
ABSTRACT
Background
Severe autoimmune hemolytic anemia complicating hereditary spherocytosis is life threatening and has not been described in a case report. Here, we report a case in which this intractable disease was treated successfully with glucocorticoids and cyclosporine.
Case presentation
A 25-year-old female patient with hereditary spherocytosis developed severe autoimmune hemolytic anemia after respiratory syncytial virus infection. Her hemoglobin level was 26 g/L and various anti-red blood cell antibodies were detected in her serum, making blood matching difficult. Glucocorticoid monotherapy was ineffective. With the addition of cyclosporine (50 mg/12 h), the patient's hemoglobin level increased significantly and the symptoms associated with anemia were greatly relieved.
Conclusion
In patients with severe autoimmune hemolytic anemia, especially when the presence of multiple anti-red blood cell antibodies and alloantibodies interferes with blood matching, a glucocorticoid-cyclosporine regimen may be tried.
Introduction
Autoimmune hemolytic anemia (AIHA) is caused by the destruction of red blood cells (RBCs) by autoimmune hemolytic mechanisms, usually mediated by anti-RBC autoantibodies with or without complement activation [Citation1–3]. It has warm, cold, and mixed reactive subtypes, according to the class and thermal amplitude of the pathogenic antibodies [Citation4]. AIHA may be primary or secondary to viral or bacterial infection, autoimmune disease, malignancy, or drug use [Citation2,Citation5]. Standard therapeutic interventions include steroid administration, splenectomy, and the use of immunosuppressive agents and monoclonal antibodies [Citation1,Citation4].
Hereditary spherocytosis (HS) encompasses a heterogeneous group of inherited anemias characterized by the presence of spherical RBCs on peripheral blood smears. It is prevalent worldwide, with a high incidence of 1:2000 in northern Europeans [Citation6] and an incidence of about 1:100,000 in the Chinese population [Citation7]. HS is characterized by anemia, jaundice, splenomegaly, and cholelithiasis [Citation8]. In addition, abnormalities in the quantity and/or quality of erythrocyte membrane proteins can lead to the reduced deformability of erythrocytes, which can easily break down and cause hemolysis [Citation9,Citation10]. Five genes are associated with HS and involved in the interaction between the erythrocyte membrane and lipid bilayer: ankyrin (ANK1); β-spectrin (SPTB); α-spectrin (SPTA1); solute carrier family 4, member 1 (SLC4A1); and erythrocyte membrane protein 4.2 (EPB42) [Citation11–13]. Mutations in one or more of these genes can cause membrane protein deficiency, leading to HS [Citation14,Citation15].
Here, we report a case of AIHA complicated with HS. Hemoglobin level of the patient was extremely low and she required emergency blood transfusion, but compatible transfusion was diffcult due to the detection of multiple erythrocyte-related antibodies in her serum. The patient's hemoglobin level increased significantly and her anemia symptoms improved after the addition of cyclosporine to glucocorticoid therapy.
Case report
In November 2021, a 25-year-old woman developed moderate to severe edema with no obvious cause, especially in both lower limbs, accompanied by abdominal distension, weakness, heart palpitations, dyspnea, inability to lie down at night, and splenomegaly. On January 29th 2022, she was admitted to the Emergency Department of China–Japan Friendship Hospital with a fever of up to 39°C. The results of laboratory tests conducted on admission are provided in . A peripheral blood smear showed mature erythrocytes of variable size, heterogeneous erythrocytes, and a count of 2 nucleated erythrocytes per 100 leukocytes. Based on the patient's decreased hemoglobin level; elevated reticulocytes, lactate dehydrogenase, bilirubin, and indirect bilirubin levels; and direct anti-human globulin test (DAT) positivity [anti-immunoglobulin (IgG) 2+ and anti-C3d 1+; ], the diagnosis of AIHA was considered. Tests performed to exclude infectious causes of the fever detected IgM antibodies against respiratory syncytial virus (RSV). It is worth noting that her platelet count decreased during the initial period of hospitalization (). This thrombocytopenia may have been mediated by immune-related factors and may have been due to the patient's hypersplenism.
Table 1. The patient's laboratory indices and clinical characteristics.
The patient had been diagnosed with HS at the Institute of Hematology and Blood Disease Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, in 2016. The diagnosis was based on symptoms and findings such as intermittent fever, hemolysis, splenomegaly, renal calculi, gallstones along with abnormal results of laboratory tests including sucrose hemolysis test (Test result: 18.3%; Reference value: 0-16.9%) and acidified glycerol lysis test (Test result: 40 s; Reference value: > 290 s). She had received frequent blood transfusions at a local hospital (about once a month between 2019 and 2021).
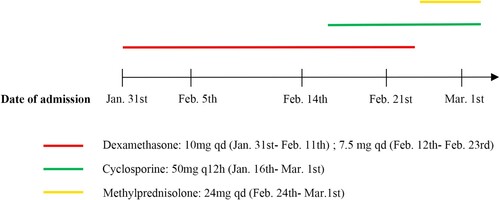
The diagnosis of AIHA complicated with HS was made, and the patient was given dexamethasone (10 mg/day intravenously) for 2 days (). She was also given 4 units of suspended RBC infusion. Blood matching was extremely difficult due to the presence of multiple (anti-f, anti-Jka and anti-N) alloantibodies in her plasma.
Figure 1. Timeline of the therapeutic intervention.
On January 30th, 2022, the patient was transferred to the Department of Hematology of China–Japan Friendship Hospital for further treatment. She underwent bone marrow aspiration and biopsy. Examination of the bone marrow cytomorphology revealed obvious erythroid line proliferation, with rubricyte and giant-like, split-phase, petal-like metarubricyte predominance. Mature RBCs were of slightly variable sizes, and some were highly pigmented and spherical; about 21% of all mature RBCs were spherical erythrocytes. Next-generation sequencing of peripheral blood from the patient was performed. The sequencing revealed heterozygous mutation of the patient's STBP gene (HS family pedigree; , ), confirming the HS diagnosis.
Figure 2. Pedigree of the hereditary spherocytosis (HS) family. Males and females are represented by squares and circles, respectively. The blank symbol represents the wild type and the half-filled symbols indicate heterozygosity. The index case is marked by an arrow.
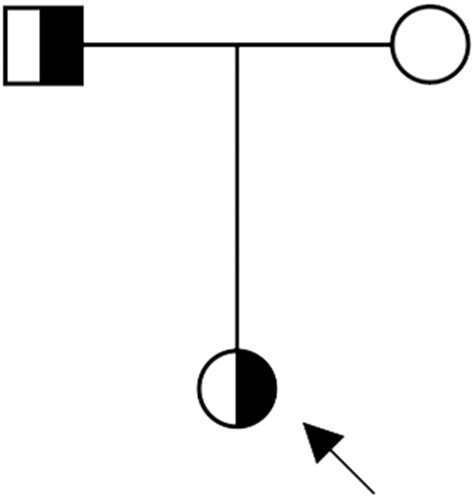
Table 2. Information on the patient's SPTB mutation.
In consideration of the AIHA diagnosis, the patient was given glucocorticoids (dexamethasone sodium phosphate, 10 mg/day intravenously from January 31st to February 11th and 7.5 mg/day intravenously from February 12th to February 23rd; methylprednisolone, 24 mg/day orally from February 24th to March 1st). Rosasta (70 mg twice weekly) was used to promote hematopoiesis. The patient's symptoms did not improve and her hemoglobin level did not rise. And to make matters worse, compatible transfusions were extremely difficult, so we decided to add the immunosuppressant cyclosporine (50 mg/12 h from February 16th to March 1st; ). The patient's platelet count increased, falling within the normal range, after glucocorticoid treatment, suggesting that its initial reduction was due to the involvement of immunological factors.
On March 1st, the patient's febrile symptoms had improved significantly and she was in good spirits. Physical examination revealed stable vital signs, yellowing of the skin and sclera, abdominal enlargement, and a palpable liver and spleen under the ribs. The patient's hemoglobin level was 80 g/L. A peripheral blood smear showed no nucleated RBC and slightly variable mature RBC size. Other laboratory test results are provided in . The patient was discharged from the hospital. Based on her treatment history and laboratory test results, the diagnosis of AIHA secondary to viral infection complicated with HS was confirmed.
Discussion
Here, we report a case of AIHA complicated with HS that was treated successfully with glucocorticoids and cyclosporine. The patient was admitted to the hospital with severe anemia and a dissatisfactory outcome of glucocorticoid treatment. She had produced multiple RBC antibodies (anti-f, anti-Jka and anti-N), making compatible blood transfusion extremely difficult. With the addition of cyclosporine to her treatment, the patient's anemia symptoms improved gradually and her hemoglobin level increased.
Antibodies to RBCs are classified as autoantibodies and alloantibodies [Citation16,Citation17]. Anti-RBC autoantibodies, detected by a DAT or indirect antiglobulin test, are often pan-reactive, making it difficult to find compatible blood. They may mask the presence of clinically significant alloantibodies, potentially resulting in a hemolytic transfusion reaction [Citation18,Citation19]. Patients with AIHA who are chronically transfused are constantly exposed to the risk of RBC alloimmunization, which occurs in 12–40% of this population [Citation20,Citation21]. In addition to the number of transfusions, longstanding infection and inflammation are associated with an increased risk of RBC antibody formation [Citation22–24]. In the case described here, the patient presented with secondary AIHA following RSV infection. The DAT (IgG and C3d) positivity and identification of anti-f, anti-Jka, and anti-N antibodies support the coexistence of the two RBC antibody types in the patient's serum.
A special feature of this case is that the patient had congenital anemia, of which AIHA is a serious and life-threatening complication [Citation25]. The diagnosis and treatment of AIHA in patients with congenital anemia are challenging. Diagnosis should be undertaken with in-depth collaboration with transfusion specialists to perform DATs using more sensitive methods (i.e. with the use of microcolumns, solid-phase testing, washing with low ionic strength solutions, and extended phenotyping) [Citation26–28]. For AIHA secondary to viral infection, antiviral treatment is required. When hemolysis is severe and persistent, treatment with steroids, immunoglobulins, or immunosuppressants is needed [Citation29, Citation30]. We chose to use cyclosporine in the present case because it is a non-myelotoxic immunosuppressant with easily controlled toxicity and it has been used successfully in cases of refractory or relapsed AIHA [Citation31–33].
This study has some limitations. Firstly, we observed only the short-term efficacy in this patient. Long-term follow-up studies should be conducted to determine the effectiveness of this treatment regimen. Secondly, it is unclear whether the cyclosporine or glucocorticoids induced the remission of the acute hemolytic event. The effects of both drugs in such patients need to be further investigated. Thirdly, our study did not measure the related blood tests of the patient's father, but only collected his medical history. For future research, to ensure its completeness, the relevant indicators of the patient's father should also be assessed to make the study more persuasive.
In summary, we report a rare case of AIHA secondary to RSV infection complicated with HS that was treated successfully with glucocorticoids and cyclosporine. This regimen may be tried to treat AIHA, especially in patients with multiple anti-RBC antibodies in their serum.
Ethics approval and consent to participate
This study was reviewed and approved by the Ethics Committee of China–Japan Friendship Hospital. Written informed consent was obtained from the patient for the publication of any potentially identifiable images and data.
Author contributions
NW and MG analyzed and interpreted the patient data and were the major contributors to the writing of the manuscript. HL interpreted the serological data. LL collected blood samples and conducted related laboratory testing. YC revised the manuscript. All authors contributed to the manuscript and approved the submitted version.
Acknowledgement
We thank Medjaden Inc. for its assistance in the preparation of this manuscript.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Jager U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the first international consensus meeting. Blood Rev. 2020;41:100648. doi:10.1016/j.blre.2019.100648
- Hill QA, Hill A, Berentsen S. Defining autoimmune hemolytic anemia: a systematic review of the terminology used for diagnosis and treatment. Blood Adv. 2019;3:1897–1906. doi:10.1182/bloodadvances.2019000036
- Barcellini W. New insights in the pathogenesis of autoimmune hemolytic anemia. Transfus Med Hemother. 2015;42:287–293. doi:10.1159/000439002
- Bass GF, Tuscano ET, Tuscano JM. Diagnosis and classification of autoimmune hemolytic anemia. Autoimmun Rev. 2014;13:560–564. doi:10.1016/j.autrev.2013.11.010
- Phillips J, Henderson AC. Hemolytic anemia: evaluation and differential diagnosis. Am Fam Physician. 2018;98:354–361. PMID: 30215915.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411–1426. doi:10.1016/S0140-6736(08)61588-3
- Wang C, Cui Y, Li Y, et al. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable Rare Dis Res. 2015;4:76–81. doi:10.5582/irdr.2015.01002
- Iolascon A, Andolfo I, Russo R. Advances in understanding the pathogenesis of red cell membrane disorders. Br J Haematol. 2019;187:13–24. doi:10.1111/bjh.16126
- King MJ, Garcon L, Hoyer JD, et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37:304–325. doi:10.1111/ijlh.12335
- He BJ, Liao L, Deng ZF, et al. Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta Haematol. 2018;139:60–66. doi:10.1159/000486229
- Steinberg-Shemer O, Tamary H. Impact of next-generation sequencing on the diagnosis and treatment of congenital anemias. Mol Diagn Ther. 2020;24:397–407. doi:10.1007/s40291-020-00478-3
- Zaninoni A, Fermo E, Vercellati C, et al. Congenital hemolytic anemias: Is there a role for the immune system? Front Immunol. 2020;11:1309. doi:10.3389/fimmu.2020.01309
- Silva R, Amarasinghe D, Perera S, et al. A systematic review on diagnostic methods of red cell membrane disorders in Asia. Int J Lab Hematol. 2022;44:248–262. doi:10.1111/ijlh.13800
- Boguslawska DM, Heger E, Listowski M, et al. A novel L1340P mutation in the ANK1 gene is associated with hereditary spherocytosis? Br J Haematol. 2014;167:269–271. doi:10.1111/bjh.12960
- Qin L, Nie Y, Zhang H, et al. Identification of new mutations in patients with hereditary spherocytosis by next-generation sequencing. J Hum Genet. 2020;65:427–434. doi:10.1038/s10038-020-0724-z
- Meulenbroek EM, Wouters D, Zeerleder SS. Lyse or not to lyse: clinical significance of red blood cell autoantibodies. Blood Rev. 2015;29:369–376. doi:10.1016/j.blre.2015.05.001
- Ahrens N, Pruss A, Kahne A, et al. Coexistence of autoantibodies and alloantibodies to red blood cells due to blood transfusion. Transfusion. 2007;47:813–816. doi:10.1111/j.1537-2995.2007.01194.x
- Raos M, Lukic M, Pulanic D, et al. The role of serological and molecular testing in the diagnostics and transfusion treatment of autoimmune haemolytic anaemia. Blood Transfus. 2022;20:319–328. doi:10.2450/2021.0235-21
- Branch DR, Petz LD. Detecting alloantibodies in patients with autoantibodies. Transfusion. 1999;39:6–10. doi:10.1046/j.1537-2995.1999.39199116888.x
- Leger RM, Garratty G. Evaluation of methods for detecting alloantibodies underlying warm autoantibodies. Transfusion. 1999;39:11–16. doi:10.1046/j.1537-2995.1999.39199116889.x
- Shirey RS, Boyd JS, Parwani AV, et al. Prophylactic antigen-matched donor blood for patients with warm autoantibodies: an algorithm for transfusion management. Transfusion. 2002;42:1435–1441. doi:10.1046/j.1537-2995.2002.00234.x
- Evers D, van der Bom JG, Tijmensen J, et al. Red cell alloimmunisation in patients with different types of infections. Br J Haematol. 2016;175:956–966. doi:10.1111/bjh.14307
- Fasano RM, Booth GS, Miles M, et al. Red blood cell alloimmunization is influenced by recipient inflammatory state at time of transfusion in patients with sickle cell disease. Br J Haematol. 2015;168:291–300. doi:10.1111/bjh.13123
- Gerritsma JJ, Oomen I, Meinderts S, et al. Back to base pairs: what is the genetic risk for red bloodcell alloimmunization? Blood Rev. 2021;48:100794. doi:10.1016/j.blre.2020.100794
- Motta I, Giannotta J, Ferraresi M, et al. Autoimmune hemolytic anemia as a complication of congenital anemias. A Case Series and Review of the Literature. J Clin Med. 2021;10: 3439. doi:10.3390/jcm10153439
- Barcellini W, Zaninoni A, Giannotta JA, et al. New insights in autoimmune hemolytic anemia: from pathogenesis to therapy stage 1. J Clin Med. 2020;9:3859. doi:10.3390/jcm9123859
- Zaninoni A, Vercellati C, Imperiali FG, et al. Detection of red blood cell antibodies in mitogen-stimulated cultures from patients with hereditary spherocytosis. Transfusion. 2015;55:2930–2938. doi:10.1111/trf.13257
- Johnson ST, Puca KE. Evaluating patients with autoimmune hemolytic anemia in the transfusion service and immunohematology reference laboratory: pretransfusion testing challenges and best transfusion-management strategies. Hematology Am Soc Hematol Educ Program. 2022;2022:96–104. doi:10.1182/hematology.2022000406
- Fattizzo B, Cantoni S, Giannotta JA, et al. Efficacy and safety of cyclosporine A treatment in autoimmune cytopenias: the experience of two Italian reference centers. Ther Adv Hematol. 2022;13:204062072210977. doi:10.1177/20406207221097780
- Hill QA, Stamps R, Massey E, et al. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. 2017;177:208–220. doi:10.1111/bjh.14654
- Baratta L, Golluscio V, Delfino M. Favorable response to cyclosporin A in a patient with steroid resistant autoimmune haemolytic anaemia. Recenti Prog Med. 2004;95:100. PMID:15074296.
- Sarper N, Caki Kilic S, Zengin E, et al. Management of autoimmune hemolytic anemia in children and adolescents: A single center experience. Turk J Haematol. 2011;28:198–205. doi:10.5152/tjh.2011.54
- Lauro A, Stanzani M, Finelli C, et al. Alemtuzumab plus cyclosporine treatment of the autoimmune hemolytic anemia in an adult bowel transplant. Case Rep Transplant. 2014;2014:262953. doi:10.1155/2014/262953