
ABSTRACT
Objectives
Cyclic thrombocytopenia (CTP) is a rare blood disorder characterized by periodic fluctuations in platelet counts. CTP usually appears in pre-menopausal women, and these fluctuations of platelets are in phase with the menstrual cycle. CTP is a heterogeneous disease, and the pathogenic mechanism is still unclear. Therefore, it harbors great significance for exploring the association of fluctuations in platelet counts with hormonal-cycle.
Materials
Firstly, we washed human platelets from healthy volunteers following the Declaration of Helsinki. Flow cytometer was employed to measure the mitochondrial inner transmembrane potential (ΔΨm) depolarization, PS exposure, P-selectin expression, and GPIIb/IIIa activation in platelets. In addition, western blot detected the related protein expression. The corresponding assay kit measured the caspase-3 and PDE3A activity. Finally, flow cytometry determined mouse platelets labeled with calcein.
Results
We find a reverse relationship between the platelet count and serum estradiol (E2) level in a CTP patient. We demonstrated that E2 induces platelet apoptosis in vitro and platelet clearance in vivo. We further discovered that E2 activates phosphodiesterase 3A, which inhibits protein kinase A (PKA), leading to PKA-mediated platelet apoptosis. Activation of PKA protected platelets from E2-induced thrombocytopenia and elevated the number of mice circulatory platelets.
Conclusions
We find that E2 induces platelet apoptosis and clearance through PDE3A-mediated PKA inhibition. Activation of PKA rescues E2-induced thrombocytopenia in mice. Thus, our study reveals a pathogenesis of E2-related CTP and suggests promising therapeutic strategies for the disease.
Introduction
Cyclic thrombocytopenia (CTP) is a rare blood disorder characterized by periodic fluctuations in platelet counts [Citation1–3]. Each cycle usually lasts three to five weeks [Citation2,Citation3]. The median nadir and peak platelet counts in this reported cases were around 10 × 109/L and 330 × 109/L [Citation2,Citation3]. Therefore, periods of life-threatening thrombocytopenia or rebound increased the risk of thrombosis found in some CTP patients [Citation1–4]. Due to the symptom of low platelet counts, CTP is often misdiagnosed as idiopathic thrombocytopenic purpura (ITP). However, distinct from ITP, CTP generally responds poorly to most treatments used successfully in ITP, such as corticosteroids, splenectomy, and intravenous immunoglobulin [Citation1,Citation2,Citation5]. The characteristic of CTP is a heterogeneous disorder. Although autoimmune platelet destruction [Citation6], megakaryocytic hypoplasia or aplasia [Citation7], infectious [Citation8], and hormonal [Citation9–11] etiology were thought to be potential mechanisms, the pathogenesis of CTP is still unclear [Citation1,Citation2]. Interestingly, CTP was more commonly found in females [Citation2], a predominance disproportionately in favor of pre-menopausal women, and these fluctuations of platelets are in phase with the menstrual cycle [Citation2,Citation9,Citation12–14], suggesting the correlation of hormonal cycle may be associated with fluctuations in platelet counts.
Platelet apoptosis has been demonstrated to determine platelet life span under physiological and pathological conditions [Citation15,Citation16]. We reported recently that PKA inhibition results in dephosphorylation of Bad at Ser155, which sequesters pro-survival BCL-XL in mitochondria leading to apoptosis [Citation15]. Therefore, activation of PKA prevented platelets from apoptosis induced by various pathophysiological stimulations [Citation15]. Estrogen and its related steroid hormones could induce apoptosis of Chinese hamster ovary cells (CHO) cells [Citation17,Citation18] and esophageal adenocarcinoma cells [Citation19]. However, it is still unclear whether estrogen is involved in platelet apoptosis.
To elucidate the possible molecular mechanism of E2 in CTP, we investigated the biological role of E2 in platelet apoptosis. Here, we described a female CTP patient whose platelet fluctuation is in phase with her menstrual cycle, and there is a reverse relationship between the platelet counts and serum E2 levels. We further find that E2 induces platelet apoptosis and clearance through PDE3A-mediated PKA inhibition in an estrogen receptor-independent manner. Activation of PKA rescues E2-induced thrombocytopenia in mice. Thus, our study reveals a pathogenesis of E2-related CTP and suggests promising therapeutic strategies for the disease.
Materials and methods
Animals
C57BL/6 wild-type (WT) mice were purchased from JOINN Laboratories (Beijing, China). Mice were six to eight weeks old, and experiments only used female mice. All animal experiments complied with the Guide of the Care and Use of Laboratory, published by the US National Institutes of Health (NIH Publication number 85-23, revised 1996), and were approved by the ethics committee of the First Affiliated Hospital of Soochow University.
Materials and methods
Details of antibodies and reagents are shown in Supplemental Information.
Methods
The female CTP patient’s hormone levels in serum were detected and analyzed by the Roche Cobas e801 electro-chemiluminescence immunoassay system (Roche Diagnostics, Mannheim, Germany). Preparation of washed human platelets from healthy volunteers following the Declaration of Helsinki [Citation20]. Platelets were prepared by the previous method [Citation15,Citation16]. Preparation of protein samples from washed human platelets as the previous method [Citation15,Citation16]. Mitochondrial inner transmembrane potential (ΔΨm) depolarization, PS exposure, P-selectin expression, and GPIIb/IIIa activation in human platelets were measured by a flow cytometer prepared by the previous method [Citation15,Citation16]. Mouse platelets were labeled with calcein and were determined by flow cytometry according to the previous method [Citation15,Citation16]. Protein expression was detected through specific antibodies by western blot. The caspase-3 activity assay follows the manufacturer’s protocol (C1116, Beyotime, China). The PDE3A activity assay follows the manufacturer’s protocol (Abcam). Platelet clearance in vivo was prepared by the previous method [Citation15,Citation16]. Details of these methods are available in the Supplementary Information.
Statistical analysis
All data are expressed as mean ± SD. Numeric data were analyzed using one-way (for a single variant) or two-way (for multiple variants) ANOVA. Two groups were compared by the two-tailed Student’s t-test. The significance of data was assessed using GraphPad Prism 7 software. Differences were considered significant at P < 0.05. The levels of significant difference are indicated as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
A female CTP patient whose platelet counts are reverse associated with serum E2 levels
A 47-year-old female was initially diagnosed as having ITP at the age of 35 years. During the early stages, the patient was hospitalized six times due to repeatedly scattered petechial hemorrhages in her double lower limbs, spontaneous teeth bleeding, and severe thrombocytopenia. At the worst of the disease, pelvic entorrhagia ever appeared and the platelets dropped to 1 × 109/L. After about three years, it appeared that the platelet count fluctuates regularly with the menstrual cycle, usually lowest at about 10 days before each menstruation, then gradually returns to normal at two to three days before menstruation. During this period, the patient had received some treatments such as high-dose dexamethasone, intravenous immunoglobulin, cyclosporine, and traditional Chinese medicine, but almost no response was observed, and cyclic thrombocytopenia always existed. Based on these initial observations, we counted her circulating platelets regularly for six months, and the data show that her platelet counts indeed fluctuated along with her menstruation which one cycle is about 28 days, and her nadir platelet count was 1–26 × 109/L, peak platelet count was 200–300 × 109/L. We further detected serum menses-related hormone levels in this patient. Her nadir platelet count comes with a maximum value of E2, while her peak platelet count comes with a relatively low E2 ((A)). The occurrence of progesterone was similar to that of E2 ((B)). In contrast, there is no marked correlation between the number of platelets with the levels of other hormones ((C–G)). These data suggest that E2 or progesterone might be involved in modulating platelet counts.
Figure 1. A reverse relationship between platelet counts and levels of serum E2 in a CTP patient. (A–G) Fluctuations of platelet (PLT) counts and serum levels of estradiol (E2) (A), progesterone (P4) (B), insulin (InS) (C), prolactin (PRL) (D), follicle-stimulating hormone (FSH) (E), luteinizing hormone (LH) (F), and testosterone (TTE) (G) of the case.
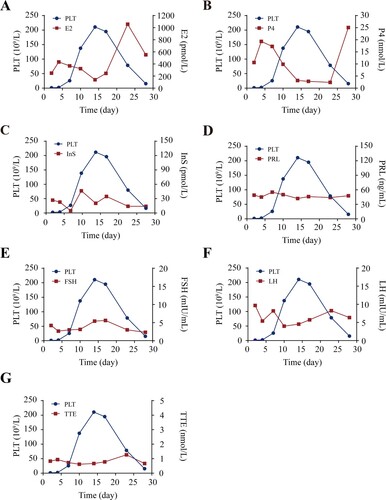
E2 induces platelets apoptosis in vitro
Estrogen-related steroid hormones were reported to induce apoptosis in Hela [Citation17] and esophageal adenocarcinoma cells [Citation19]. Recently, we demonstrated that platelet apoptosis determines platelet lifespan and peripheral number [Citation15,Citation16]. Thus, there is the possibility that estrogen-related steroid hormones incur platelet apoptosis leading to thrombocytopenia in CTP patients. To further examine this hypothesis, washed human platelets incubate with E2 in vitro. We found that E2 dose-dependently induced mitochondrial inner transmembrane potential (ΔΨm) depolarization ((A)), which may incur mitochondrial-mediated endogenous programmed apoptosis in platelets [Citation15,Citation21–23]. We further found that E2 elevated the total caspase-3 activity ((B)) and induced caspase-3 cleavage, which indicates activation of caspase-3 ((C)) in platelets. The increased activity of caspase-3 would destroy the plasma membrane, leading to phosphatidylserine (PS) externalization [Citation15,Citation21–23]. Indeed, E2 induced PS exposure on platelets in a dose-dependently manner ((D)). However, E2 did not lead to platelet P-selectin externalization and PAC-1 binding (Supplementary Figure S1(A,B)). These data demonstrate that E2 induces apoptosis but not activation in platelets. In addition, progesterone did not lead to ΔΨm depolarization and PS externalization (Supplementary Figure S2), excluding the role of progesterone in platelet apoptosis. Therefore, these results suggest that E2 induces platelet apoptosis which may contribute to CTP.
Figure 2. E2 induces platelet apoptosis in vitro. (A) Representative flow cytometric figures (left) and quantification (right) of Δψm depolarization of washed human platelets incubated with E2 or vehicle at 37°C for 96 h (mean ± SD, n = 5). *P < 0.05, **P < 0.01, ***P < 0.001 compared with control, one-way ANOVA. (B and C) Caspase-3 activity analysis (B) and western blot analysis of caspase-3 cleavage (C) in washed human platelets incubated with E2 or vehicle at 37°C for 96 h (mean ± SD, n = 5). **P < 0.01, ***P < 0.001 compared with control, one-way ANOVA. (D) Representative flow cytometric figures (left) and quantification (right) of PS externalization of platelets incubated with E2 or vehicle at 37°C for 96 h (mean ± SD, n = 5). *P < 0.05, **P < 0.01, ***P < 0.001 compared with control, one-way ANOVA.
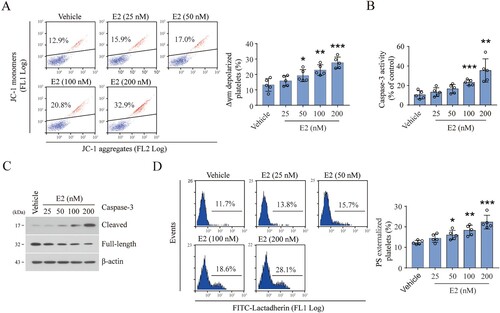
E2 induces platelet apoptosis by inhibiting PKA activity in an estrogen receptor-independent manner
Next, we explored the mechanism for E2-induced platelet apoptosis. Recently, we reported that inhibition of PKA dephosphorylates Bad Ser155, which releases BAK and BAX, resulting in mitochondrial-mediated platelet apoptosis [Citation15]. We found that PKA activity was dose-dependently reduced by E2, as indicated by dephosphorylation of the PKA substrate vasodilator-stimulated phosphoprotein (VASP) ((A)). Bad Ser155 was dose-dependently dephosphorylated by E2, but the other apoptotic proteins, such as BCL-XL, BAX, and BAK, were not varied by E2 ((A)). These data suggest that E2 reduces PKA activity leading to platelet apoptosis. In support of this, adenylate cyclase activator forskolin revised E2-induced dephosphorylation of VASP ((B)). Consequently, E2-induced ΔΨm depolarization and PS exposure were markedly reduced by forskolin ((C,D)). These results suggest that E2 induces platelet apoptosis via inhibition of PKA activity.
Figure 3. E2 induces platelet apoptosis by inhibiting PKA activity. (A and B) Western blot analysis for the levels of indicated proteins of washed human platelets incubated with E2 or vehicle at 37°C for 96 h. Data are representative of five separate experiments. Quantification (right) of phospho-Bad levels (mean ± SD, n = 5). ***P < 0.001 compared with control, one-way ANOVA. (C–E) Washed human platelets were incubated with forskolin (FSK) or vehicle at 37°C for 10 min and further incubated with E2 (100 nM) or vehicle for 96 h (n = 5). Representative western blots (left) and quantification (right) of phosphor-VASP levels (mean ± SD, n = 5). *P < 0.05, ***P < 0.001 compared with control, one-way ANOVA (C). Representative flow cytometric figures (left) and quantification (right) of Δψm depolarization (D), and representative flow cytometric figures (left) and quantification (right) of PS externalization (E) are shown (mean ± SD, n = 5). *P < 0.05, **P < 0.01 compared with control, one-way ANOVA.
E2 was reported to regulate intercellular signals through the E2 receptor (ER) or G protein-coupled estrogen receptor (GPER) in karyocytes [Citation24,Citation25]. We found that the expression of ERβ (the primary receptor of E2 on platelet [Citation26]), phosphorylated ERβ, or GPER did not change after E2 treatment (Supplementary Figure S3(A)). Furthermore, the GPER inhibitor G15 did not rescue E2-induced PKA inhibition (Supplementary Figure S3(B)). These data suggest that the effect of E2 on platelet is not through ER or GPER.
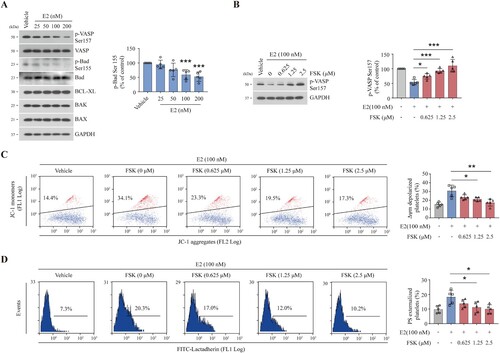
E2 inhibits PKA activity through the regulation of phosphodiesterase
In Hela cells, E2 was found to induce apoptosis by binding directly to phosphodiesterase 3A (PDE3A), an enzyme that is known to hydrolyze intracellular cyclic AMP to reduce PKA activity, and the mechanism was presumed to stabilize Schlafen-12 protein turnover [Citation17]. To investigate the effect of E2 on PDE3A in platelets, we detected the protein level and activity of PDE3A. We found that the PDE3A protein level did not change after E2 treatment (Supplementary Figure S3(C)). However, PDE3A activity was dose-dependently elevated by E2 ((A)), suggesting that E2 activates PED3A in platelets. Since PDE3A is a strong PKA inhibitor, inhibition of PDE3A should enhance PKA activity. Indeed, we found that E2-reduced PKA activity was elevated by PDE3A inhibitors ((B)). Correspondingly, E2-induced apoptotic events were markedly reduced by PDE3A inhibitors ((C,D)). These data suggest that E2 induces platelet apoptosis through PDE3A-mediated PKA inhibition ((E)).
Figure 4. E2 inhibits PKA activity through the regulation of PDE3A. (A) PDE3A activity analysis of washed human platelets incubated with E2 or vehicle at 37°C for 96 h (mean ± SD, n = 5). *P < 0.05, **P < 0.01, compared with control, one-way ANOVA. (B–D) Washed human platelets were incubated with milrinone (2.5 µM) and cilostazol (2.5 µM) or vehicle at 22°C for 20 min and further incubated with E2 (100 nM) or vehicle at 37°C for 96 h (n = 5). Representative western blots (left) and quantification (right) of phosphor-VASP levels (mean ± SD, n = 5). **P < 0.01, ***P < 0.001 compared with control, one-way ANOVA (B). Quantifications of Δψm depolarization (C) and PS externalization (D) are shown (mean ± SD, n = 5). **P < 0.01 compared with control, one-way ANOVA. (E) Schematic representation of proposed mechanism for E2-induced platelet apoptosis. E2 reduces PKA activity by activating PDE3A. The decrease of PKA activity results in dephosphorylation of BAD at Ser155 which leads to mitochondria-dependent apoptosis.
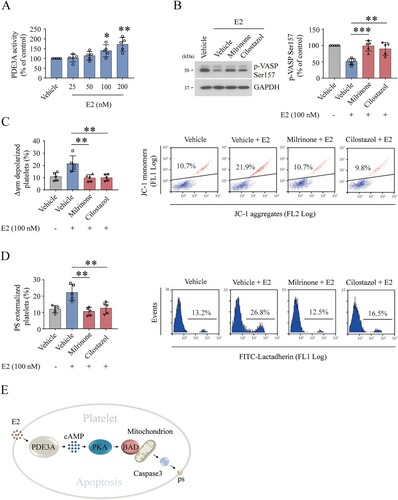
Activation of PKA protects the E2-treated platelets from clearance in mice
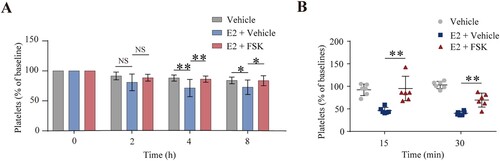
To further verify E2-induced platelet clearance in vivo, we intravenously injected E2 into mice. E2 incurred platelet clearance in mice ((A)). This finding, therefore, demonstrates the correlation of E2 with thrombocytopenia in vivo. In support of this finding, some CTP patients responded to danazol therapy [Citation1]. Now that E2 induces platelet apoptosis through PKA inhibition, activation of PKA may rescue E2-induced platelet clearance. Firstly, to verify this hypothesis, the washed mouse platelets were pre-incubated with forskolin and E2 in vitro, then the pre-treated platelets were injected into mice. We found that forskolin protected platelets from E2-induced clearance ((B)). Secondly, forskolin was injected into mice intraperitoneal, followed by E2 through the eye socket veins. We found that forskolin rescued E2-induced platelet clearance ((A)). These results indicate that activation of PKA inhibits E2-induced platelet clearance in vivo, thus providing a promising therapeutic strategy for E2-related CTP.
Figure 5. Forskolin rescued the E2-induced thrombocytopenia in mice. (A) WT mice were intraperitoneally injected with forskolin (5 mg/kg) or vehicle 30 min earlier, followed by E2 (1.3 mg/kg) through the eye socket veins. Whole blood was collected from the eye socket veins of the mice at 0 (baseline, before the injection), 2, 4, and 8 h after injection. Platelet counts were performed with Mindray BC-500 Vet Hematologic Analyzer. Data are represented as the mean of the normalized percentage of platelets (time point 0 = 100%) ± SD (n = 9). *P < 0.05, **P < 0.01, two-way ANOVA. (B) Washed mouse platelets were incubated with forskolin (2.5 µM) or vehicle at 22°C for 20 min and further incubated with E2 (100 nM) or vehicle at 22°C for 72 h. The platelets were labeled with calcein and injected into WT mice. The percentage of calcein-labeled platelets remaining in circulation was determined by flow cytometry. Data are represented as the mean of the normalized percentage of calcein-labeled platelets (time point 0 = 100%) ± SD (n = 6). **P < 0.01, two-way ANOVA.
Discussion
In this study, we report a female CTP patient whose platelet counts are reverse associated with her serum E2 levels. We further demonstrated that E2 induces platelet apoptosis by inhibiting PKA activity. Activation of PKA prevents platelet clearance induced by E2 in mice, which suggests promising therapeutic strategies for thrombocytopenia caused by E2.
CTP is a kind of rare thrombocytopenia, and the pathogenesis is still unclear [Citation27]. In the female CTP patient, we unexpectedly found that her serum E2 or progesterone (P4) levels were inversely associated with her platelet counts, suggesting a possible role of E2 and P4 in the pathogenesis of CTP. Therefore, we investigated the association of E2 and P4 with thrombocytopenia and found that E2 but not P4 could induce platelet apoptosis and clearance. The reasons why P4 could not induce platelet apoptosis may be as follows: Ⅰ. The function of progesterone is mainly in the ovary, endometrium, and other tissues, while the effect on platelets is relatively weak. Ⅱ. Progesterone may bind less to platelets, thus lacking the necessary properties to induce platelet apoptosis. Therefore, progesterone has little effect on platelet apoptosis. Although estrogen and its related steroid hormones had been reported to induce apoptosis of CHO cells [Citation17], this is the first finding that E2 could induce apoptosis in platelets. However, interestingly, previous studies did not report apoptotic markers in estradiol analogs-treated platelets [Citation28]. Considering that estradiol analogs were incubated with platelets for a relatively shorter time in this research, the discrepancy seems to be due to experimental differences.
Next, we sought to identify the molecular mechanism underlying thrombocytopenia induced by E2. Our previous study demonstrated that PKA plays a role in platelet apoptosis aroused by various physiopathological factors [Citation15]. Indeed, in this study, E2 induced PKA inhibition. Forskolin markedly reduced E2-induced apoptotic events by activating PKA. A previous study reports that estrogen-like hormones could induce apoptosis by binding to PDE3A [Citation17]. We found that E2 increased PDE3A activity in a dose-dependent manner, and the PDE3A inhibitors directly increased PKA activity and reduced platelet apoptosis. Therefore, our results indicated that E2 induces platelet apoptosis through PDE3A-mediated PKA inhibition.
Unlike the experiments in vitro, it takes a shorter time for E2 to initiate clearance in vivo, suggesting that shear stress of blood flow or other reasons contribute to accelerating the destruction and activation of platelets in vivo. More importantly, the PKA activator (forskolin) rescued platelet clearance induced by E2, thus, providing a promising therapeutic strategy for E2-related thrombocytopenia. As known, fluctuations in serum E2 levels are common in healthy women. However, it is still unknown why only this case presented thrombocytopenia. Further work is needed to disclose the mystery.
Conclusions
We report a case whose platelet fluctuation is in phase with her menstrual cycle, and there is a reverse relationship between the platelet counts and serum E2 levels. We further find that E2 induces platelet apoptosis and clearance through PDE3A-mediated PKA inhibition. Activation of PKA rescues E2-induced thrombocytopenia in mice. Thus, our study reveals a pathogenesis of E2-related CTP and suggests promising therapeutic strategies for the disease.
Supplemental Material
Download MS Word (627.1 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Kyrle PA, Eichinger S. How I manage cyclic thrombocytopenia. Blood. 2021;137(2):178–184. doi:10.1182/blood.2020008218
- Go RS. Idiopathic cyclic thrombocytopenia. Blood Rev. 2005;19(1):53–59. doi:10.1016/j.blre.2004.05.001
- Şumnu A, Diz-Küçükkaya R. Cyclic thrombocytopenia: a case report. Turk J Haematol. 2010;27(3):196–199. doi:10.5152/tjh.2010.28
- Connor DE, Joseph JE. Cyclic thrombocytopenia associated with marked rebound thrombocytosis and fluctuating levels of endogenous thrombopoietin and reticulated platelets: a case report. Am J Hematol. 2012;87(1):120–122. doi:10.1002/ajh.22186
- Langlois GP, Arnold DM, Potts J, et al. Cyclic thrombocytopenia with statistically significant neutrophil oscillations. Clin Case Rep. 2018;6(7):1347–1352. doi:10.1002/ccr3.1611
- Brey O, Garner EP, Wells D. Cyclic thrombocytopenia associated with multiple autoantibodies. Br Med J. 1969;3(5667):397–398. doi:10.1136/bmj.3.5667.397
- Hoffman R, Briddell RA, van Besien K, et al. Acquired cyclic amegakaryocytic thrombocytopenia associated with an immunoglobulin blocking the action of granulocyte-macrophage colony-stimulating factor. N Engl J Med. 1989;321(2):97–102. doi:10.1056/NEJM198907133210207
- Harvey JW, Simpson CF, Gaskin JM. Cyclic thrombocytopenia induced by a Rickettsia-like agent in dogs. J Infect Dis. 1978;137(2):182–188. doi:10.1093/infdis/137.2.182
- Tomer A, Schreiber AD, McMillan R, et al. Menstrual cyclic thrombocytopenia. Br J Haematol. 1989;71(4):519–524. doi:10.1111/j.1365-2141.1989.tb06312.x
- Oh H, Nakamura H, Yokota A, et al. Serum thrombopoietin levels in cyclic thrombocytopenia. Blood. 1996;87(11):4918. doi:10.1182/blood.V87.11.4918a.bloodjournal87114918a
- Menitove JE, Pereira J, Hoffman R. Cyclic thrombocytopenia of apparent autoimmune etiology. Blood. 1989;73(6):1561–1569. doi:10.1182/blood.V73.6.1561.1561
- Cohen T, Cooney DP. Cyclic thrombocytopenia case report and review of literature. Scand J Haematol. 1974;12(1):9–17. doi:10.1111/j.1600-0609.1974.tb00174.x
- Wahlberg P, Nyman D, Ekelund P, et al. Cyclical thrombocytopenia with remission during lynestrenol treatment in a woman. Ann Clin Res. 1977;9(6):356–358.
- Chen G, Chen L, Qin X, et al. Cyclic thrombocytopenia related to menstrual cycle: a case report and literature review. Int J Clin Exp Med. 2014;7(10):3595–3598.
- Zhao L, Liu J, He C, et al. Protein kinase A determines platelet life span and survival by regulating apoptosis. J Clin Invest. 2017;127(12):4338–4351. doi:10.1172/JCI95109
- Chen M, Yan R, Zhou K, et al. Akt-mediated platelet apoptosis and its therapeutic implications in immune thrombocytopenia. Proc Natl Acad Sci U S A. 2018;115(45):E10682–E10691. doi:10.1073/pnas.1808217115
- Li D, Chen J, Ai Y, et al. Estrogen-Related hormones induce apoptosis by stabilizing schlafen-12 protein turnover. Mol Cell. 2019;75(6):1103–1116.e9. doi:10.1016/j.molcel.2019.06.040
- Lewis-Wambi JS, Jordan VC. Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res. 2009;11(3):206. doi:10.1186/bcr2255
- Sukocheva OA, Wee C, Ansar A, et al. Effect of estrogen on growth and apoptosis in esophageal adenocarcinoma cells. Dis Esophagus. 2013;26(6):628–635. doi:10.1111/dote.12000
- Wu Y, Dai J, Zhang W, et al. Arsenic trioxide induces apoptosis in human platelets via C-Jun NH2-terminal kinase activation. PLoS One. 2014;9:e86445. doi:10.1371/journal.pone.0086445
- Li S, Wang Z, Liao Y, et al. The glycoprotein Ibα–von Willebrand factor interaction induces platelet apoptosis. J ThrombHaemost. 2010;8(2):341–350. doi:10.1111/j.1538-7836.2009.03653.x
- Leytin V. Apoptosis in the anucleate platelet. Blood Rev. 2012;26(2):51–63. doi:10.1016/j.blre.2011.10.002
- Tang WH, Stitham J, Jin Y, et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation. 2014;129(15):1598–1609. doi:10.1161/CIRCULATIONAHA.113.005224
- du Rusquec P, Blonz C, Frenel JS, et al. Targeting the PI3K/Akt/mTOR pathway in estrogen-receptor positive HER2 negative advanced breast cancer. Ther Adv Med Oncol. 2020;12:175883592094093. doi:10.1177/1758835920940939
- Hua K, Feng W, Cao Q, et al. Estrogen and progestin regulate metastasis through the PI3 K/AKT pathway in human ovarian cancer. Int J Oncol. 2008;33(5):959–967.
- Tong MH, Jiang H, Liu P, et al. Spontaneous fetal loss caused by placental thrombosis in estrogen sulfotransferase-deficient mice. Nat Med. 2005;11(2):153–159. doi:10.1038/nm1184
- Connor DE, Joseph JE. Cyclic thrombocytopenia associated with marked rebound thrombocytosis and fluctuating levels of endogenous thrombopoietin and reticulated platelets: a case report. Am J Hematol. 2012;87(1):120–122. doi:10.1002/ajh.22186
- Repsold L, Pretorius E, Joubert AM. Ex vivo apoptotic and autophagic influence of an estradiol analogue on platelets. Exp Hematol Oncol. 2015;5:18. doi:10.1186/s40164-016-0048-z