
ABSTRACT
Background: T-prolymphocytic leukemia (T-PLL) is an aggressive hematologic malignancy. A portion of patients can be cured with alemtuzumab induction followed by allogeneic hematopoietic stem cell transplant, but patients who relapse after transplant have a poor prognosis, and there is no standard of care.
Methods: We report a case of a 64-year-old man with relapsed JAK3-mutant T-PLL following allogeneic transplant who was treated with ruxolitinib and venetoclax.
Results: Treatment with ruxolitinib and venetoclax resulted in a partial response including stabilization of the peripheral lymphocyte count, improvement in thrombocytopenia, decrease in splenomegaly, and a numerical reduction in the percentage of bone marrow involved by T-PLL. The combination was well tolerated with the exception of neutropenic infections.
Conclusion: This case adds to the growing body of literature supporting venetoclax and rituximab as a viable treatment option for relapsed/refractory T-PLL with JAK-STAT alterations.
Introduction
T-prolymphocytic leukemia (T-PLL) is an aggressive T-cell malignancy characterized by rapid proliferation of mature post-thymic T-cells. Treatment with the anti-CD52 monoclonal antibody, alemtuzumab, has improved clinical outcomes, but responses are short-lived, and patients will inevitably relapse [Citation1]. Long-term survival is possible with the use of allogeneic hematopoietic stem cell transplant, and up to a third of patients can survive free of disease four years post-transplant [Citation2–4]. The prognosis of patients with relapsed disease following allogeneic stem cell transplant is poor, and there is no established standard of care. Re-treatment with alemtuzumab with or without a purine analog is often considered, but outcomes remain unsatisfactory [Citation5].
An understanding of the molecular mechanisms involved in T-PLL leukemogenesis may result in rational approaches to treatment selection [Citation6,Citation7]. Recurrent genetic and molecular abnormalities are common in T-PLL including rearrangements involving the T-cell leukemia 1 (TCL1) protooncogene on chromosome 14, deletions or inactivating mutations of ATM and TP53, and activating mutations in the IL2RG-JAK1-JAK3-STAT5B axis, among others [Citation8–12]. JAK1 and JAK3 mutations occur in 8% and 30% and represent an attractive therapeutic target due to the clinical availability of JAK inhibitors [Citation10]. Indeed, case reports have demonstrated responses to JAK inhibition in T-PLL [Citation12–14], although the presence of a JAK1 or 3 mutation does not appear to strongly correlate to drug sensitivity ex vivo [Citation6]. Additionally, in vitro drug sensitivity testing has identified the BCL2 inhibitor, venetoclax, as a possible active agent in T-PLL, and clinical responses in humans have been reported, although responses to venetoclax as monotherapy have been inadequate [Citation15,Citation16].
We report a case of relapsed T-PLL with JAK3 mutations that achieved partial response with the addition of ruxolitinib to venetoclax after disease progression on venetoclax monotherapy.
Case presentation
A 64-year-old man with no significant medical comorbidities was diagnosed with T-prolymphocytic leukemia after leukocytosis was incidentally discovered on a complete blood count. After a six-month period of observation, he developed rapidly progressive leukocytosis and underwent treatment with the anti-CD52 monoclonal antibody alemtuzumab intravenously. He attained complete response (CR) and underwent consolidative therapy with a 10/10 matched unrelated peripheral blood stem cell transplant with Flu/Mel/TBI reduced-intensity conditioning (melphalan 100 mg/m2 day −6, fludarabine 40 mg/m2/day on days −5 to −2, total body irradiation 200 cGy day −1). He received graft-versus-host (GVH) prophylaxis with cyclophosphamide (50 mg/kg day +3 and +4), tacrolimus, and mycophenolate. Day +30 and day +100 bone marrow evaluations demonstrated ongoing CR and >98% donor chimerism.
Routine bone marrow biopsy one-year post-transplant showed small lymphoid aggregates consisting of CD4+/CD8- T-cells positive for CD123 and TCL1 occupying 10% of cellular elements, consistent with recurrent T-PLL. Donor lymphocyte infusion (DLI) was performed on two occasions with escalating CD34 cell doses but was unsuccessful in recapturing response.
Three months after the second DLI, the patient developed worsening leukocytosis (WBC 28,200/µL, lymphocytes 16,600/µL). He reported abdominal discomfort, and his spleen was palpable below the costal margin. Bone marrow evaluation demonstrated 50% T-PLL involvement. 34% of T cells were double positive for CD4 and CD8. Fluorescence in-situ hybridization (FISH) analysis demonstrated a rearrangement involving the TCL1a locus (14q32). Next generation sequencing demonstrated mutations in JAK3 (M511I, variant frequency 20.2%; V674A, variant frequency 11.6%). Alterations in STAT5B, ATM, and TP53 were not observed. Treatment with venetoclax (ramp up from 50 mg to 400 mg over 4 days) was initiated (), but the leukocytosis worsened, and cytoreductive therapy with hydroxyurea was added. After three weeks, venetoclax and hydroxyurea were discontinued and the patient was admitted to the hospital for salvage therapy with cladribine and alemtuzumab (cladribine 5 mg/m2 IV day 1–5; alemtuzumab 10 mg IV x1, followed by 30 mg IV 3x/week). Although treatment resulted in a significant reduction in the peripheral white blood count, bone marrow biopsy on day 25 demonstrated 70% involvement by T-PLL. In addition, treatment was complicated by CMV reactivation (day 11) treated with letermovir, and severe oral HSV requiring admission (day 30). Alemtuzumab was discontinued due to safety concerns and for lack of efficacy.
Figure 1. Annotated treatment summary.
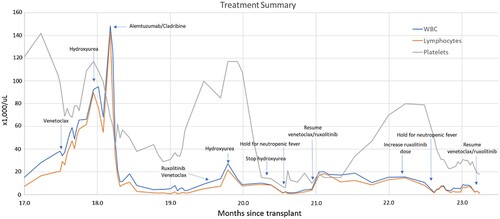
Following recovery, venetoclax (100 mg daily with dose escalation to 600 mg daily over 4 days) in combination with the JAK2 inhibitor, ruxolitinib (10 mg BID) was initiated. At the time of therapy initiation, WBC was 14,000/µL, lymphocyte count was 7,300/µL, hemoglobin was 7.3 g/dL (normal 13–17 g/dL), platelets were 85,000/µL (normal 150–450,000/µL), alkaline phosphatase was 447units/L (normal 45–115 units/L), and aspartate aminotransferase (AST) was 86 units/L (normal 5–34 units/L). Due to initial increase in WBC (27,200/µL) on day 4, the venetoclax dose was increased to 800 mg daily, ruxolitinib was increased to 20 mg BID, and hydroxyurea 1000 mg TID was added to treatment. Hydroxyurea was tapered off by day 22. On day 26, the patient was admitted for neutropenic fever attributed to possible pneumonia, and venetoclax and ruxolitinib were held.
Venetoclax 800 mg daily and ruxolitinib 10 mg BID were resumed on day 39. After resuming venetoclax and ruxolitinib, the patient achieved a partial response to treatment according to T-PLL International Study Group (ISG) consensus criteria [Citation17] with reduction of circulating lymphocyte count from 18,400 to 8,200/µL, improvement in platelets from 15,000 to 80,000/µL, and resolution of and splenomegaly (, months 21–22.5). The patient was profoundly neutropenic at the start of treatment, and ANC transiently improved to greater than 1,000/µL starting day 50. His abdominal discomfort and appetite improved, and constitutional symptoms resolved.
Peripheral blood cytometry showed persistence of T-PLL, and ruxolitinib was increased to 20 mg BID on day 80. On day 91, the patient was admitted with neutropenic fever due to respiratory syncytial virus and treatment was again held. Bone marrow biopsy on day 99 demonstrated 60% involvement by T-PLL, overall with decreased disease burden compared to 70% T-PLL marrow involvement post alemtuzumab. Treatment with ruxolitinib 5 mg BID was resumed on day 108, and venetoclax 400 mg daily was resumed on day 109. The patient remained on treatment while under the care of the authors.
Discussion
Patients with relapsed T-PLL following allogeneic hematopoietic stem cell transplant have a poor prognosis, and no standard treatment options exist. We report a case of a patient with T-PLL who experienced several months of clinical benefit with a combination of venetoclax and ruxolitinib following relapse after allogeneic transplant. Treatment resulted in reduction in spleen size, improvement in liver function abnormalities, improvement in platelet and neutrophil count, and slight decrease in the amount of marrow involvement by T-PLL. Criteria for partial response were met according to T-PLL ISG criteria. Treatment was complicated by neutropenic infections requiring hospitalization, although it should be noted that the patient was severely neutropenic prior to the start of treatment.
Following relapse, treatment with venetoclax monotherapy was unsuccessful in reducing a rapidly rising lymphocyte count. Salvage treatment with alemtuzumab and cladribine was able to reduce the circulating lymphocyte count but did not reduce the disease burden within the bone marrow. The addition of ruxolitinib to venetoclax was able to maintain a stable circulating lymphocyte count for a period of several months with associated clinical benefit.
JAK inhibitors are an attractive therapeutic option in T-PLL due to both the frequency of activating mutations in the JAK-STAT pathway and the clinical availability of several FDA-approved JAK inhibitors. Ruxolitinib was chosen for this patient because of our familiarity with its use in other hematologic disorders and our ability to procure the medicine off-label. However, ruxolitinib has relatively low affinity to JAK3 (IC50 3.3 nM for JAK1, 2.8 nM for JAK2, and 323 nM for JAK3) [Citation18]. In an ex vivo study, T-PLL samples both with and without JAK1 and JAK3 mutations were sensitive to ruxolitinib [Citation6]. Degryse et al. demonstrated that expression of JAK1 is essential for JAK3 mutant signaling through STAT5B providing a rationale for the activity of ruxolitinib despite its low affinity to JAK3 [Citation19]. Tofacitinib, a pan-JAK inhibitor approved for several autoimmune disorders, has a higher affinity for JAK3 than ruxolitinib (IC50, 1 nM) [Citation18], and may also be a suitable candidate for future research in T-PLL; responses in humans have been reported [Citation13]. In contrast, pacritinib has a high IC50 for JAK1 [Citation20] and may not be an ideal candidate for study in T-PLL.
Since ruxolitinib was added after failure of venetoclax, it is not known whether the combination functioned in a synergistic fashion or whether benefit was primarily due to ruxolitinib alone. Potential synergy between ruxolitinib and venetoclax has been demonstrated in various xenograph models. For instance, Karjalainen et al. showed that ruxolitinib restored sensitivity of acute myeloid leukemia (AML) to venetoclax by counteracting tumor stroma-induced resistance [Citation21]. Additionally, in a T-acute lymphoblastic leukemia (T-ALL) model, ruxolitinib and venetoclax demonstrated potential therapeutic benefit via simultaneous inhibition of IL7-Rα ligand-independent signaling and BCL2, which functions as downstream mediator of the IL7R survival mechanism [Citation22].
In human T-PLL cells, Herbaux et al. demonstrated using a BH3 profiling assay that T-PLL cells are relatively unprimed for apoptosis compared to CLL cells, which potentially explains the lack of efficacy of venetoclax as a single agent [Citation23]. Pretreatment with ruxolitinib selectively increased priming for apoptosis in a BCL-2 dependent manner, and the combination of ruxolitinib and venetoclax was highly active in cells with JAK/STAT pathway alterations.
Along with his preclinical work, Herbaux reported his experience treating two patients with venetoclax and ruxolitinib [Citation23]; one patient achieved partial response and a venetoclax-pretreated patient achieved table disease. He later reported a series of 15 patients treated with the venetoclax/ruxolitinib combination [Citation24]. In patients with JAK/STAT pathway mutations, 10 of 12 patients had a partial response. Two-thirds of patients experienced transient lymphocytosis shortly after starting ruxolitinib, which matches our experience.
In summary, outcomes in relapsed T-PLL following allogeneic hematopoietic stem cell transplant are poor. Venetoclax in combination with ruxolitinib resulted in a several month period of clinical benefit. The combination was well tolerated with the exception of expected cytopenias and resulting neutropenic infections. Our report adds to a small but growing body of literature supporting the use of venetoclax and ruxolitinib in refractory T-PLL. Further studies evaluating the clinical utility of this combination are warranted.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Dearden CE, Matutes E, Cazin B, et al. High remission rate in T-cell prolymphocytic leukemia with CAMPATH-1H. Blood. 2001;98:1721–1726. doi:10.1182/blood.V98.6.1721
- Wiktor-Jedrzejczak W, Drozd-Sokolowska J, Eikema DJ, et al. EBMT prospective observational study on allogeneic hematopoietic stem cell transplantation in T-prolymphocytic leukemia (T-PLL). Bone Marrow Transplant. 2019 Sep;54(9):1391–1398. doi:10.1038/s41409-019-0448-x
- Dholaria BR, Ayala E, Sokol L, et al. Allogeneic hematopoietic cell transplantation in T-cell prolymphocytic leukemia: a single-center experience. Leuk Res. 2018;67:1–5. doi:10.1016/j.leukres.2018.01.009
- Krishnan B, Else M, Tjonnfjord G, et al. Stem cell transplantation after alemtuzumab in T-cell prolymphocytic leukaemia results in longer survival than after alemtuzumab alone: a multicentre retrospective study. Br J Haematol. 2010 Jun;149(6):907–910. doi:10.1111/j.1365-2141.2010.08134.x
- Khot A. Where do we currently stand with T-cell prolymphocytic leukemia? Leuk Lymphoma. 2019;60:563–565. doi:10.1080/10428194.2018.1551543
- Andersson EI, Putzer S, Yadav B, et al. Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and mutation profiling. Leukemia. 2018 Mar;32(3):774–787. doi:10.1038/leu.2017.252
- Shi Z, Yu J, Shao H, et al. Exploring the molecular pathogenesis associated with T-cell prolymphocytic leukemia based on a comprehensive bioinformatics analysis. Oncol Lett. 2018 Jul;16(1):301–307. doi:10.3892/ol.2018.8615
- Lopez C, Bergmann A, Paul U, et al. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br J Haematol. 2016 Apr;173(2):265–273. doi:10.1111/bjh.13952
- Stengel A, Kern W, Zenger M, et al. Genetic characterization of T-PLL reveals two major biologic subgroups and JAK3 mutations as prognostic marker. Genes Chromosomes Cancer. 2016 Jan;55(1):82–94. doi:10.1002/gcc.22313
- Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124:1460–1472. doi:10.1182/blood-2014-03-559542
- Bergmann AK, Schneppenheim S, Seifert M, et al. Recurrent mutation of JAK3 in T-cell prolymphocytic leukemia. Genes Chromosomes Cancer. 2014 Apr;53(4):309–316. doi:10.1002/gcc.22141
- Greenplate A, Wang K, Tripathi R, et al. Genomic profiling of T-cell neoplasms reveals frequent JAK1 and JAK3 mutations with clonal evasion from targeted therapies. JCO Precis Oncol. 2018;2:1–16. doi:10.1200/PO.17.00019
- Li G, Waite E, Wolfson J. T-cell prolymphocytic leukemia in an adolescent with ataxia-telangiectasia: novel approach with a JAK3 inhibitor (tofacitinib). Blood Adv. 2017 Dec 18;1(27):2724–2728. doi:10.1182/bloodadvances.2017010470
- Wei M, Koshy N, van Besien K, et al. Refractory T-cell prolymphocytic leukemia with JAK3 mutation: in vitro and clinical synergy of tofacitinib and ruxolitinib. Blood. 2015;126:5486. doi:10.1182/blood.V126.23.5486.5486
- Boidol B, Kornauth C, van der Kouwe E, et al. First-in-human response of BCL-2 inhibitor venetoclax in T-cell prolymphocytic leukemia. Blood. 2017 Dec 7;130(23):2499–2503. doi:10.1182/blood-2017-05-785683
- Hampel P, Parikh S, Call T, et al. Venetoclax treatment of patients with relapsed T-cell prolymphocytic leukemia. Blood Cancer J. 2021 Mar;11(3):47. doi:10.1038/s41408-021-00443-1
- Staber P, Herling M, Bellido M, et al. Consensus criteria for diagnosis, staging, and treatment response assessment of T-cell prolymphocytic leukemia. Blood. 2019;134(14):1132–1143. doi:10.1182/blood.2019000402
- Furumoto Y, Gadina M. The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. BioDrugs. 2013 Oct;27(5):431–438. doi:10.1007/s40259-013-0040-7
- Degryse S, de Bock C, Cox L, et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood. 2014;124:3092–3100. doi:10.1182/blood-2014-04-566687
- Duenas-Perez A, Mead A. Clinical potential of pacritinib in the treatment of myelofibrosis. Ther Adv Hematol. 2015 Aug;6(4):186–201. doi:10.1177/2040620715586527
- Karjalainen R, Pemovska T, Popa M, et al. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood. 2017 Aug 10;130(6):789–802. doi:10.1182/blood-2016-02-699363
- Senkevitch E, Li W, Hixon J, et al. Inhibiting Janus Kinase 1 and BCL-2 to treat T cell acute lymphoblastic leukemia with IL7-Rα mutations. Oncotarget. 2018 Apr 27;9(32):22605–22617. doi:10.18632/oncotarget.25194
- Herbaux C, Kornauth C, Poulain S, et al. BH3 profiling identifies ruxolitinib as a promising partner for venetoclax to treat T-cell prolymphocytic leukemia. Blood. 2021;137(25):3495–3506. doi:10.1182/blood.2020007303
- Herbaux C, Poulain S, Roos-Weil D, et al. Preliminary study of ruxolitinib and venetoclax for treatment of patients with T-cell prolymphocytic leukemia refractory to, or ineligible for alemtuzumab. Blood. 2021;138(Supplement 1):1201. doi:10.1182/blood-2021-149228