
ABSTRACT
Background
Transient abnormal myelopoiesis (TAM) is characterized by leukocytosis with increased circulating megakaryoblasts that harbor N-terminal truncating mutations in the GATA1 gene. Approximately 10% of affected patients experience early death.
Observations
A 2-month-old boy with Down syndrome was diagnosed with TAM and followed without treatment. Although the blasts in the peripheral blood disappeared, liver failure progressed. A pathological examination revealed liver fibrosis, and double-immunostaining for full-length GATA1 and CD42b identified megakaryocytes with a GATA1 mutation.
Conclusions
This simple and cost-effective method can be applied in routine practice to detect TAM blasts during assessment in a TAM crisis.
Introduction
Transient abnormal myelopoiesis (TAM) is a hematological disorder that exclusively affects approximately 10% of neonates with Down syndrome (DS) and is characterized by transient appearance of immature megakaryoblasts in the peripheral blood, liver, spleen, and other organs.[Citation1] Most cases have a favorable clinical course with spontaneous remission, but approximately 10% of patients die from multiorgan failure followed by severe hepatic fibrosis and cardiopulmonary failure at an early age.[Citation1] Recent studies on TAM identified hyperleukocytosis, ascites, prematurity, cholestasis, liver fibrosis, renal dysfunction, and disseminated intravascular coagulation (DIC) as risk factors for early death.[Citation2–5] Detection of these risk factors and provision of early treatments are essential. However, the symptoms of patients with TAM vary, and even severe cases can present normal blood cell counts and low blast counts. Therefore, risk stratification and determination of necessary treatments can be challenging.
Clinical diagnosis of TAM is based detection of blasts that exhibit morphological and immunological features of the megakaryocytic lineage in peripheral blood smears. Bone marrow examination is not generally useful for the diagnosis of TAM, and the most sensitive diagnostic marker is GATA-binding factor 1 (GATA1) mutations.[Citation6] GATA1 is an essential transcription factor for erythropoiesis and megakaryopoiesis.[Citation7] The GATA1 mutations are typically located in exon 2 and surrounding gene regions that contain the translation start site, resulting in the formation of N-terminal-truncated short GATA1 protein (GATA1s) and the absence of full-length GATA1 protein (GATA1f). Recently, Lee and colleagues demonstrated the potential of new immunohistochemical staining options for detection of TAM blasts using a commercially available monoclonal antibody (mAb) against GATA1f.[Citation7,Citation8] In one of their studies, they showed that the bone marrow megakaryocytes of all 15 examined patients with myeloid proliferative disorder-related DS (MPD-DS) lacked GATA1f expression.[Citation7] As targeted sequencing is the most common method for detection of GATA1 mutations, this new pathological method could provide a simple and valuable alternative for diagnostic examination in MPD-DS.
Herein we report the case of an infant with TAM complicated by liver fibrosis but without blasts in the peripheral blood. Our pathological examination involving double-immunostaining for GATA1f and CD42b was able to detect megakaryocytic blasts with a GATA1 mutation in a liver biopsy specimen.
Patient presentation
The patient was a male neonate born by standard vaginal delivery at 39 weeks of gestation with a birth weight of 3,036 g. The patient had no family history of chromosomal or hereditary diseases. At birth, the neonate presented with distinct facial dysmorphism, and karyotype analysis confirmed DS (47, XY, + 21). Laboratory data on day 1 were as follows: white blood cell (WBC) count, 55,000 /μL with 25% blasts; platelet count, 33,000 /μL. Based on these findings, the patient was diagnosed with DS accompanied by TAM. The patient did not have organomegaly, cholestasis, hepatopathy, skin rash, or pericardial and pleural effusions, and was classified as a low-risk group of TAM.[Citation4] The WBC count gradually normalized without treatment, but cholestasis, thrombocytopenia, and anemia progressively worsened at 2 months of age, and the patient was referred to our hospital.
On admission (day 56), a physical examination revealed jaundice, slight fever, and hepatosplenomegaly. Laboratory data were as follows: WBC count, 5,600 /μL with 28% blasts; platelet count, 33,000 /μL; hemoglobin, 13.2 g/dL; alanine transaminase, 332 IU/L; aspartate transaminase, 172 IU/L; total bilirubin, 10.4 mg/dL; direct bilirubin (D-Bil), 7 mg/dL; alkaline phosphatase, 483 IU/L; γ-glutamyl transferase, 60 IU/L; prothrombin time, 14 s; activated partial thromboplastin time, 48.2 s; international normalized ratio, 1.21; ammonia, 82 µg/dL. Liver fibrosis was confirmed by high levels of serum markers: procollagen type III peptide, 349 ng/mL; type IV collagen, 115 ng/mL. A flow cytometric analysis of the peripheral blood revealed co-expression of stem cell markers (CD34 and CD117), myeloid markers (CD33), and platelet glycoproteins (CD41 and CD61), as well as aberrant expression of CD56 and CD7. After obtaining informed consent from the patient’s parents, direct sequencing for GATA1 mutations was performed in a peripheral blood sample. A mutation was detected in exon 2 of the GATA1 gene (c.90_91delAG, p.Val32Phefs*7). Thus, the patient was diagnosed with TAM complicated by severe liver fibrosis, and received treatment with supportive liver therapy and fat-soluble vitamins, but not chemotherapy for TAM. Subsequently, the blasts in the peripheral blood disappeared on day 79. However, hyperbilirubinemia worsened, and repeated blood and platelet transfusions were required (). On day 105, a needle liver biopsy was performed, and prednisone treatment (2 mg/kg) was started, but liver fibrosis progressed. Despite our recommendation for an early liver transplantation, the parents rejected further treatment and strongly desired for the patient to receive medical treatment at home. Finally, the patient died on day 166.
Figure 1 . Clinical course of the patient and changes in bilirubin, white blood cell count, and blast ratio in the peripheral blood. WBC: white blood cell; BMA: bone marrow aspiration; T-Bil: total bilirubin; D-Bil: direct bilirubin.
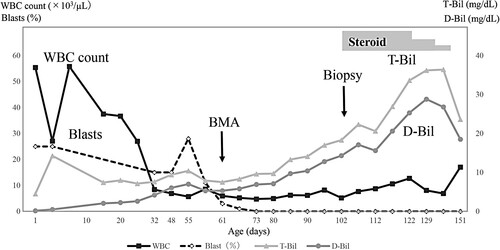
A histological examination of the liver biopsy specimen showed bridging fibrosis at the margins of the hepatic lobules, fibrosis around individual hepatocytes, and structural disorder of the hepatic cords, indicating severe hepatic fibrosis. Sporadic formation of erythroblastic islands was also observed, and megakaryocytes appeared, confirming extramedullary hematopoiesis (A, B). Of interest, double-immunohistochemical staining for CD42b and GATA1f revealed the presence of megakaryocytes with a GATA1 mutation (A, B). Thus, the hepatic pathology findings showed a moderate-to-severe degree of hepatic fibrosis and megakaryocytes with a GATA1 mutation.
Figure 2 . Histopathological examination of the liver biopsy specimen. (A) Azan staining reveals liver cord atrophy with fibrosis around individual hepatocytes, indicating severe hepatic fibrosis. (B) Hematoxylin and eosin staining shows erythroblastic islands and sporadic megakaryocytes in the liver tissue.
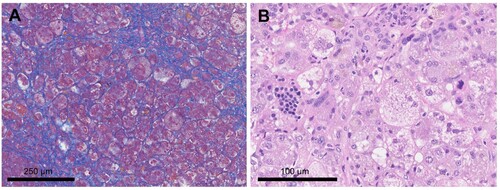
Figure 3 . Immunohistochemical examination of the liver biopsy specimen and positive control. (A) Double-immunostaining shows that GATA1 is negative in the nuclei of CD42b-positive megakaryocytes (arrow), but positive in the nuclei of erythroblasts found in erythroblastic islands within the liver (arrowhead). (B) Double-immunostaining of the normal bone marrow, as a positive control, shows that GATA1 is positive in the nuclei of CD42b-positive megakaryocytes as well as in the nuclei of background erythroblasts.
Discussion
We report a fatal pediatric case with peripheral blasts, anemia, thrombocytopenia, jaundice, and hepatosplenomegaly diagnosed as low-risk TAM. Although the TAM blasts disappeared, liver failure progressed, and the patient was diagnosed with liver fibrosis consisting of megakaryocytic blasts with a GATA1 mutation by pathological findings.
The GATA1 gene has six exons, and its mutations are concentrated in and around exon 2, which contains the translation start site. In TAM, nonsense mutations in exons 2 and 3 prevent the production of GATA1f and result in the production of N-terminal-truncated GATA1s. This feature can be determined by observation of GATA1 mutant megakaryocytes on immunohistochemical staining using a mAb raised against a 13-amino-acid peptide that is present in GATA1f but absent in GATA1s.[Citation7,Citation8] While immunohistochemical staining with the anti-GATA1f mAb was positive in the nuclei of megakaryocytes and erythroblasts in the bone marrow of both normal individuals and patients with other childhood myeloid neoplasms without DS, the staining was negative in the nuclei of megakaryocytes in patients with MPD-DS, including TAM, due to the presence of GATA1s.[Citation7,Citation8]
In the present case, double-immunohistochemical staining was performed on a liver biopsy specimen using an anti-CD42b antibody (MM2/174; Leica) as a megakaryocyte marker and an anti-GATA1f mAb (D52H6; Cell Signaling Technology). CD42b-positive megakaryocytes in the liver biopsy specimen did generally not contain GATA1f, indicating that TAM-derived atypical megakaryocytes with GATA1s were present in the liver where TAM-associated liver fibrosis had occurred (C). As no previous pathological reports have demonstrated the existence of megakaryocytes with a GATA1 mutation in the liver of patients with TAM-associated hepatic fibrosis, this is the first report of such a case.
Severe liver disease is one of the consistent and critical risk factors for early death in patients with TAM. Massey et al.[Citation2] demonstrated that transaminase levels were significantly higher in infants with TAM who died early than in survivors, and all infants with TAM who died early had evidence of liver failure and DIC. Klusmann et al.[Citation3] reported that high WBC count (>100,000 /µL), preterm delivery, bleeding diatheses, and loss of spontaneous remission were significantly associated with early death, and all seven patients with hepatic fibrosis died. In Japan, Muramatsu et al.[Citation4] reported that early gestational age (EGA), WBC count, and D-Bil were independent predictors of poor outcomes, and devised a simple risk stratification system that combined EGA and WBC count to predict poor outcomes.
Meanwhile, Park et al.[Citation9] indicated that all DS patients could develop liver disease, regardless of the risk of early death, during the clinical course of TAM. In their report, the patients with liver disease had elevated D-Bil levels that further increased even though blasts in the peripheral blood had decreased or disappeared. Therefore, there may be no correlation between the percentage of peripheral blood blasts and the development of liver disease. Careful evaluation of liver function and early detection of liver disease development are recommended for all patients with TAM.
Although the pathogenesis of liver fibrosis in patients with TAM is unknown, a previous study revealed that megakaryocytic blasts in TAM invaded the liver tissue and induced fibrotic changes through the production of transforming growth factor-β1 and platelet derived growth factor subunit[Citation10]. These findings suggest that residual blasts in the liver may continue to release cytokines that cause fibrotic changes and lead to the progression of liver fibrosis, regardless of the presence of peripheral blood blasts. In the present patient, a new pathological assessment method involving double-immunostaining for GATA1f and CD42b in a liver biopsy specimen was useful for evaluation of liver fibrosis and detection of TAM blasts. Further studies are required to investigate the potential of the method for prediction of liver fibrosis and prognosis.
In conclusion, we have reported a patient who was initially diagnosed with low-risk TAM but gradually developed liver fibrosis despite the absence of peripheral blood blasts. We performed a liver biopsy for evaluation of liver fibrosis and confirmed the existence of megakaryocytes with a GATA1 mutation in the liver by double-immunostaining with anti-GATA1f and anti-CD42b antibodies. This method using commercially available antibodies is simple and cost-effective, and can be incorporated into routine pathologic practice for detection of megakaryocytic blasts in TAM patients.
Data access statement
The datasets and analysis performed during the current study are available from the corresponding author upon reasonable request.
Ethical statement
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and approved by the ethics committee or institutional review board of each institution (Central ethics committee of Gifu University).
Consent to participate
The patient’s legal guardians gave their informed consent to be included.
Consent for publication
The patient’s legal guardians provided written informed consent for the publication of this study.
Acknowledgements
The authors thank Alison Sherwin, PhD, from Edanz (https://jp.edanz.com.ac) for editing a draft of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Watanabe K. Recent advances in the understanding of transient abnormal myelopoiesis in Down syndrome. Pediatr Int. 2019 Mar;61(3):222–229. doi:10.1111/ped.13776. Epub 2019 Mar 4. PMID: 30593694.
- Massey GV, Zipursky A, Chang MN, et al. Weinstein HJ; children’s oncology group (COG). A prospective study of the natural history of transient leukemia (TL) in neonates with Down syndrome (DS): children’s oncology group (COG) study POG-9481. Blood. 2006 Jun 15;107(12):4606–4613. doi:10.1182/blood-2005-06-2448. Epub 2006 Feb 9. PMID: 16469874.
- Klusmann JH, Creutzig U, Zimmermann M, et al. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood. 2008 Mar 15;111(6):2991–2998. doi:10.1182/blood-2007-10-118810. Epub 2008 Jan 8. PMID: 18182574; PMCID: PMC2265448.
- Muramatsu H, Kato K, Watanabe N, et al. Risk factors for early death in neonates with Down syndrome and transient leukaemia. Br J Haematol. 2008 Aug;142(4):610–615. doi:10.1111/j.1365-2141.2008.07231.x. Epub 2008 May 28. PMID: 18510680.
- Gamis AS, Alonzo TA, Gerbing RB, et al. Natural history of transient myeloproliferative disorder clinically diagnosed in Down syndrome neonates: a report from the children’s oncology group study A2971. Blood. 2011 Dec 22;118(26):6752–6759; quiz 6996. doi:10.1182/blood-2011-04-350017. Epub 2011 Aug 17. PMID: 21849481; PMCID: PMC3245202.
- Tunstall O, Bhatnagar N, James B, et al. Wright M; British society for haematology. guidelines for the investigation and management of transient leukaemia of Down syndrome. Br J Haematol. 2018 Jul;182(2):200–211. doi:10.1111/bjh.15390. Epub 2018 Jun 19. PMID: 29916557.
- Lee WY, Weinberg OK, Evans AG, et al. Loss of full-length GATA1 expression in megakaryocytes is a sensitive and specific immunohistochemical marker for the diagnosis of myeloid proliferative disorder related to Down syndrome. Am J Clin Pathol. 2018 Mar 7;149(4):300–309. doi:10.1093/ajcp/aqy001. PMID: 29481579; PMCID: PMC5848381.
- Lee WY, Weinberg OK, Pinkus GS. GATA1 is a sensitive and specific nuclear marker for erythroid and megakaryocytic lineages. Am J Clin Pathol. 2017 Apr 1;147(4):420–426. doi:10.1093/ajcp/aqx018. PMID: 28340113; PMCID: PMC5848378.
- Park MJ, Sotomatsu M, Ohki K, et al. Liver disease is frequently observed in Down syndrome patients with transient abnormal myelopoiesis. Int J Hematol. 2014 Feb;99(2):154–161. doi:10.1007/s12185-013-1487-5. Epub 2013 Dec 14. PMID: 24338744.
- Hattori H, Matsuzaki A, Suminoe A, et al. High expression of platelet-derived growth factor and transforming growth factor-beta 1 in blast cells from patients with Down syndrome suffering from transient myeloproliferative disorder and organ fibrosis. Br J Haematol. 2001 Nov;115(2):472–475. doi:10.1046/j.1365-2141.2001.03093.x. PMID: 11703351.