
ABSTRACT
Introduction
B-cell acute lymphoblastic leukemia (B-ALL) is the most prevalent malignant tumor affecting children. While the majority of B-ALL patients (90%) experience successful recovery, early relapse cases of B-ALL continue to exhibit high mortality rates. MZ1, a novel inhibitor of Bromodomains and extra-terminal (BET) proteins, has demonstrated potent antitumor activity against hematological malignancies. The objective of this study was to examine the role and therapeutic potential of MZ1 in the treatment of B-ALL.
Methods
In order to ascertain the fundamental mechanism of MZ1, a sequence of in vitro assays was conducted on B-ALL cell lines, encompassing Cell Counting Kit 8 (CCK8) assay, Propidium iodide (PI) staining, and Annexin V/PI staining. Western blotting and quantitative real-time polymerase chain reaction (qRT-PCR) were employed to examine protein and mRNA expression levels. Transcriptomic RNA sequencing (RNA-seq) was utilized to screen the target genes of MZ1, and lentiviral transfection was employed to establish stably-expressing/knockdown cell lines.
Results
MZ1 has been observed to induce the degradation of Bromodomain Containing 4 (BRD4), Bromodomain Containing 3 (BRD3), and Bromodomain Containing 2 (BRD2) in B-ALL cell strains, leading to inhibited cell growth and induction of cell apoptosis and cycle arrest in vitro. These findings suggest that MZ1 exhibits cytotoxic effects on two distinct molecular subtypes of B-ALL, namely 697 (TCF3/PBX1) and RS4;11 (MLL-AF4) B-ALL cell lines. Additionally, RNA-sequencing analysis revealed that MZ1 significantly downregulated the expression of Cyclin D3 (CCND3) gene in B-ALL cell lines, which in turn promoted cell apoptosis, blocked cell cycle, and caused cell proliferation inhibition.
Conclusion
Our results suggest that MZ1 has potential anti-B-ALL effects and might be a novel therapeutic target.
Introduction
Acute lymphoblastic leukemia (ALL), a highly treatable disease, is the most prevalent malignancy among children [Citation1]. B-ALL accounts for approximately 80% of all pediatric ALL cases, representing approximately 25% of all pediatric tumors [Citation2]. Although B-ALL is considered the most common pediatric neoplasm, it can also affect older individuals [Citation3]. Over the past four decades, significant efforts have been made to improve the remission rates of childhood ALL, which currently stand at 90–100% [Citation4]. However, the prognosis for adult ALL is considerably worse. Despite these advancements, ALL relapse remains fatal for some patients [Citation5]. Thus, further advancements and innovative therapeutic strategies are necessary to attain superior outcomes and mitigate treatment-related adverse effects.
The BET proteins, namely the bromodomain and extra-terminal domain proteins, are recognized as regulators of gene expression that play a role in tumorigenesis. These proteins serve multiple functions in cancer [Citation6], including acting as a family of epigenetic readers that are specifically designed to identify acetylated lysine side chains of histones, such as H3K27ac marks and associated transcription factors (TFs) [Citation7]. Additionally, they are responsible for aberrant chromatin remodeling and erroneous gene transcription. Given the widespread occurrence of epigenetic dysregulation in tumors, targeting BET proteins has emerged as a promising therapeutic strategy.
The BET family, consisting of four members including testis-specific BRDT, BRD 2, 3, and 4, is headed by BRD4, the sole generally expressed BET protein [Citation8]. Prior research has demonstrated BRD4's ability to regulate normal cell cycle and development [Citation9], with overexpression leading to accelerated cell cycle progression, in vitro invasion, metastasis, and migration of cancer cells [Citation8]. The inhibition of bromodomains by BET inhibitors, particularly BRD4 inhibitors, results in the disruption of BET proteins function. Consequently, small molecule inhibitors targeting the BET family have proven efficacious against hematological malignancies [Citation10–12].
The identification of small molecule inhibitors of the BET family has presented a new avenue for pharmacological intervention in cancer treatment [Citation13]. Among these inhibitors, JQ1 has been intensively researched as a monovalent BRD4 inhibitor, however, its clinical application is limited due to its reversible binding characteristics and short half-life [Citation14,Citation15]. In contrast, the development of Proteolysis targeting chimeric (PROTAC) molecules represents a novel class of compounds that can recruit E3 ligases and facilitate proteasome degradation of BRD4 [Citation16]. The PROTAC technology represents a significant advancement over conventional medications [Citation17]. For example, the PROTAC BET degrader MZ1, derived from JQ1, exhibits the capacity to degrade BRD2, BRD3 and BRD4. Furthermore, MZ1 demonstrates more potent protein degradation activities in comparison to JQ1 and dBET1 (other PROTAC BRD4 inhibitor). MZ1 also exhibits significant cytotoxicity against various malignancies, including colorectal cancer [Citation18], and triple-negative breast cancer [Citation19]. However, the precise role and mechanism of MZ1 in B-ALL remain inadequately understand.
In order to investigate the therapeutic efficacy of MZ1 on B-ALL and its underlying mechanism, two distinct B-ALL cell lines (697 and RS4;11) were utilized to assess the cytotoxic impact of MZ1. These cell lines represent disparate molecular subtypes (TCF3/PBX1, and MLL-AF4, respectively). Our studies found that MZ1 can induce the degradation of BET proteins in B-ALL cell lines in vitro, thus inhibiting cell growth, inducing apoptosis and cell cycle arrest. These findings suggest that MZ1 has cytotoxic effects on B-ALL cell lines. In addition, RNA sequencing analysis and functional experiments showed that downregulation of CCND3, the target gene of MZ1, can promote apoptosis, block cell cycle, and inhibit cell proliferation. These results suggest that MZ1 may serve as a promising therapeutic intervention for B-ALL.
Methods
Cell culture
The Cell Bank of the Chinese Academy of Sciences was approached to obtain the following cells: SUP-B15, NALM-6, RS4;11, and 697, which were kept in a humidified incubator within 37°C/5% CO2. An Roswell Park Memorial Institute 1640 (RPMI-1640) medium (cat. No. 11875093, GIBCO, Life Technologies, USA) that contains 1% penicillin–streptomycin (cat. No. V900929, Millipore, Billerica, MA, USA), and 10% of fetal bovine serum (FBS) (cat. No. 04-001-1A, Biological Industries, CT, USA) was used as the culture medium. Mycoplasma assessment was performed routinely.
Public data
The Cancer Cell Line Encyclopedia (CCLE) was searched for information on BRD4 mRNA expression patterns in various cancer cell types (https://depmap.org).
Measuring cell viability
MZ1 (cat. No. HY-101838, MedChemExpress, USA) was dissolved in 100% dimethyl sulfoxide (DMSO) at a concentration of 10 mmol, and placed in a refrigerator at −80°C. Seeding of B-ALL cells was conducted in 96-well plates at a density of 2 × 105 cells/well, and respectively managed with variable MZ1, JQ1 (cat. No. HY-13030, MedChemExpress, USA), and dBET1 (cat. No. HY-107425, MedChemExpress, USA) degrees for 48 h, then, the density of 3 × 104 cells per well was seeded in 96-well plates, the experimental group was added with MZ1, and the control group was added with equal volume 0.05% DMSO for 7 days. Cell viability was measured by CCK8 (cat. No. CK04, Dojindo Molecular Technologies, Tokyo, Japan) by a Microplate reader (Thermo Fisher Scientific) to measure the 450 nm absorbance. Experiments were done in triplicate and repeated for ≥ 3 independent times. Calculations of the half-maximum inhibitory concentration (IC50) values were performed using GraphPad Prism 8.3. (GraphPad Software Inc., San Diego, CA, USA).
Measuring cell apoptosis
As described before [Citation20], the B-ALL cells were seeded at the density of 1 × 106 cells per well in 6-well plates, treated with MZ1 in 1 μM and 2 μM for 48 h. On the other hand, B-ALL cells were transduced with CCND3 short hairpin RNA (shRNA) for 4 days. Treated B-ALL cells were collected, and processed for labeling with Annexin-V-fluorescein isothiocyanate and PI, with the aid of the FITC-Annexin V apoptosis kit (cat. No. 556420, BD Biosciences, Franklin Lakes, NJ, USA). Furthermore, apoptosis was quantification using the Beckman Gallios™ flow cytometer (Beckman, Krefeld, Germany) was approached.
Cell cycle analysis
The B-ALL cells were seeded in 6-well plates at a density of 2 × 106 cells per well, exposed to different concentrations (0.125 μM and 0.25 μM) of MZ1 for 48 h. On the other hand, four days after transduction with CCND3 shRNA, collecting B-ALL cells required a brief fixation in 70% ethanol at 4°C overnight, followed by 15 min at room temperature in 0.5 mL PI/RNase Staining Buffer (cat. No. 550825, BD PharmingenTM, San Diego, CA, USA). The Modfit software was used to evaluate data collected from a Beckman GalliosTM flow cytometer (Beckman, Krefeld, Germany) to determine where cells were in their life cycles.
Western blotting analysis
The B-ALL cells were seeded in 6-well plates at a density of 1 × 106 cells per well, and collected after adding MZ1 at different concentrations (i.e. 0.125, 0.25, 0.5, 1, 2 μM) and treating them continuously for 48 h. Cells were collected after 1 μM MZ1 treated B-ALL cells at different times (i.e. 8, 16, 24, 32, 48 h). MG132 (cat. No. 474787, Sigma-Aldrich, St. Louis, MO, USA) was used as a proteasome inhibitor with effective and reversible proteasome-relevant activities. Following treatment with different concentrations of MG132 and 0.25 μM MZ1 for 12 h, cellular lysis of B-ALL cells occurred, which was followed by collecting and quantifying the total proteins by centrifugation and the Pierce BCA Kit (cat. No. A53226, Thermo Fisher Scientific) [Citation20]. The boiled protein samples with loading buffer (cat. No. B1012-100, Applygen Technologies, China) were isolated using SDS-PAGE gel, and transferred over polyvinylidene fluoride (PVDF) membranes. For 2 h, the membranes were immersed in skim milk (5% w/v), after which they were treated with 1 ry and 2 ry antibodies for 12 and 1 h, respectively. The following antibodies were used: BRD2 (cat. No. 5848S, Cell Signaling Technology, Boston, MA, USA), BRD3 (cat. No. 11859-1-AP, Proteintech, Chicago, IL, USA), BRD4 (cat. No. 13440S, Cell Signaling Technology, Boston, MA, USA), Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (cat. No. MA3374, Millipore, USA), Von Hippel Lindau (VHL) (cat. No. 68547S, Cell Signaling Technology, Boston, MA, USA), CCND3(cat. No. 183338, Abcam, Cambridge, UK), Cyclin-dependent kinases 4 (CDK4) (cat. No. 12790S, Cell Signaling Technology, Boston, MA, USA), Cyclin-dependent kinases 6 (CDK6) (cat. No. 3136S, Cell Signaling Technology, Boston, MA, USA), Myeloid cell leukemia-1 (MCL-1) (cat. No. 32078, Abcam, Cambridge, UK) and Poly ADP-ribose polymerase (PARP) (cat. No. 9542, Cell Signaling Technology, Boston, MA, USA). We also used AI600image (GE, USA) for protein band detection and analysis.
Lentivirus preparation and infection
Lentiviral vectors were commercially constructed by IGE Biotechnology, Ltd. (China) to knockdown or over-express the targeted genes. VHL-targeted was 5’-CCGGGCTCAACTTCGACGGCGAGCCCTCGAGGGCTCGCCGTCGAAGTTGAGCTTTTTGAATT-3’. CCND3#1-targeted sequence was 5’- CCGGGCACATGATTTCCTGGCCTTCCTCGAGGAAGGCCAGGAAATCATGTGCTTTTTGAATT-3’. CCND3#2-targeted sequence was 5’- CCGGCAGACCAGCACTCCTACAGATCTCGAGATCTGTAGGAGTGCTGGTCTGTTTTTGAATT-3’. The lentivirus transfer plasmids together with envelop plasmid pMD2.G (cat. No. 12259, Addgene, Cambridge, USA) and packaging plasmid psPAX2 (cat. No. 12260, Addgene, Cambridge, USA) were co-transported into 293FT cells and replaced with fresh medium following six hours. The viral supernatant used to infect B-ALL cell lines was gathered following transfection by two days passing within a 0.45 μm filter. After 48 h, stable cell lines were obtained after puromycin (cat. No. P7255, Sigma-Aldrich, USA) selection.
RNA-Sequencing and analysis
Following extraction of total RNA with TRIzol® reagent (cat. No. 15596026, Invitrogen, CA, USA), reverse transcription into cDNA, and sequencing to create a library, we compared B-ALL cells treated with MZ1 (n = 2) and DMSO (n = 2) for 24 h. Novogene Bioinformatics Technology Co., Ltd. sequenced the RNA in this study using RNA-seq technology (Beijing, China). The unclean reads were filtered out and mapped using the Hierarchical Indexing for Spliced Alignment of Transcripts 2 (HISAT2) program. Using the FPKM technique (Fragments Per Kilobase of exon Model per Million aligned fragments), the relative abundance of each gene was determined. Absolute log2(fold change) > 1 and adjusted P value < 0.05 and were used to determine differentially regulated transcripts using the DESeq2 R/Bioconductor tool. The Java GSEA Desktop Application (www.broadinstitute.org) was used to perform gene set enrichment analysis (GSEA). Data from the RNA-seq experiments performed in this research are available in the GEO library (www.ncbi.nlm.nih.gov, entry code: GSE 217540).
Quantitative real-time polymerase chain reaction (qRT-PCR) expression analysis
qRT-PCR analysis was carried out using LightCycler® 480 SYBR Green I Master mix (cat. No. 04707516001; Roche, Penzberg, Germany) with a Light cycler 480 Real-Time System (Roche, Penzberg, Germany), according to the standard protocol. mRNA relative expression values of target gene were estimated by the 2ΔΔCt approach, using GAPDH as internal reference. The primer sequences utilized within the current investigation include GAPDH: forward, TGCACCACCAACTGCTTAG and reverse: GATGCAGGGATGATGTTC; CCND3: forward, TACCCGCCATCCATGATCG and reverse, AGGCAGTCCACTTCAGTGC; ERG: forward, CGTGCCAGCAGATCCTACG and reverse, GGTGAGCCTCTGGAAGTCG; BCL11A: forward, CGCCAGAGGATGACGATTGTT and reverse, CCAGGCGTGGGGATTAGAG; ANP32A: forward, CACCTCAATCGCAAACTTACCA and reverse, AACACATTTTCTCGGTAGTCGTT; ETS1: forward: GATAGTTGTGATCGCCTCACC and reverse, GTCCTCTGAGTCGAAGCTGTC; CXCR4: forward, ACTACACCGAGGAAATGGGCT and reverse, CCCACAATGCCAGTTAAGAAGA.
Statistical analysis
This was conducted by the GraphPad Prism software as previously mentioned. Furthermore, the Mann–Whitney u test or the student's t-test was employed to evaluate the significant differences within groups. Significance was considered when a p-values was ≤ 0.05.
Results
MZ1 promotes the degradation of BET proteins in B-ALL cell lines
Data obtained from the CCLE dataset ((A)) indicated that BRD4 mRNA exhibited uniform expression across various diseases. A comparative analysis of BRD4 expression in different cancers revealed a higher expression in T-ALL, CML, and B-ALL cell lines. Western blotting analysis of BRD 4, 3, and 2 proteins expression demonstrated their ubiquitous expression in four B-ALL cell lines (697, SUP-B15, NALM-6, and RS4;11) ((B)) with the highest expression observed in RS4;11 cells, suggesting a potential association of BET proteins with leukemia pathogenesis. 697 and RS4;11 with the optimal cellular states were selected for the following studies. First, the PROTAC BRD4 inhibitor MZ1 was used to treat B-ALL cell lines. Western blot analysis revealed almost complete degradation of BRD4 protein after MZ1 treatment ((C)). Furthermore, the administration of MZ1 resulted in a simultaneous reduction of both BRD2 and BRD3 expression levels ((C)). These findings suggested that MZ1 treatment leads to a degradation of BET proteins in B-ALL cell lines.
Figure 1. MZ1 inhibits the expression of BET protein in B-ALL cells and the effect is better than those of JQ1 and dBET1. (A) The mRNA expression of BRD4 was characterized in different cancer cell lines. High expression of BRD4 was observed in B-ALL cell lines. (generated from the web site: https://depmap.org/portal/download/). (B) Basal BET protein levels in B-ALL cells, 697, NALM-6, SUP-B15, and RS4;11 cells. (C) Western blot results showed that MZ1 induced BET proteins degradation, PARP cleavage in 697 and RS4;11 cells. (D) Cell viability of 697 and RS4;11 cells treated with different concentrations of MZ1 for 48 h. Values of IC50 have been estimated. (E) Cell viability of 697 and RS4;11 cell lines after treatment with MZ1, JQ1 and dBET1 for 48 h. Cell viability measuring experiment was tested at least three times independently. Western blot experiment was tested one time independently. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
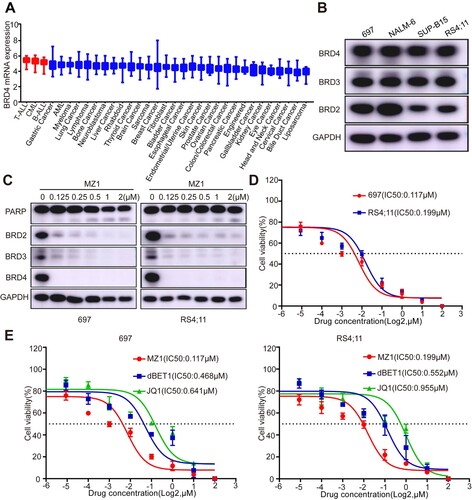
The sensitivity of MZ1 was observed in B-ALL cell lines, with IC50 values of 0.117 μM and 0.199 μM for 697 and RS4;11, respectively ((D)). Additionally, MZ1 exhibited a more favorable impact on inhibiting the proliferation of 697 and RS4;11 cell lines compared to other BRD4 inhibitors, JQ1 and dBET1 ((E)). These results demonstrate that MZ1 is more potent than conventional BRD4 inhibitors.
MZ1 inhibits cell growth, arrested cell cycle and induces apoptosis of B-ALL cell lines
Furthermore, treatment with MZ1 resulted in a significant decrease in the number of 697 and RS4;11 cells in the white slice ((A)). In addition, following a 7-day treatment with MZ1, the viability of 697 and RS4;11 cell lines exhibited a more pronounced decrease in a time-dependent manner compared to DMSO-treated cells ((B)). This suggests that MZ1 effectively suppresses viability of B-ALL cell lines.
Figure 2. MZ1 inhibits cell growth, blocked cell cycle and promoted apoptosis of B-ALL cells. (A) Morphology of 697 and RS4;11 cells treated with DMSO or MZ1. (B) Cell growth curves of 697 and RS4;11 cells treated with DMSO or MZ1 for 7 days. (C) Flow cytometry was performed to calculate the cell cycle distribution by PI staining. The result revealed that 697 and RS4;11 cells were synchronized in G1/S phase and the percentage of cell population in G1 phase increased significantly after treatment with MZ1 for 48 h. (D) Dose-dependent increase in the apoptotic cells was observed in B-ALL cells after treatment with MZ1 for 48 h. Flow cytometry experiment was independently performed at least three times. Western blot experiment was tested one time independently. **p < 0.01, ***p < 0.001, ****p < 0.0001.
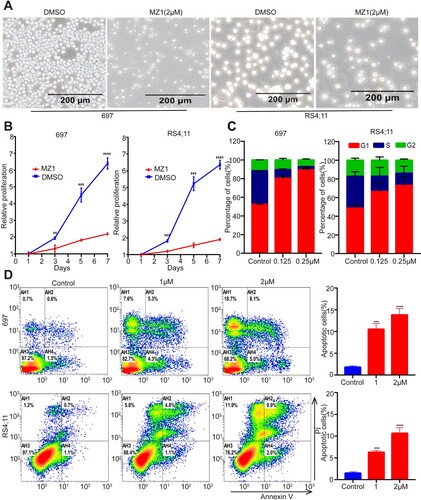
Given the crucial role of the BET family in cell cycle regulation, as demonstrated by recent studies [Citation21], we proceeded to evaluate the impact of MZ1 treatment on the cell cycle distribution of B-ALL cell lines. Notably, compared to DMSO, the G1 phase ratio was significantly elevated 48 h after MZ1 treatment, while the ratios of G2 and S phase were reduced ((C)). The results of this study indicate that MZ1 has the ability to impede the proliferation of B-ALL cells by inducing cell cycle arrest in the G1 phase. Furthermore, the use of flow cytometry to analyze Annexin V/PI staining demonstrated a dose-dependent increase in apoptotic cells after 48 h of MZ1 treatment compared to the DMSO control group ((D)). Additionally, a dose-dependent increase in PARP cleavage was observed ((C)). As expected, PARP cleavage appears gradually with time by treatment with 1 μM MZ1 in B-ALL cell lines (Figure S1). In conclusion, these findings suggest that MZ1 has the potential to induce cell cycle arrest and promote apoptosis of B-ALL cell lines.
MZ1 induced degradation of BET proteins through VHL-mediated ubiquitination
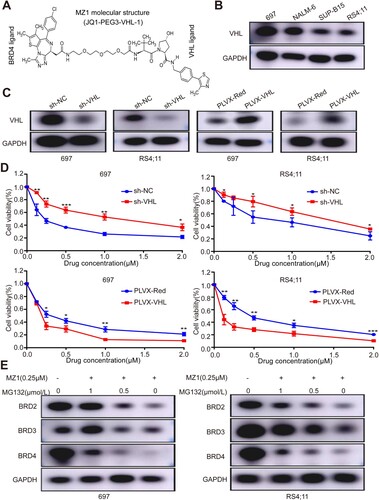
MZ1, a selective PROTAC inhibitor, facilitates the degradation of target proteins through the ubiquitin-mediated protein degradation system. VHL functions as a ligand for the E3 ubiquitin ligase and is a constituent of MZ1. ((A)). It facilitates interactions between MZ1 and proteins belonging to the BRD family. The efficacy of VHL-overexpression and knockdown plasmids was demonstrated in RS4;11 and 697 cells, and western blotting analysis confirmed the widespread presence of VHL in four B-ALL cell lines ((B and C)). Knockdown of VHL was observed to diminish MZ1 activity in the CCK8 assay, whereas overexpression of VHL significantly enhanced MZ1 activity ((D)). These findings indicate that VHL serves as a valuable tool in the management of MZ1. To investigate the role of the proteasome in MZ1-induced BET family proteins degradation, we used MG132 (a proteasome inhibitor) to evaluate proteasome activity, the effect of MG132 treatment on MZ1 treated B-ALL cell lines was examined with western blot to detect the protein levels of BRD2, BRD3 and BRD4. The results revealed that MG132 inhibited the breakdown of BET proteins in a dose-dependent manner ((E)). These results suggested that MZ1 regulates the degradation of BET proteins through VHL-mediated ubiquitination.
Figure 3. MZ1 degradation of BET protein depends on VHL in B-ALL cells. (A) Schematic diagram of the bifunctional PROTAC molecule (MZ1). (B) VHL protein expression in B-ALL cells by Western blot analysis. (C) 697 and RS4;11 cells were transfected with sh-VHL or PLVX-VHL lentivirus, and the expression level of VHL was detected by Western blot. (D) Downregulation of VHL significantly inhibited the MZ1 activity in 697 and RS4;11 cell lines. In contrast, overexpression of VHL increased the MZ1 activity in 697 and RS4;11 cell lines. (E) After MZ1(0.25 μΜ) and different concentrations of MG132 treated with 697 and RS4;11 cells for 12 h, western blot assay showed that MG132 inhibited BRD2, BRD3 and BRD4 protein degradation in a dose-dependent manner. Cell viability measuring experiment was tested in triplicate and independently performed at least three times. Western blot experiment was tested one time independently. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CCND3 as a critical target of MZ1
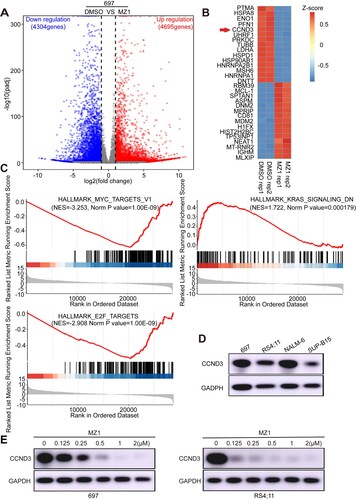
RNA-seq technology was utilized to analyze and screen genes exhibiting altered expression levels in MZ1-treated 697 cells, thereby facilitating further exploration of MZ1's functional mechanism. A total of 8,999 differentially expressed genes (4,695 upregulated genes and 4,304 downregulated genes) were identified in MZ1-treated 697 cells, with |log2fold change | > 1 and an adjusted p < 0.05, compare to DSMO ((A)). The heatmap depicts the expression of dysregulated genes that are associated with multiple signaling mechanisms, with the top 15 downregulated and upregulated genes being selected ((B)). Additionally, GSEA plots of RNA-Seq data from MZ1-treated 697 cells revealed genes enrichment in HALLMARK_MYC_TARGETS-V1, HALLMARK_KRAS_SIGNALING_DN, and HALLMARK_E2F_TARGETS signaling pathways ((C)). To validate the altered expression of genes observed in RNA-seq result, we employed qRT-PCR to identify genes that were either up-regulated or down-regulated in MZ1-treated 697 cells (Figure S2A). Through screening and functional analysis of genes with expression alterations, we found a significant reduction in the mRNA expression of the CCND3 gene. It was reported that CCND3 is indispensable for the maintenance of B-cell acute lymphoblastic leukemia [Citation22]. Notably, CCND3 exhibited increased expression levels in 697 and NALM-6 cell lines compared to RS4;11 and SUP-B15 cell lines ((D)). The resulting downregulation of CCND3 was observed in a dose-dependent manner in MZ1-treated 697 and RS4;11 cell lines ((E)). These findings suggest that CCND3 may serve as a crucial target of MZ1.
Figure 4. CCND3 is a downstream effector of MZ1 in B-ALL cell lines. (A) Volcano plot of differentially expressed genes identified in 697 cells between MZ1-treatment and DMSO-control group. Genes highlighted in red were upregulated, and those highlighted in blue were downregulated. (B) Heat-map view displayed the top downregulated genes(log2FC > 1, p < 0.05) in 697 cells treated with 1 μM MZ1 for 24 h, including CCND3. (C) GSEA plots displayed gene enrichment in HALLMARK_MYC_TARGETS-V1, HALLMARK_KRAS_SINGNALING_DN and HALLMARK_E2F_TARGETS signaling pathways in 697 cells treated with MZ1. (D) Western blot experiment showed different expression of CCND3 protein in B-ALL cells. (E) Western blot analysis showed that MZ1 treatment downregulated CCND3 in a dose-dependent manner in 697 and RS4;11 cells.
Downregulation of CCND3 expression in B-ALL cells inhibits cell proliferation by arresting cell cycle, and promoting cell apoptosis
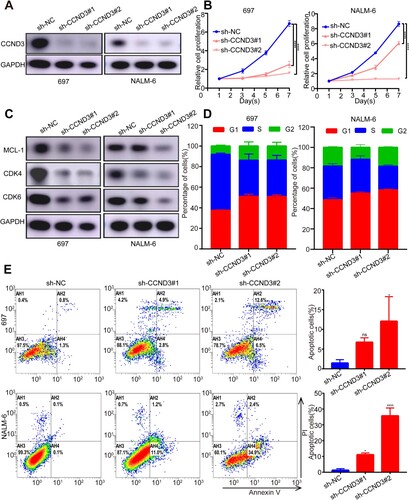
To investigate the potential involvement of CCND3, specific sh-RNAs targeting CCND3 were transfected into 697 and NALM-6 cell lines. Further qRT-PCR and western blotting analysis revealed that CCND3 was knocked down at both the RNA level and the protein level ((A) and S2B). Notably, the knockdown of CCND3 resulted in a significant inhibition of proliferation in both 697 and NALM-6 cell lines ((B)), as evidenced by the white slice (Figure S2C), when compared to the scramble cells. Western blotting results indicate that the downregulation of CDK4, CDK6, and MCL-1 proteins was significant following CCND3 knockdown ((C)). Additionally, cell cycle analysis revealed that CCND3 knockdown led to cell cycle arrest at G1 phase, while also decreasing the ratios of G2 and S phases ((D)). Furthermore, the observed reduction in MCL-1 protein levels and increased apoptosis in 697 and NALM-6 cell lines upon CCND3 knockdown, as illustrated in (E), suggests that aberrant CCND3 expression plays a crucial role in cell proliferation and apoptosis of B-ALL cell lines.
Figure 5. CCND3 knockdown causes cell cycle arrest and apoptosis, and suppresses cell proliferation. (A) Western blot verified the knockdown of CCND3 by sh-RNAs (sh-CCND3#1 and sh-CCND3#2) in 697 and NALM-6 cells. (B) CCK8 assays showed that compared to the scramble cells, CCND3 knockdown significantly inhibited the proliferation of 697 and NALM-6 cells. (C) Protein expression levels of CDK4, CDK6 and MCL-1. (D) Cell cycle profile measured by PI staining showed that CCND3 knockdown caused cell cycle arrest at G1 phase. (E) Apoptosis by Annexin V staining showed that knockdown of CCND3 could increase apoptosis in 697 and NALM-6 cells, compared to the scramble cells. Flow cytometry experiment and cell viability measuring experiment were independently performed at least three times. Western blot experiment was tested one time independently. *p < 0.05, **p < 0.01, ****p < 0.0001.
Discussion
B-ALL is a prevalent hematologic cancer that arises from the abnormal proliferation of precursor B (pre-B) or transformed progenitor B (pro-B) lymphocytes [Citation3], affecting both pediatric and adult populations [Citation20]. This cancer encompasses over 20 subtypes, including TCF3/PBX1 and MLL-AF4, each exhibiting distinct genetic alterations associated with chromatin regulation, kinase signaling, cell-cycle regulation, and lymphoid progressio [Citation23]. The prognosis for B-ALL is typically unfavorable in cases involving mixed lineage leukemia rearrangements (MLLr) and patients under the age of one year [Citation24]. However, advancements in standard chemotherapy, risk stratification, supportive care, and minimal residual disease (MRD) monitoring have led to notable improvements in the overall 5-year survival rate of childhood ALL, exceeding 90% in recent studies [Citation23,Citation25]. In contrast, adult B-ALL is characterized by a cure rate below 50% [Citation26].
Premature relapse, therapy resistance, and chemotherapy-induced adverse events continue to be the leading causes of mortality in leukemia [Citation27]. Furthermore, the efficacy of conventional chemotherapy in treating B-ALL has reached a plateau, necessitating the development of innovative therapeutic approaches to enhance the prognosis of this disease [Citation23]. Targeted therapeutic agents hold promise in improving the outcomes of B-ALL subtypes, particularly in younger patients and those with subtypes that are currently unresponsive to standard chemotherapy [Citation25]. Therefore, we need to comprehensively investigate the occurrence and development of B-ALL to potentially identify new small molecules that might help in developing enhanced targeted therapeutics.
BRD4, a chromatin-binding protein, clusters in super-enhancer areas linked with oncogenes like CDK6, Bcl-2, and c-Myc [Citation9,Citation10,Citation28], and regulates cell cycle, growth, and transcription [Citation9]. Our findings are consistent with other studies, which showed that BRD4 is overexpressed in other hematologic malignancies (including colorectal cancer, breast cancer, and acute leukemia), and can be recognized as a poor prognostic marker [Citation18,Citation19,Citation28–30]. Similarly, BRD4 overexpression was observed in ALL [Citation20], which was also indicated in our investigation as BRD4 is highly expressed in B-ALL cell lines compared with other cancer cell lines and subtypes. Our study found that BRD4 was expressed in all four B-ALL cell lines, the expression level of RS4;11 was the highest, and the expression levels of the other three cell lines were basically the same, so we selected 697 and RS4;11 with the optimal cellular states to conduct follow-up experiments to prove the potential applicability of BRD4 as a new therapeutic target of B-ALL.
JQ1 is the first- generation-specific inhibitor of BET. J. Ott et al. reported that JQ1 could reduce B-ALL cell lines viability and decrease their cytogenetic risk, confirming the therapeutic potential of JQ1 in B-ALL [Citation31]. However, the clinical application was significantly limited because of the prolonged administration-related resistance and JQ1 short half-life. The novel approach PROTACs consists of a single protein of interest (POI) bound ligand, another E3 ubiquitin ligase-recruiting ligand, and a linker that connects both. The PROTAC technology hijacks the ubiquitin-proteasome system (UPS) to degrade a POI, and in vivo degradation studies have demonstrated that PROTACs can reduce targeted protein in various tissues including solid tumors. MZ1 is the refined PROTAC that was built with E3 recruiter VH032 and a target engagement warhead JQ1. VHL is responsible for the recruitment of target proteins, and the effectively increased ubiquitin-proteasome mediated degradation of target proteins. Low concentrations of MZ1 can effectively and quickly induce reversible, persistent, and unexpected selective removal of BRD4, with a low probability of inducing adverse events (e.g. natural VHL substrate stabilization) [Citation32]. Our study found that MZ1 can exert cytotoxic effects at a lower concentration than JQ1 and other PROTAC BRD4 inhibitor (dBET1), which is consistent with previous studies.
In our study, we found that MZ1 successfully inhibits the proliferation of B-ALL cell lines containing a wide variety of oncogenic ‘drivers,’ including the relapsed B-ALL line 697 with a TCF3-PBX1 rearrangement, and MLL-AF4-harboring line RS4;11. Our data show that after being exposed to MZ1, protein concentrations of BRD 2, 3, and 4 all decline. At the same time, MZ1 can halt the cell cycle, promote cell death, and suppress cell growth, the results presented in this investigation support the use of MZ1 as a new therapy approach for B-ALL. Overexpression of VHL in vitro can increase MZ1 inhibition in B-ALL cell lines while reduction of VHL can reduce MZ1 inhibition in these lines. Not only that, but it turns out that the commonly used proteasome inhibitor MG132 can substantially counteract the breakdown of BRDs brought on by MZ1. The current findings corroborated previous findings that MZ1 activity is dependent on VHL and proteasome mRNA.
To further identify genes involved in the downstream pathways affected by MZ1, RNA-seq was used to compare the MZ1-treated transcriptomes of B-ALL cell lines to those that were not. BRD4 is closely related to cell cycle regulation [Citation7,Citation9]. Interestingly, we found that suppression of BET proteins by MZ1 can induce a state of multiple down-regulated genes (PTMA, HSPA8, ENO1, PFN1 and CCND3). qRT-PCR showed that CCND3 gene expression was most significantly down-regulated among down-regulated genes, therefore, we speculate that MZ1 plays a biological function by down-regulating CCND3 transcription. the western blot assay showed that the CCND3 protein was downregulated by MZ1 at different concentrations in both two B-ALL cell lines. Meanwhile, Western blotting verified the expression of CCND3 in four B-ALL cell lines, of which NALM-6 and 697 were the highest, so we selected these cell lines for subsequent experiments to prove the role of CCND3 in B-ALL. CCND3-encoding protein performs its cell cycle regulator function by arresting the cells in the G0/G1 phase [Citation33]. It binds to and stimulates CDK4/6, which cumulatively induces p-Rb phosphorylation, leading to cell cycle progression [Citation34]. Several studies have shown that CCND3 might be a potential therapeutic target for T-ALL, AML, MCL and B-ALL [Citation34–38]. We also found that the knockdown of CCND3 could reduce MCL-1, CDK4, and CDK6 proteins level, which indicates what was previously reported in the literature. Previous investigations indicated the significant B-ALL apoptosis and growth arrest due to CCND3 knockdown, particularly in cell lines RS4;11, and NALM-6, containing MLL-AF4, and ETV6-PDGFR translocations, respectively. CCND3 carcinogenic effect of additional B-ALL subtypes (like 697 with a TCF3-PBX1 rearrangement) was investigated in this study. These results are consistent with previous observations [Citation37,Citation38]. We also found that CCND3 knockdown leads to cell cycle arrest which is vital for B-ALL survival.
Conclusion
The B-ALL cell line exhibits a diverse range of molecular subtypes, which can impede the efficacy of anticancer drugs, However, the utilization of the PROTAC technology enables MZ1 to exert its anticancer activity by down-regulating the expression of BET family proteins. Additionally, MZ1 effectively inhibits the growth of the B-ALL cell line, induces cellular apoptosis, arrests cell cycle progression, and reduces CCND3 expression. These findings suggest that MZ1 holds great promise as a therapeutic intervention for the treatment of B-ALL.
Ethics approval and consent to participate
This study was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Supplemental Material
Download JPEG Image (1.9 MB){kind=link}
Supplemental Material
Download JPEG Image (210.3 KB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available on reasonable request from the corresponding author. RNA seq and original data have been submitted to the GEO database with Accession Number GSE 217540. https://www.ncbi.nlm.nih.gov/geo.
Additional information
Funding
References
- Neerav Shukla MMLS, MD. Blinatumomab for treatment of children with high-risk relapsed B-cell acute lymphoblastic leukemia. JAMA. 2021;325(9):830–832.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30.
- Rajakumar SA, Papp E, Lee KK, et al. B cell acute lymphoblastic leukemia cells mediate RANK-RANKL–dependent bone destruction. Sci Transl Med. 2020;12(561):eaba5942.
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
- Zanetti C, Kumar R, Ender J, et al. The age of the bone marrow microenvironment influences B-cell acute lymphoblastic leukemia progression via CXCR5-CXCL13. Blood. 2021;138(19):1870–1884.
- Stathis A, Bertoni F. Bet proteins as targets for anticancer treatment. Cancer Discov. 2018;8(1):24–36.
- Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12(7):465–477.
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discovery. 2014;13(5):337–356.
- Devaiah BN, Gegonne A, Singer DS. Bromodomain 4: a cellular Swiss army knife. J Leukocyte Biol. 2016;100(4):679–686.
- Peirs S, Frismantas V, Matthijssens F, et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia. 2017;31(10):2037–2047.
- Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol. 2015;6(3):128–141.
- Andrieu GP, Kohn M, Simonin M, et al. PRC2 loss of function confers a targetable vulnerability to BET proteins in T-ALL. Blood. 2021;138(19):1855–1869.
- Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073.
- Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528.
- Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J Hematol Oncol. 2020;13(1):50.
- Lai AC, Toure M, Hellerschmied D, et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew Chem, Int Ed. 2016;55(2):807–810.
- Bondeson DP, Mares A, Smith IED, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11(8):611–617.
- Otto C, Schmidt S, Kastner C, et al. Targeting bromodomain-containing protein 4 (BRD4) inhibits MYC expression in colorectal cancer cells. Neoplasia. 2019;21(11):1110–1120.
- Noblejas-López M, Nieto-Jimenez C, Burgos M, et al. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. 2019;38(1):383.
- Ma L, Wang J, Zhang Y, et al. BRD4 PROTAC degrader MZ1 exerts anticancer effects in acute myeloid leukemia by targeting c-Myc and ANP32B genes. Cancer Biol Ther. 2022;23(1):1–15.
- Devaiah BN, Singer DS. Two faces of brd4: mitotic bookmark and transcriptional lynchpin. Transcription. 2013;4(1):13–17.
- Cooper AB, Sawai CM, Sicinska E, et al. A unique function for cyclin D3 in early B cell development. Nat Immunol. 2006;7(5):489–497.
- Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105(11):2524–2539.
- Sanjuan-Pla A, Bueno C, Prieto C, et al. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood. 2015;126(25):2676–2685.
- Ratti S, Lonetti A, Follo MY, et al. B-ALL complexity: is targeted therapy still a valuable approach for pediatric patients? Cancers (Basel). 2020;12(12)):3498.
- Jabbour E, Pui C-H, Kantarjian H. Progress and innovations in the management of adult acute lymphoblastic leukemia. JAMA Oncol. 2018;4(10):1413–1420.
- Lopez-Millan B, Sanchéz-Martínez D, Roca-Ho H, et al. NG2 antigen is a therapeutic target for MLL-rearranged B-cell acute lymphoblastic leukemia. Leukemia. 2019;33(7):1557–1569.
- Knoechel B, Roderick JE, Williamson KE, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. 2014;46(4):364–370.
- Latif A-L, Newcombe A, Li S, et al. BRD4-mediated repression of p53 is a target for combination therapy in AML. Nat Commun. 2021;12(1):241.
- Peter B, Eisenwort G, Sadovnik I, et al. Brd4 degradation blocks expression of MYC and multiple forms of stem cell resistance in Ph+ chronic myeloid leukemia. Am J Hematol. 2022;97(9):1215–1225.
- Ott CJ, Kopp N, Bird L, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–2852.
- Li K, Crews CM. PROTACs: past, present and future. Chem Soc Rev. 2022;51(12):5214–5236.
- Chi Y, Huang S, Liu M, et al. Cyclin D3 predicts disease-free survival in breast cancer. Cancer Cell Int. 2015;15(1)):89.
- Pikman Y, Alexe G, Roti G, et al. Synergistic drug combinations with a CDK4/6 inhibitor in T-cell acute lymphoblastic leukemia. Clin Cancer Res. 2017;23(4):1012–1024.
- Boldrin E, Gaffo E, Niedermayer A, et al. MicroRNA-497/195 is tumor suppressive and cooperates with CDKN2A/B in pediatric acute lymphoblastic leukemia. Blood. 2021;138(20):1953–1965.
- Mao X, Liang S-b, Hurren R, et al. Cyproheptadine displays preclinical activity in myeloma and leukemia. Blood. 2008;112(3):760–769.
- Martín-Garcia D, Navarro A, Valdés-Mas R, et al. CCND2 and CCND3 hijack immunoglobulin light-chain enhancers in cyclin D1− mantle cell lymphoma. Blood. 2019;133(9):940–951.
- Ketzer F, Abdelrasoul H, Vogel M, et al. CCND3 is indispensable for the maintenance of B-cell acute lymphoblastic leukemia. Oncogenesis. 2022;11(1):1.