation11]. For instance, pyrimethamine or pimozide have been shown to, respectively, inhibit the phosphorylation of STAT3 and STAT5 in different hematopoietic cancers [Citation12,Citation13].
SH-4-54, a most potent nonphosphorylated STAT inhibitor, bind strongly to STAT3 protein (KD = 300nM) and STAT5 protein (KD = 464nM). Recently, the critical functions of SH-4-54 in numerous tumors have been illustrated. In all cases, SH-4-54 alone or in combination with other drugs decreased the growth and survival of solid cancers or leukemic cells, and resensitized resistant cancer cells to chemotherapy. For the effect of SH-4-54 on malignant tumors, SH-4-54 was found to potently kill glioblastoma brain cancer stem cells and control glioma tumor growth in vivo by exhibiting blood–brain barrier permeability [Citation14]. Other studies have demonstrated that SH-4-54 induced temozolomide-resistant glioblastoma cells apoptosis via triggering respiratory chain dysfunction and mitochondrial STAT3 translocation [Citation15]. SH-4-54 is also attractive for treatment-resistant human breast cancer [Citation16]. Hindupur SV et al. illustrated that SH-4-54 reduces cell proliferation of bladder cancer cells both in vitro and in vivo [Citation17]. Moreover, they showed a dose-dependent increase in the percentage of apoptotic cells which is in line with prior reports [Citation18,Citation19]. Nevertheless, the roles of SH-4-54 in regulating the development of CML remain unclear.
Here, we found that SH-4-54 dose and time dependently inhibited the proliferation of cell lines K562 and K562R. In addition, SH-4-54 significantly induced apoptosis of CML cells. We further demonstrated that SH-4-54 down-regulates STAT3/5 expression in CML cells. In this study, we report SH-4-54 as a unique inhibitor STAT3/5 oncogenic pathway that was effective against IM-resistant CML cells. These findings support its potential therapeutic application in STAT3- and STAT5-related malignancies.
Methods
Reagents and drugs
SH-4-54 (MedChemExpress, New York, NJ, USA), a novel small molecule STAT3/5 inhibitor [Citation20], was reconstituted in dimethyl sulfoxide (DMSO) at a 20 mM stock. Individual aliquots were stored at −20°C. Imatinib mesylate (STI571) was purchased from MCE. Stock solutions of 10 mM were prepared by dissolving in DMSO and stored in aliquots at −20°C. Fresh working solutions were prepared in RPMI 1640 medium for immediate use. All antibodies used in this study were obtained from Cell signaling Technology (Beverly, MA, USA).
Chronic myeloid leukemia cells lines
The IM-sensitive K562 cells used in the present study are preserved in our laboratory. The IM-resistant cell lines K562R cells were obtained from American Type Culture Collection (ATCC, CRL-3344). Both cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS), 2 mM glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco, USA) at 37°C, 5% CO2. K562R cells were cultured with 1 μM IM to maintain their drug-resistant status. The medium was changed every 3 days.
Cell viability assay
Cell viability was evaluated with a Cell Counting Kit-8 (CCK-8, Dojindo, Japan) according to the manufacturer’s protocols. Briefly, cells were seeded into 96-well plates at a density of 3 × 103cells per well and treated with IM or SH-4-54 at different concentrations and times. Then, 10 μL CCK-8 was added to each well and incubated for 4 h at 37°C. The optical density at 450 nm was measured using microplate reader (Bio-Rad Co, USA). The concentration required to reduce cell viability by 50% (IC50) was calculated with the curve-fitting algorithm of the GraphPad software.
Evaluation of apoptosis
Detection of apoptosis was performed using the Annexin V-FITC and propidium iodide (PI) detection kit (BD Biosciences, USA). Briefly, K562 and K562R cells were treated with IM or SH-4-54 for 24 h, washed with PBS, resuspended in the binding buffer, and incubated with Annexin V-FITC and PI for 30 min at room temperature in the dark. The apoptotic cell population was analyzed using flow cytometry (Calibur, BD, USA). The cells were considered to be early apoptotic (Annexin V+/PI-) or late apoptotic (Annexin V+/PI+).
Western blot analysis
After IM or SH-4-54 treatment for the indicated time, cells were lysed in RIPA lysis buffer (Cell signaling Technology) with protease inhibitor. Protein concentrations were measured by BCA assay. Equal amounts of protein were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. After being blocked with 5% skim milk, the membranes were incubated with primary antibodies as follows at 4°C overnight: STAT3, p-STAT3, STAT5, p-STAT5, GAPDH (Danvers, MA, USA). Subsequently, the membranes were washed and incubated with specific secondary antibodies (Danvers, MA, USA). Protein band detection was performed with GE Image Quant LAS4000 mini.
mRNA transcriptome sequencing
K562 and K562R cells were treated with DMSO (control) or SH-4-54 (20 μM for 24 h) and duplicates of 3 control samples and 3 test samples were processed for mRNA sequencing. RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). The library preparations were sequenced on an Illumina Novaseq platform and 150 bp paired-end reads were generated.
Statistical analysis
GraphPad Prism software ver. 6.0 was used for statistical analysis. All data were expressed as the mean ± standard deviation (SD) from at least three independent experiments. Comparisons between two groups were analyzed using Student’s t-test. Statistical analysis of multiple groups was performed by one-way analysis of variance (ANOVA). Statistical significance was established as *p < 0.05.
Results
Effect of SH-4-54 on cell proliferation
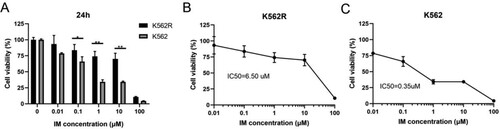
Initial experiments were carried out to determine that the K562R cells we used in this study are IM-resistant CML cells. The IM-sensitive (K562) and the equivalent IM-resistant (K562R) cells were treated with various concentrations of IM for 24 h (ranging from 0.01 to 100 μM). Their cell viability was assessed by the CCK-8 assay. The results showed that IM preferentially inhibits the proliferation of IM-sensitive K562 cells. We calculated the IC50 (50% inhibition concentration) to be 0.35 μmol/L in K562 cells and 6.5 μmol/L in K562R cells, which indicated that K562R cells have a markedly higher IC50 compared with K562 cells (). The IM resistance fold-change of the K562R cells was 18.57. Therefore, the K562R cell line was resistant to IM, which confirmed the efficacy of the IM-resistant cell model.
Figure 1. The inhibitory effect of IM in K562 and K562R cells. Imatinib mesylate (IM)-sensitive K562 and IM-resistant K562 cells (K562R) were treated with IM for DMSO as control for 24 h. Cell viability was determined by CCK-8 assays. (A) Statistical analysis of cell viability from K562 and K562R cells, respectively. * p < 0.05; **** p < 0.0001. (B–C) IM decreases the viability of K562 and K562R cells over concentration. 50% inhibition of cell viability (IC50) values was calculated with GraphPad software. Data are presented as mean ± SD of three independent experiments (n = 3) in triplicates.
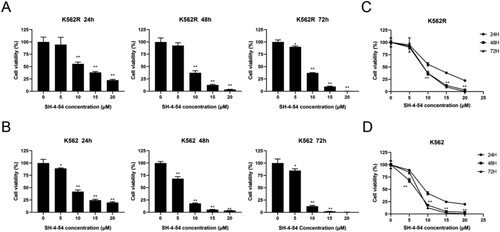
To investigate the inhibitory effects of SH-4-54, the CML cell lines K562 and K562R were incubated with four concentrations (5, 10, 15, 20 μM) of SH-4-54 for 24, 48, and 72 h, and cell viability was analyzed using CCK-8 assay. Our data showed that SH-4-54 inhibited the growth of CML cell lines with increasing concentration (p < 0.05) ((A–B)). When comparing with the same concentrations at 24 h, the inhibitory effect of SH-4-54 on the proliferation of CML cells was also significantly enhanced (p < 0.05) ((C–D)). As shown in , these results suggested that SH-4-54 could suppress K562 cell viability in dose and time-dependent manners and was also effective against imatinib-resistant K562R cells.
Figure 2. Growth inhibition effects of SH-4-54 on CML cells. Cell viability was measured by CCK-8 assays after treatment of K562R(A) and K562(B) cells with increasing concentrations of SH-4-54 or DMSO as control for different durations. Results are expressed as mean ± SD, and the error bars represent the SD of at least three independent experiments. (A–B) Representative bar graphs * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001 vs. control group. (C–D) Representative line graphs *** p < 0.001; **** p < 0.0001 vs. the same concentrations for 24 h.
SH-4-54 induces the apoptosis of CML cells
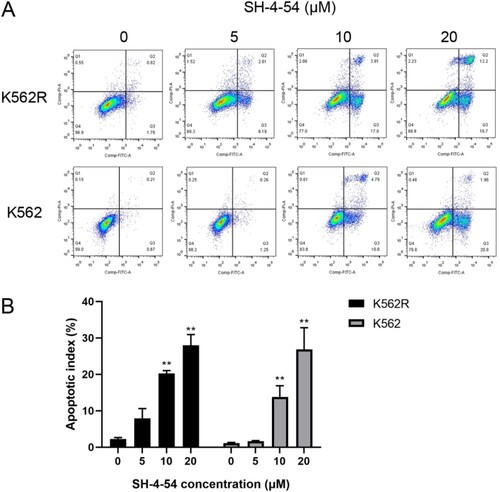
To further clarify whether the antiproliferation effects of SH-4-54 in CML cells are associated with cell apoptosis, K562 and K562R cells were treated with 0, 5, 10, and 20 μM of SH-4-54, following which cells were labeled with Annexin V-FITC and PI. The apoptotic rates of the cells were measured using flow cytometric analysis. The results revealed that K562 cells did not undergo apoptosis with 5 μM SH-4-54. Similarly, K562R cells showed no or minimal signs of apoptosis with the same as SH-4-54. However, 10 or 20 μM SH-4-54 dramatically induced apoptosis of K562 and K562R cells compared with control (). Interestingly, we observed that SH-4-54 mainly induced early apoptosis of K562 cells. Moreover, this effect was evident in early and late apoptosis for K562R cells. In addition, the rates of apoptosis in the K562R cells seem to be higher compared with those in the K562 cells, which need to be validated by further experiments. These data manifested that SH-4-54 overcame imatinib resistant and significantly induced cell apoptosis of CML cell lines.
Figure 3. SH-4-54 induced apoptosis in CML cell lines. K562 and K562R cells treated for 24 h with SH-4-54 and DMSO as control were stained with Annexin V/PI to determine the percentages of apoptotic cells. One representative experiment is shown (A). The histogram (B) presents the percentage of apoptotic cells (n = 3 in triplicates, data are mean ± SD, ** p < 0.01 vs. control group). Apoptotic cells are positive for Annexin V (Annexin V + and PI- as early apoptosis, Annexin V + and PI + as late apoptosis).
SH-4-54 regulates STAT3 and STAT5 signaling in CML cells
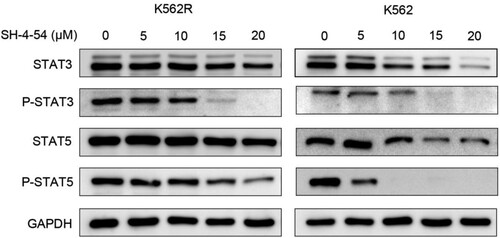
To elucidate the molecular pathogenesis by which SH-4-54 induces apoptosis in CML cells, we evaluate the ability of SH-4-54 to inhibit the phosphorylation of BCR::ABL1 downstream targets STAT3 and STAT5, which have been implicated in the signal transduction and antiapoptotic signaling stemming from constitutive activation of the BCR::ABL1 [Citation21]. K562 and K562R cells were treated with increasing concentrations of SH-4-54 for 3 h and total cell extracts were prepared to check the levels of STAT3/5 and pSTAT3/5 by Western blot. As shown in , SH-4-54 effectively inhibited the phosphorylation of STAT3 and STAT5 and the effect was dose-dependent. This inhibition was more prominent in K562 cells. Overall, deregulation of proteins in CML cell proliferation signal transduction pathway by SH-4-54 contributes to apoptosis.
Figure 4. Effects of SH-4-54 on the expression of proteins downstream of BCR-ABL in CML cells. K562 and K562R cells were treated with 0, 5, 10, 15, 20 μM SH-4-54 for 3 h. Then, whole-cell lysates were extracted to assess the levels of STAT3, pSTAT3, STAT5, pSTAT5 by Western blot analysis. GAPDH was used as a loading control. The results are representative of three determinations with identical results.
Effect of SH-4-54 on gene expression profile of CML cells
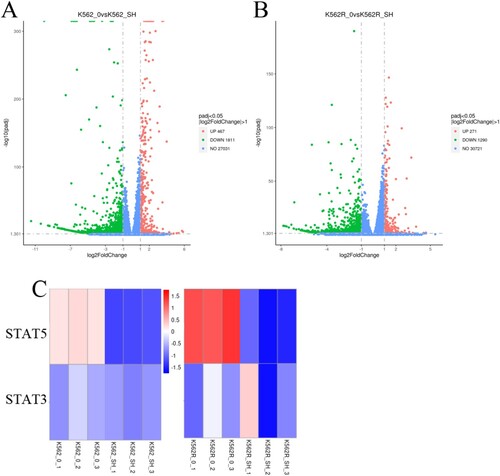
To further elaborate the effect of SH-4-54 on gene expression profile of CML cells, we next conducted next-generation sequence (NGS) to analyze possible SH-4-54 induced alterations in the mRNA transcriptome of CML cell. To this end, K562 and K562R cells were either treated with DMSO (control) or exposed to 20 μM SH-4-54 for 24 h prior to the collection of total RNA. A total of 467 genes were upregulated and 1811 genes were downregulated in the K562 cell line. Similarly, the expression levels of 271 genes were increased, whereas 1290 genes were decreased in K562R cell line ((A–B)). The altered genes are complex and involve multiple signaling pathways, including the JAK-STAT pathway. As shown in (C), STAT3 and STAT5 were significantly reduced in the K562 and K562R cell lines, which further confirmed that SH-4-54 regulated the JAK-STAT signaling pathway of CML cells from the gene level.
Figure 5. Effects of SH-4-54 on gene expression profile of CML cells. (A-B) Volcano plots of gene expression differences of K562 and K562R cells treated with control or for 20 μM SH-4-54 for 24 h. (C) Heat map of mRNA expression profiles from control (DMSO-treated) and SH-4-54 treated (20 μM, 24 h) K562/K562R cells. The expression of each mRNA corresponds to the average of 3 replicates. Also shown is the correlation of the log fold expression changes of differential expressed genes between two groups. For the SH-4-54 group, transcripts with >2-fold decrease in are shown in green, while those with >2-fold increase are shown in red.
Discussion
Targeted therapies have become a hot topic of research for the treatment of cancers. Identification of key oncogenic drivers has given rise to the development of drugs that have markedly improved the prognosis of cancer patients. The discovery of TKIs represents the best example of the targeted treatment against CML. However, severe adverse effects and resistance often occur in CML patients treated by TKIs. Therefore, novel targets for the treatment of CML need to be sought.
STAT3 and STAT5 are important effectors of cellular transformation. They also contribute to drug resistance in hematopoietic cancers and are now well recognized as common targets in cancer treatment. Previous studies have documented elevated levels of STAT5 in IM-resistant CML patients and K562R cells [Citation22]. The activation of STAT5 contributes to BCR::ABL1-triggered CML via four key mechanisms which are to stimulate cells proliferation, to enhance cell viability by upregulation of anti-apoptotic genes, to counteract TKI-induced cell death and to increase the possibility of acquiring BCR::ABL1 mutations [Citation23,Citation24]. The importance of STAT5 in CML is further emphasized by findings that STAT5 is upregulated during disease progression. Furthermore, STAT5 activity is associated with poor prognosis.
During the last 10 years, many studies have been conducted on STAT3 and CML. JAK2/STAT3 signaling is constitutively hyperactivated in CML which promotes CML cells growth, leukemia stem cells survival, and drug resistance, thus disease evolution [Citation25–27]. Earlier experiments found that bone marrow stromal cells stimulated STAT3 phosphorylation in a BCR::ABL1 independent manner, inducing IM resistance. More interestingly, targeting STAT3 and STAT5 could overcome the resistance of CML cells to IM [Citation28]. Collectively, these data indicate that directly target STAT3 and/or STAT5 might provide new opportunities for CML therapy. Noteworthy, several reports indicated that SH-4-54 could strongly bind to STAT3 and STAT5 protein, effectively suppress STAT3/5 phosphorylation and their downstream transcriptional targets. In the present study, we demonstrated that high concentrations of SH-4-54 reduced the expression of STAT3 and STAT5 in K562 and K562R cells. Besides, SH-4-54 inhibited the phosphorylation of these two proteins in a concentration-dependent manner. Using NGS, we obtained mRNA expression profiles in SH-4-54 treated K562 and K562R cells and identified differentially expressed mRNAs. Among these, STAT3 and STAT5 were markedly downregulated. The above results were consistent with previous research. So we hypothesized that SH-4-54 had an inhibitory effect on CML cells.
Compound SH-4-54 showed the best cytotoxicity to temozolomide-resistant glioblastoma cells and it has also been reported to play an important role in neuroblastoma [Citation29]. In this study, we examined the antitumor effects of SH-4-54 on CML cell lines. SH-4-54 reduced cell viability in a time- and concentration-dependent manner in K562 and K562R cells. SH-4-54 cytotoxic effects correlated with a significant induction of apoptosis. We presumed that SH-4-54-induced apoptosis is triggered by protein degradation. The results of western blot analysis showed that the downstream proteins of BCR::ABL1, such as STAT3 and STAT5, decreased after SH-4-54 treatment; moreover, the phosphorylation of both proteins was inhibited in a dose-dependent manner. In summary, our data confirmed that SH-4-54 suppressed the biological activity of CML cells in vitro. The killing of SH-4-54 on K562R cells proved that SH-4-54 could overcome IM resistance in CML. However, more studies are needed to investigate the clinical implication of SH-4-54 in TKI-resistant CML patients. Together, these data provide preliminary preclinical rationale for using SH-4-54 to treat CML.
Tumors are genetically diverse and drug-resistant seems inevitable. To enhance the efficiencies or to overcome the limitations of monotherapy options, combination therapies are necessary. Over the years, accumulating evidence has shown that STAT3 is associated with chemotherapy resistance in various cancers [Citation30,Citation31]. Moreover, STAT5 was also demonstrated to play a crucial role in the maintenance of LSCs from resistant CML patients [Citation32,Citation33]. In this study, we found that SH-4-54 could inhibit phosphorylation of STAT3 and STAT5 proteins, thereby inhibiting proliferation and inducing cell apoptosis of IM-sensitive and resistant CML cell lines. It remains to be investigated the possibility of combining SH-4-54 with TKI so as to reinforce their respective antileukemic activities. This finding is important in the clinical setting in the future. Last but not least, the optimization of combination therapies using SH-4-54 with TKI or other key players will be required to find the right combination that safely unlocks drug resistance in CML patients.
Taken together, our results indicate that SH-4-54 may inhibit proliferation and induce apoptosis of CML cells via suppressing the activation of STAT3/5 simultaneously. In conclusion, SH-4-54 could overcome imatinib resistant and may be a promising treatment option for CML therapy.
Conclusion
SH-4-54 could suppress CML cell viability in dose and time-dependent manners and significantly induce cell apoptosis of CML cell lines. SH-4-54 effectively inhibited the phosphorylation of STAT3 and STAT5 and the effect was dose-dependent. Overall, SH-4-54 could overcome imatinib resistant and may be a promising treatment option for CML therapy.
Data accessibility statement
The raw data, supporting the conclusions of this manuscript, will be made available by contacting the corresponding author.
Acknowledgements
The authors would like to thank the support and encouragement of the members in the Department of Hematology, The Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–984. doi:10.1038/s41375-020-0776-2
- Al Hamad M. Contribution of BCR-ABL molecular variants and leukemic stem cells in response and resistance to tyrosine kinase inhibitors: a review. F1000Res. 2021;10:1288. doi:10.12688/f1000research.74570.1
- Igelmann S, Neubauer HA, Ferbeyre G. STAT3 and STAT5 activation in solid cancers. Cancers. 2019;11(10):1428. doi:10.3390/cancers11101428
- Larrue C, Heydt Q, Saland E, et al. Oncogenic KIT mutations induce STAT3-dependent autophagy to support cell proliferation in acute myeloid leukemia. Oncogenesis. 2019;8(8):39. doi:10.1038/s41389-019-0148-9
- Wingelhofer B, Maurer B, Heyes EC, et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia. 2018;32(5):1135–1146. doi:10.1038/s41375-017-0005-9
- Dumas PY, Naudin C, Martin-Lanneree S, et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica. 2019;104(10):2017–2027. doi:10.3324/haematol.2018.205385
- Minieri V, De Dominici M, Porazzi P, et al. Targeting STAT5 or STAT5-regulated pathways suppresses leukemogenesis of Ph+ acute lymphoblastic leukemia. Cancer Res. 2018;78(20):5793–5807. doi:10.1158/0008-5472.CAN-18-0195
- Katerndahl CDS, Heltemes-Harris LM, Willette MJL, et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat Immunol. 2017;18(6):694–704. doi:10.1038/ni.3716
- Klein K, Witalisz-Siepracka A, Maurer B, et al. STAT5B (N642H) drives transformation of NKT cells: a novel mouse model for CD56(+) T-LGL leukemia. Leukemia. 2019;33(9):2336–2340. doi:10.1038/s41375-019-0471-3
- Mi T, Wang Z, Bunting KD. The cooperative relationship between STAT5 and reactive oxygen species in leukemia: mechanism and therapeutic potential. Cancers. 2018;10(10.
- Brachet-Botineau M, Polomski M, Neubauer HA, et al. Pharmacological inhibition of oncogenic STAT3 and STAT5 signaling in hematopoietic cancers. Cancers. 2020;12(1):240. doi:10.3390/cancers12010240
- Nelson EA, Sharma SV, Settleman J, et al. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget. 2011;2(6):518–524. doi:10.18632/oncotarget.296
- Nelson EA, Walker SR, Weisberg E, et al. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117(12):3421–3429. doi:10.1182/blood-2009-11-255232
- Liu HM, Guo CL, Zhang YF, et al. Leonurine-repressed miR-18a-5p/SOCS5/JAK2/STAT3 axis activity disrupts CML malignancy. Front Pharmacol. 2021;12:657724. doi:10.3389/fphar.2021.657724
- Cui P, Wei F, Hou J, et al. STAT3 inhibition induced temozolomide-resistant glioblastoma apoptosis via triggering mitochondrial STAT3 translocation and respiratory chain dysfunction. Cell Signal. 2020;71:109598. doi:10.1016/j.cellsig.2020.109598
- Linher-Melville K, Nashed MG, Ungard RG, et al. Chronic inhibition of STAT3/STAT5 in treatment-resistant human breast cancer cell subtypes: convergence on the ROS/SUMO pathway and its effects on xCT expression and system xc- activity. PloS one. 2016;11(8):e0161202. doi:10.1371/journal.pone.0161202
- Hindupur SV, Schmid SC, Koch JA, et al. STAT3/5 inhibitors suppress proliferation in bladder cancer and enhance oncolytic adenovirus therapy. Int J Mol Sci. 2020;21(3):1106. doi:10.3390/ijms21031106
- Shao D, Ma J, Zhou C, et al. STAT3 down-regulation induces mitochondria-dependent G2/M cell cycle arrest and apoptosis in oesophageal carcinoma cells. Clin Exp Pharmacol Physiol. 2017;44(3):413–420. doi:10.1111/1440-1681.12708
- Tsujita Y, Horiguchi A, Tasaki S, et al. STAT3 inhibition by WP1066 suppresses the growth and invasiveness of bladder cancer cells. Oncol Rep. 2017;38(4):2197–2204. doi:10.3892/or.2017.5902
- Haftchenary S, Luchman HA, Jouk AO, et al. Potent targeting of the STAT3 protein in brain cancer stem cells: a promising route for treating glioblastoma. ACS Med Chem Lett. 2013;4(11):1102–1107. doi:10.1021/ml4003138
- Warsch W, Grundschober E, Berger A, et al. STAT5 triggers BCR-ABL1 mutation by mediating ROS production in chronic myeloid leukaemia. Oncotarget. 2012;3(12):1669–1687. doi:10.18632/oncotarget.806
- Cheng Y, Hao Y, Zhang A, et al. Persistent STAT5-mediated ROS production and involvement of aberrant p53 apoptotic signaling in the resistance of chronic myeloid leukemia to imatinib. Int J Mol Med. 2018;41(1):455–463.
- Warsch W, Grundschober E, Sexl V. Adding a new facet to STAT5 in CML: multitasking for leukemic cells. Cell Cycle. 2013;12(12):1813–1814. doi:10.4161/cc.25116
- Xu X, Zhang X, Liu Y, et al. BM microenvironmental protection of CML cells from imatinib through Stat5/NF-kappaB signaling and reversal by Wogonin. Oncotarget. 2016;7(17):24436–24454. doi:10.18632/oncotarget.8332
- Zhao M, Yang C, Chai S, et al. Curcumol and FTY720 synergistically induce apoptosis and differentiation in chronic myelomonocytic leukemia via multiple signaling pathways. Phytotherapy Research: PTR. 2021;35(4):2157–2170. doi:10.1002/ptr.6968
- Cheng L, Tang Y, Chen X, et al. Deletion of MBD2 inhibits proliferation of chronic myeloid leukaemia blast phase cells. Cancer Biol Ther. 2018;19(8):676–686. doi:10.1080/15384047.2018.1450113
- Stella S, Tirro E, Conte E, et al. Suppression of survivin induced by a BCR-ABL/JAK2/STAT3 pathway sensitizes imatinib-resistant CML cells to different cytotoxic drugs. Mol Cancer Ther. 2013;12(6):1085–1098. doi:10.1158/1535-7163.MCT-12-0550
- Gleixner KV, Schneeweiss M, Eisenwort G, et al. Combined targeting of STAT3 and STAT5: a novel approach to overcome drug resistance in chronic myeloid leukemia. Haematologica. 2017;102(9):1519–1529. doi:10.3324/haematol.2016.163436
- Memarzadeh K, Savage DJ, Bean AJ. Low UBE4B expression increases sensitivity of chemoresistant neuroblastoma cells to EGFR and STAT5 inhibition. Cancer Biol Ther. 2019;20(12):1416–1429. doi:10.1080/15384047.2019.1647049
- Zhao C, Li H, Lin HJ, et al. Feedback activation of STAT3 as a cancer drug-resistance mechanism. Trends Pharmacol Sci. 2016;37(1):47–61. doi:10.1016/j.tips.2015.10.001
- Zhou X, Ren Y, Liu A, et al. WP1066 sensitizes oral squamous cell carcinoma cells to cisplatin by targeting STAT3/miR-21 axis. Sci Rep. 2014;4:7461.
- Casetti L, Martin-Lanneree S, Najjar I, et al. Differential contributions of STAT5A and STAT5B to stress protection and tyrosine kinase inhibitor resistance of chronic myeloid leukemia stem/progenitor cells. Cancer Res. 2013;73(7):2052–2058. doi:10.1158/0008-5472.CAN-12-3955
- Prost S, Relouzat F, Spentchian M, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature. 2015;525(7569):380–383. doi:10.1038/nature15248