
ABSTRACT
The process of erythropoiesis is complex and involves the transfer of cells from the yolk sac to the fetal hepar and, ultimately, to the bone marrow during embryonic development. Within the bone marrow, erythroid progenitor cells undergo several stages to generate reticulocytes that enter the bloodstream. Erythropoiesis is regulated by various factors, with erythropoietin (EPO) synthesized by the kidney being the promoting factor and hepcidin synthesized by the hepar inhibiting iron mobilization. Transcription factors, such as GATA and KLF, also play a crucial role in erythropoiesis. Disruption of any of these factors can lead to abnormal erythropoiesis, resulting in red cell excess, red cell deficiency, or abnormal morphological function. This review provides a general description of erythropoiesis, as well as its regulation, highlighting the significance of understanding the process for the diagnosis and treatment of various hematological disorders.
Introduction
Erythropoiesis is a process by which red blood cells (erythrocytes) are produced in the marrow. It involves the differentiation of erythroid progenitor cells into mature RBCs, which transport oxygen from the lungs to the body’s tissues. erythropoiesis is a complex process that is strictly regulated by varieties of factors, including hormones, cytokines, and growth factors.
In recent years, there have been vital advances in our awareness of the molecular mechanisms that govern erythropoiesis. These advances have led to the positive result of new therapeutic strategies for the treatment of erythropoietic disorders. In the article, we review the issues of embryonic erythropoiesis, the cytokines involved in erythropoiesis, signaling cascades, and possible problems in erythropoiesis.
The normal physiological course of erythropoiesis
Erythropoiesis in embryos
In embryos, erythropoiesis occurs in a different location than in adults. During embryonic development, erythropoiesis initially takes place in the yolk sac, and later shifts to the hepar and spleen, before ultimately transitioning to the marrow [Citation1].
The Yolk sac is the original site of erythropoiesis during the first few weeks of embryonic development [Citation2], and it’s a short-lived membrane which is gradually absorbed during development [Citation3]. Blood islands, which contain primitive erythroid progenitor cells, form in the mesoderm layer of the yolk sac. These progenitor cells differentiate into erythroblasts, which produce embryonic hemoglobin (α2ϵ2) [Citation4]. α2ϵ2 is composed of a pair of alpha and a pair of epsilon globin chains and has a stronger affinity for oxygen than adult hemoglobin.
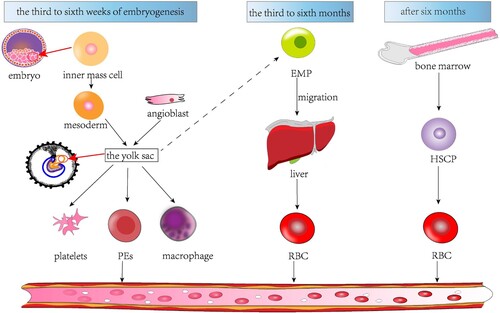
Around 6–8 weeks of gestation, erythropoiesis shifts to the hepar and spleen [Citation5], as these organs become capable of producing erythropoietin (EPO), the hormone that stimulates erythropoiesis. Studies show that EMP produced in the mouse yolk sac migrates to the hepar and colonizes [Citation6], and hematopoietic stem cells also migrate to the hepar [Citation7]. The hepar becomes the primary site of erythropoiesis from around weeks 10–28 of gestation, while the spleen mainly produces RBCs in the second trimester.
By the end of the second trimester, erythropoiesis begins to shift to the marrow, which becomes the initial site of erythropoiesis by the time of birth (). At this stage, fetal hemoglobin (HbF) is the primary type of hemoglobin produced, which possesses a stronger affinity for oxygen than adult hemoglobin (HbA) and helps to facilitate oxygen transport across the placenta. After birth, the output of HbF gradually decreases and is replaced by the output of HbA.
Figure 1. An overview of erythropoiesis in the embryo. Erythropoiesis in the embryo can be divided into three stages. The first stage is the third to sixth week of embryogenesis, during which the yolk sac, formed by mesodermal cells and angioplastic cells, produces platelets, PEs, and macrophages, with the erythrocyte-containing PEs entering the circulation. The second stage is the third to sixth month of embryonic development, during which EMPs produced by the yolk sac migrate to the liver and proliferate and differentiate into red cells in the liver. The third stage is after six months of embryonic development, during which the hematopoietic center is located in the bone marrow, and hematopoietic stem cells differentiate into red cells and enter the circulation.
Fetal hematopoiesis is regulated by multiple factors. For example, ferritin IRP2 regulates iron infusion from the maternal placenta [Citation8], and EpoR can lengthen the early stages of the erythrocyte cycle by increasing red cell cycle length [Citation9]. Furthermore, growth factors (SCF) are critical for erythropoiesis and are critical for maintaining normal adult erythrocyte numbers [Citation10].
Erythropoiesis first occurs in the yolk sac, then metastasizes to the fetal liver, and finally resides in the bone marrow after birth. Switching between these sites is associated with changes in globin expression and oxygen-binding affinity of erythrocytes. When erythrocyte proliferation metastasizes from the yolk sac to the fetal liver, a primordial to final erythroblastic transformation occurs. Fetal to adult globin conversion occurs when erythropoiesis is transferred from the fetal liver to the bone marrow. Fetal globin has a higher oxygen affinity than adult globin and can better extract oxygen from maternal blood. These spatial and temporal switches during erythropoiesis allow the developing embryo/fetus to adapt to the changing oxygen requirements during development [Citation2].
Erythropoiesis in bone marrow
Erythropoiesis in bone marrow is the process by which progenitor cells become mature red blood cells (RBCs), about 20 billion erythrocytes emerge in the marrow every day [Citation11]. This course is regulated and controlled by a heterogeneous network of signaling pathways and transcription factors.
In the bone marrow, erythroid progenitor cells differentiate into erythroblasts, which undergo several rounds of cell division and synthesis of hemoglobin, the protein that brings oxygen in RBCs. The cells gradually reduce their size and nucleus, and increase their cytoplasmic volume, ultimately becoming reticulocytes.
The output of erythroblasts is regulated by a number of cytokines and growth factors, including erythropoietin (EPO), stem cell factor (SCF), and interleukin-3 (IL-3), among others. EPO, which is produced primarily by the kidneys in response to low oxygen levels in the body, excites the differentiation and proliferation of erythroid progenitor cells.
The process of erythropoiesis in the bone marrow is also affected by the microenvironment, or niche, in which the cells remain. This niche consists of various cell types, containing osteoblasts, endothelial cells, stromal cells, as well as extracellular matrix components [Citation12].
Osteoblasts, which are bone-forming cells, have been shown to regulate erythropoiesis through the output of cytokines and growth factors, as well as direct cell-to-cell contacts with erythroid progenitor cells. Endothelial cells also play a role in regulating erythropoiesis by producing cytokines and growth factors, and by providing a source of oxygen and other nutrients. Stromal cells, which are mesenchymal cells that support hematopoiesis, also contribute to the regulation of erythropoiesis by producing growth factors and providing a supportive niche for erythroid progenitor cells ().
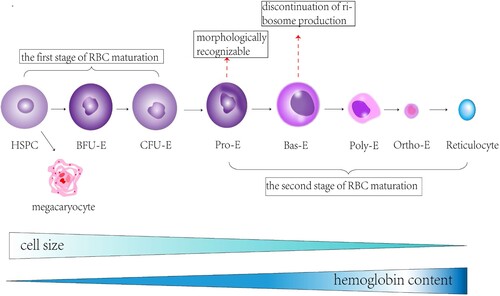
Figure 2. An overview of erythropoiesis in the bone marrow. The formation of reticulocytes from hematopoietic stem cells (HSPC) in the bone marrow takes about seven steps. During the first stage of erythropoiesis, hematopoietic stem cells produce megakaryocytes in addition to erythroid bursting-forming units (BFU-E)and erythroid colony-forming units(CFU-E). The second stage of erythropoiesis is marked by the production of proerythroblasts (Pro-E), the first erythroid cells to be morphologically recognizable. The pro-E then produces basophils (Bas-E), which cease to produce ribosomes during basophilization. Bas-E subsequently produces polychromic phage (Poly-E), Poly-E subsequently produces positively stained erythrocytes (Ortho-E), and Ortho-E eventually produces reticulocytes. During erythropoiesis, the size of the cells decreases continuously, and the intracellular hemoglobin content gradually increases.
Terminal enucleation is the process by which the nucleus is extruded from late erythrocytes before they mature into reticular cells. This is a unique feature of mammalian erythrocyte production [Citation13]. Some congenital dysplasia of erythropoietic anemia (CDAs) and myelodysplastic syndrome (MDS) may have a pathological phenomenon of erythrocyte enucleation disorder. In MDS, abnormal erythrocyte islands and poor erythrocyte segregation may indicate impaired peripheral enucleation. In CDA type I, caused by CDAN1 or C15orf41 mutations, the encoded proteins are thought to have roles in chromatin remodeling during nuclear extrusion. In CDA type II caused by SEC23B mutations, SEC23B may be involved in mesomeric formation during cell division, and its dysfunction can lead to impaired enucleation [Citation14]. In CDA type III, caused by a KIF23 mutation, the mutant protein disrupts cytokinesis through effects on the mitotic spindle apparatus. In these cases, the molecular mechanisms of impaired enucleation are not fully understood, but may involve protein dysfunction involved in the regulation of chromatin remodeling, cell division, and erythrocyte island formation.
Erythroblast island
Erythroblast island refers to a unique microenvironment in the marrow where erythroblasts, the precursors of erythrocytes, interact with macrophages. The erythroblast island is symbolized by a central macrophage surrounded by a circle of developing erythroblasts.
Macrophages within the erythroblast island play an important role in supporting erythropoiesis, the course by which progenitor cells differentiate into mature erythrocytes. Macrophages provide various factors such as transferrin, iron, cytokines, and growth factors to support erythropoiesis [Citation12].
The erythroblasts in the erythroblast island depend on the macrophages for survival and differentiation. Macrophages provide adhesion molecules and phagocytose the extruded nuclei and organelles from erythroblasts. Additionally, macrophages produce growth factors and cytokines like erythropoietin, stem cell factor, and insulin-like growth factor-1, which promote erythropoiesis and the proliferation of erythroblasts.
The close association between macrophages and erythroblasts within the erythroblast island creates a supportive microenvironment that stimulates the efficient output of functional erythrocytes. Dysfunction or disruption of this microenvironment can lead to impaired erythropoiesis and anemia. Stress erythropoiesis is highly dependent on signals such as EPO/EPOR/JAK2/STAT5, as well as activation signals in erythroid islets such as BMP4/SMAD5 and integrin signals [Citation15].
Stages of RBC maturation and differentiation
Erythropoiesis is a multi-stage process that typically lasts for approximately 14 days. It includes the differentiation of hematopoietic stem cells into erythroid progenitor cells and megakaryocytes. Erythroid progenitor cells consist of erythroid bursting forming units (BFU-E) and erythroid colony forming units (CFU-E), which are a continuous stage of erythroid progenitor cells [Citation16]. These cells then differentiate into erythroid precursors of varying morphologies.
The earliest recognizable erythroid precursor cells are pro-erythroblasts (pro-E), which mark the beginning of the second stage of red blood cell (RBC) maturation. Pro-E cells develop into basophilic (Bas-E), polychromatic (Poly-E), and orthochromatic erythrocytes (Ortho-E) in a sequential manner, ultimately leading to the formation of reticulocytes that lack a nucleus [Citation17]. During this process, the size of the cells and nuclei gradually decreases while the accumulation of hemoglobin increases [Citation1].
Late-stage erythropoiesis is characterized by the loss of the nucleus, loss of surface biomarkers, and the formation of a strong and extendable cytoskeleton. These changes allow for the final maturation and differentiation of erythroid precursors to fully mature erythrocytes [Citation18].
Biomarkers of erythropoiesis
Studies utilizing single-cell genomics have demonstrated that the output of erythrocytes (RBCs) is an ongoing process characterized by varying cellular states and the expression of different genes [Citation16]. The progenitor cells, BFU-E and CFU-E, are highly enriched in the umbilical cord and bone marrow as well as peripheral blood, as indicated by the expression of CD34, CD105, CD36, CD71, and CD45RA markers. By using CD34/CD105 and GPA/CD105 spectra, all four progenitor stages and five terminal erythroid differentiation stages can be identified [Citation19].
Further differentiation of human erythroid precursors can be classified by surface expression of the transferrin receptor (CD71) and glycoprotein A (CD235a). Additionally, the expression of Integrin α4 (CD49D) and band three protein has been found to promote the resolution of multifarious stages of erythroid precursor differentiation [Citation1]. A combination of GYPA (CD235A) and CD105 expression or the gain of band3/SLC4A1 (CD233) expression and loss of CD49d can be used to track erythroid maturation [Citation16].
Biomarkers of erythropoiesis in human and mouse is totally different. In humans, miR-451 and miR-144 are highly expressed during erythropoiesis and play an important role in promoting erythrocyte differentiation. They are located in the same gene cluster and are regulated by the red transcription factor GATA1. miR-486 is also upregulated during erythropoiesis and collaborates with GATA1 as a regulator of erythrocyte differentiation. miR-221 and miR-222 are down-regulated during the differentiation of human hematopoietic progenitor cells into red blood cells. miR-320 is associated with the expression of CD71 protein, a marker of reticular cell maturation in human sickle cell anemia. CD71 and CD36 in early progenitor cells, glycoprotein A in late progenitor cells/precursors, and CD235a in mature red blood cells are key markers of human erythropoiesis.
In mice, as in humans, miR-451 and miR-144 are key red blood cell biomarkers in mice that promote erythropoiesis when overexpressed. miR-150 is down-regulated during erythrocyte terminal differentiation in mice. miR-221, miR-222 and miR-223 were down-regulated during erythropoiesis in mice. let-7 miRNA is upregulated during mouse erythrocyte maturation [Citation20]. In mice, Ter119, CD71, and HE4 are markers of progenitor cells and erythrocytes, and Ter119 is a marker of mature red blood cells. In addition to EPO, SCF and IL-3 are key cytokines that regulate early erythrocyte production in mice. Similar to humans, TGF-β signaling also negatively regulates late erythropoiesis in mice. Transcription factors such as GATA-1 and KLF1 are key mouse erythroid regulators [Citation21].
Regulatory mechanisms of erythropoiesis
Exogenous regulation and control of erythropoiesis
Varieties of external elements can activate essential downstream signaling pathways that regulate the differentiation, proliferation, and survival of erythrocytes. One such factor is IL-3, which can stimulate the proliferation of early progenitors, containing BFU-E. Additionally, KIT ligands can bind to KIT (CD117), promoting the proliferation of BFU-E, CFU-E, and erythroblasts [Citation16].
Wnt signaling also plays a part in the regulation of erythropoiesis, particularly in the terminal differentiation of erythrocytes in myeloid sinusoidal endothelial cells, as well as in the maturity of reticulocytes and the overall function of red blood cells and hematopoietic cells in mesenchymal stromal cell populations [Citation22].
Regulation of erythropoiesis by EPO
EPO is a glycoprotein that plays a crucial role in adjusting erythrocyte quality and hematopoiesis in translocation and formation of erythrocyte transcriptional programs [Citation11]. During stress erythropoiesis, EPO quickly improves the appearance of the anti-apoptotic regulator BCL-XL in early erythrocytes by STAT5 and inhibits the pro-apoptotic proteins BIMsponse to alternations in tissue oxygenation [Citation23]. EPO is produced by renal stromal cells in adults and mainly by the hepar in embryos. EPO contributes to hematopoiesis of hematopoietic stem and progenitor cells (HSPC) by promoting erythroid proliferation and survival from CFU-E, which rapidly decreases with the terminal maturation of the cell [Citation16].
EPO induces erythropoiesis by binding to EPO receptor (EPOR) on precursors of erythroid cells in the marrow, promoting their survival, proliferation, and differentiation. EPOR is expressed primarily on CFU-E, erythrocytes, and early basophils. Binding to EPO prevents apoptosis of these erythroid progenitors [Citation24].
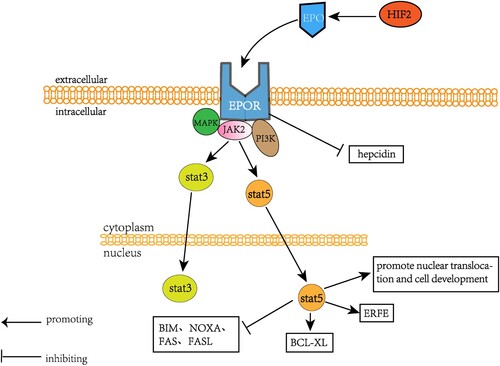
The EPO/EPOR pathway stimulates erythroid development by activating the JAK2/STAT3/STAT5 pathway, which aids in Nucl, NOXA, Fas, and FasL [Citation25]. EPO also stimulates erythroid precursor cells in the marrow and spleen to quickly produce erythroferrone (ERFE) by STAT5 [Citation26].
Activation of STAT3 by EPO/EPO receptors leads to increased expression of genes involved in red blood cell progenitor cell survival (Bcl-xL), proliferation, and differentiation. A key target of STAT3 is the transcription factor GATA-1, which regulates the expression of many red blood-cell specific genes, including those involved in hemoglobin production. STAT3 is rigidly and briefly regulated during erythropoiesis through EPO-mediated JAK2 activation, followed by rapid feedback inhibition through SOCS protein, phosphatase, and proteasome degradation to prevent overactivity. This allows STAT3 to induce erythroid gene expression in a precise manner to match the physiological requirements of red blood cell production [Citation11].
JAK2 inactivation leads to anemia, whereas JAK2 mutations lead to raised erythrocyte quantity and erythrocytosis. EPO and EPOR play critical roles in erythrocyte quality control, and their dysregulation can result in various hematological disorders.
During hypoxia, the highly conserved hypoxia-inducible factor (HIF) signaling pathway is activated, and the first target of HIF is EPO. EPO levels increase by 1000-fold in response to tissue hypoxia. In mice, HIF2a specifically activates EPO, which is required for EPO expression in the kidney and hepar. Destruction of HIF2a leads to anemia and hematopoietic defects. EPO binds to EPOR in the hepar and inhibits the expression of hepcidin, which can also be inhibited by EPO-derived factors [Citation27]. Osteoblasts can also generate EPO to promote erythropoiesis under stable conditions of constitutive HIF. Exogenous EPO and the formation of bone have a connection with raised bone marrow vascular density and angiogenesis in many repair models. EPO directly stimulates osteogenesis by targeting mesenchymal stromal cells and hematopoietic stem cells, and indirectly stimulates osteoblast differentiation by irritating HSCs to secrete bone morphogenetic proteins. EPO also directly stimulates osteoblast differentiation toward mature osteoclasts via Jak2 and PI3K pathways, resulting in an according decline in EPOR transcript levels [Citation11] ().
Figure 3. An overview of the mechanism by which EPO regulates erythropoiesis. Hypoxia-inducible factor 2 (HIF-2) induces EPO production, which in combination with EPOR activates MAPK, JAK2, and PI3K signaling and directly inhibits the expression of hepcidin. JAK2 subsequently activated both STAT3 and STAT5 signaling, in which STAT5 activated erythroid receptor (ERFE) and antiapoptotic regulator BCL-XL, promoting the development of nuclear translocation NRD. Stat5 also inhibited BIM, NOXA, Fas, and FasL.
Regulation of erythropoiesis and iron metabolism by the ERFE-ferritin-transferritin axis
The regulation of iron metabolism and erythropoiesis is crucial, and it also involves the ERFE-ferritin-transferrin axis [Citation16]. Approximately two-thirds of the body’s iron is stored in RBCs [Citation28]. RBCs require 20–25 mg of iron daily, which is supplied by iron transport proteins that contain only 3 milligrams of iron at any given moment and are replaced every several hours [Citation29]. Since the human body cannot excrete iron, the balance between iron intake, transport, utilization, and storage must be tightly regulated [Citation30]. Iron required by RBCs is obtained largely from macrophages phagocytosing old RBCs, and is supplied daily by 20–25 mg of iron, which is absorbed in the intestine by 1–2 mg (0.05% of total iron content in the body) to make up the iron loss due to intestinal epithelial cell loss and a small amount of blood loss in the skin [Citation31].
Non-heme iron can be absorbed from the lumen by the divalent metal transport protein 1 (DMT1) located at the apical surface of epithelial cells in the duodenum by the duodenal cytochrome B reductase (DCYTB), which reduces iron to ferrous metal, and heme iron is absorbed more efficiently in the gut than non-heme iron [Citation32]. Absorbed iron can be utilized by intestinal cells, kept in ferritin, or transported to the circulation by iron a basolateral membrane of the intestinal cells [Citation33]. Iron absorption and tissue distribution are regulated mainly by the interaction of the hepar hormones hepcidin and ferritin. Ferritin exerts its function through three common forms: ferritin coordinated by the side chains of proteins, ferritin in the heme loop, and ferritin in the iron-sul cluster. The iron homeostasis system is mainly affected by erythrocytes (erythrocytes and their precursors in erythropoiesis-producing organs), two storage locations (hepatocytes in the hepar, macrophages in the spleen and hepar), plasma transporting iron between tissues and organs, and absorptive intestinal cells in the duodenum [Citation34].
Erythroferrone (ERFE) is a protein synthesized and secreted by red blood cells in the marrow and extramedullary regions, and is induced by EPO output. ERFE belongs to the C1Q tumor necrosis factor-related protein family, and acts directly in the hepar to decrease hepcidin expression. Unlike EPO, which can stimulate erythropoiesis in both stress and homeostatic conditions, ERFE only responds to stress erythropoiesis. ERFE inhibits hepcidin expression, increases the supply of iron, and helps maintain iron balance during periods of high erythropoietic demand [Citation26].
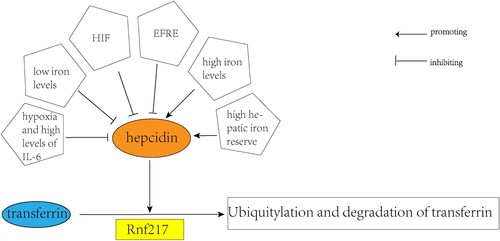
Ferritin is synthesized in the hepar and upon binding to iron, it undergoes ubiquitination and gets degraded in the lysosomes. This process reduces the transfer of ferritin to the cell membrane, keeps iron inside the cell, and lowers the amount of iron in the blood. The regulation of ferritin by endocytosis is similar to general ligand-induced endocytosis in vivo. This process is triggered by transporter conformational changes induced by ferritin, leading to ubiquitin of the lysine-rich cytoplasmic fraction that links the two six-helix regions of ferritin. Ferritin is then targeted for lysosomal and proteasomal degradation through ubiquitination, which is facilitated by Rnf217, an important E3 ubiquitin ligase [Citation26,Citation34].
Hepcidin is regulated by various factors including plasma iron concentration (majorly relying on the contact of ferritin with the transferrin receptors TFR1 and TFR2), hepar iron reserve, systemic inflammation signals transmitted majorly by IL-6 to hepatocytes, and erythroid activity signals transmitted mainly through ERFE concentration [Citation34]. Excessive iron can be toxic and cause oxidative DNA damage [Citation35], and in patients with iron overload, hepcidin levels are elevated. The combination of hepcidin and ferritin results in the rapid degradation of hepcidin. When systemic iron levels are low, this results in decreased expression of hepcidin and raised iron mobilization. Hypoxia inhibits hepcidin expression, and the hypoxia-induced hepcidin inhibition, unrelated to EPO and erythrocyte output, is mediated by hypoxia-mediated degradation of C/EBPA, a key transcription factor required for hepcidin basal expression. Hepar hypoxia is a strong inhibitor of hepcidin expression during hepar inflammation and IL-6 output. HIF has a hand in the suppression of hepcidin, and animal studies have shown that HIF activation in the mouse hepar decreases hepcidin expression and increases ferritin in the intestine and macrophages [Citation27]. Stressful and ineffective erythropoiesis can also inhibit ferritin. GDF15 and TWSG1, members of the transforming growth factor B superfamily, are considered to be pathological inhibitors of hepcidin in ineffective erythropoiesis., GDF15 and TWSG1 inhibit hepcidin expression [Citation26] ().
Figure 4. Mechanism of the action of hepcidin. Various systemic factors regulate the expression of hepcidin. Plasma iron levels and liver iron reserve levels are factors promoting ferritin expression. In contrast, systemic hypoxia, low iron levels, high IL-6 levels in the inflammatory state, and HIF and ERFE were inhibitory factors of hepcidin. Ferritin can cooperate with the E3 ubiquitin ligase Rnf217 for ubiquitylation and degradation of ferritin.
HIF2α is activated in the intestine when there are low levels of iron in the epithelium, leading to an increase in hepcidin-mediated degradation of ferritin. During iron deficiency, PHD activity is reduced, which further enhances the expression of HIF2α. Although intestinal iron levels do not change during erythropoiesis, the decreased oxygen levels in the intestine result in HIF2α activation. Hepcidin exhibits bacteriostatic effects on several bacteria and fungi [Citation27].
The transferrin (Tf) complex consists of two lobes, each of which can bind one iron atom. The degree of Tf saturation reports the body’s iron capacity, which is typically between 20% and 45% in healthy human beings [Citation36]. Transferrin is the only iron transport protein known so far. Stable transferrin binding to the cell membrane stimulates iron absorption by the duodenum, enhances iron recovery by macrophages from old red blood cells and other cells, and allows the transfer of iron contained in the hepar [Citation26]. Most transferrin molecules are bound with hepcidin most of the time, and this mechanism is quickly adjustable when the concentration of hepcidin is reduced. In addition to hepcidin, inflammatory stimulation in the form of Toll-like receptor ligands straightly inhibits cellular ferritin mRNA quantity and reduces cellular plasma iron output [Citation34].
The transferrin (Tf) complex binds one iron molecule and has two receptors, TFR1 and TFR2. TFR1 mediates the internalization of Tf in the iron-transfer pathway, and it can bind partially saturated transferritin, as well as iron in both the light and heavy chains of ferritin. The affinity of TFR1 for c-valve and n-valve saturated transferritin is only 5 and 6 times that of Apo-Tf, respectively. At physiological pH levels, Tf is discharged from TFR1 to the serum, where it can combine with iron again [Citation37]. The IRE/IRP system increases TFR1 expression. The IRE/IRP system functions to regulate cellular iron homeostasis, ensuring that cells can increase TfR1 expression when the iron supply is insufficient, thereby enhancing iron absorption and utilization efficiency. Once the cellular iron level reaches an appropriate level, IRP dissociates from the IRE, leading to a decrease in TfR1 expression to prevent excessive iron uptake. When the cellular iron level is low, IRP binds to the IRE (iron-responsive element) located in the 3′ UTR (untranslated region) of TfR1 mRNA, stabilizing its structure and promoting the stability and translation of TfR1 mRNA. As a result, the expression level of TfR1 increases, allowing cells to more effectively uptake iron [Citation38]. TFR1 is overexpressed in differentiated erythroid cells. Nuclear endosomes including Holo-Tf-TFR1 approach the mitochondria and transfer iron to the organelle by rapid and transient contact with the mitochondria, restraining the cytoplasmic iron pool without triggering the adjustment of the IR/IRP system [Citation36].
Tfr2 is a transmembrane glycoprotein that acts as a receptor for circulating iron. It’s similar to the classical transferrin receptor Tfr1. Tfr2 plays an important part in regulating iron homeostasis by controlling hepcidin levels. Loss-of-function mutations in Tfr2 result in low levels of hepcidin, excess circulating iron, and hemochromatosis [Citation39], while mutations in hepcidin cause severe hereditary iron overload [Citation40].
Unlike TFR1, TFR2 is not adjusted by the IR/IRP system. TFR2 expression is instead regulated post-translationally by its ligand, Holo-Tf [Citation36]. TFR2 protects ferrous plasma Tf from lysosomal degradation and is a part of the erythroid progenitor EpoR complex, critical for valid transfer of EpoR to the cell surface and final differentiation.
Hepatic TfR2 stimulates iron signaling to ferritin to repress further iron influx into the blood when iron is present. In contrast, erythroid TfR2 limits Epo sensitivity to prevent overdose erythropoiesis. Under iron-deficient conditions, TfR2 becomes unstable, leading to hepatic TfR2 downregulation, which inhibits iron signaling to ferritin, stimulates cellular iron excretion, and increases iron supply to RBC. Reduction of erythroid TfR2 promotes Epo’s sensitivity to erythropoiesis and contributes to the suppression of hepcidin by ERFE [Citation41].
Transcriptional control of erythropoiesis
RBC output is regulated by intrinsic transcription factors that regulate the differentiation of erythroid cells. Among these transcription factors, GATA1 plays an important part in directing the differentiation of erythroid cells towards RBC lineage. Another key transcription factor involved in RBC output is KLF1. Mutations in GATA1 and KLF1 are associated with various types of anemia in humans [Citation1]. These transcription factors activate and suppress regulatory networks involved in RBC output and have different functions in this process [Citation16].
Transcriptional regulation of GATA in erythropoiesis
The GATA transcription factor, which contains zinc-finger DNA binding domains, is critical for various biological processes, including hematopoiesis [Citation42]. GATA factors interact with specific regulatory elements to mediate transcriptional changes [Citation43]. GATA-1 is essential for the existence and differentiation of progenitors of red blood cells, while GATA-2 adjusts the survival and proliferation of hematopoietic stem and progenitor cells [Citation12]. GATA1 has three functional domains, including an N-terminal activation domain and two homologous Zn finger domains located in the C-terminal region [Citation44]. Various cofactors, such as FOG-1/ZFPM1, nuclear remodeling bodies, and deacetylase (NURD) complexes, promote the binding of GATA1 to regulatory sites [Citation16].
During erythroid cell development, GATA factor switching occurs, whereby GATA2 protein levels are decreased and GATA1 protein levels are raised. GATA1 targets several genes that are regulated by GATA2 in hematopoietic stem and progenitor cells [Citation43]. In erythroid cells, GATA1 occupies both the promoter and enhancer, but the enhancer activity decreases during differentiation, with GATA1 predominantly binding to the promoter. Enhancers play a more significant role in the early stages of cell differentiation, while promoters are more critical in the terminal stages.
In mouse erythroid progenitors, the GATA2 transcription factor increases the production of the stem cell cytokine receptor KIT, while GATA1 is responsible for reducing KIT expression to promote terminal differentiation. However, in human erythroid progenitors, high levels of KIT expression require GATA1 binding on erythroid-specific SE.
GATA1 is regulated by various factors at the post-transcriptional level. HSP27 and HSP70 members of the heat shock protein family, along with ribosomal proteins, promote the ubiquitination and proteasomal degradation of GATA1. HSP70 also plays a role in preventing the caspase-3-mediated intranuclear division of GATA1, while ribosomal proteins regulate the normal translation of GATA1 [Citation45]. Additionally, there is an interaction between GATA1 and p53, which inhibits RNA Pol I transcription by restricting the RNA Pol I complex from assembling on the rDNA facilitator. In cancer, the loss of function of p53 and mass output of ribosomes may lead to the inhibition of GATA1 by p53 [Citation17]. On the other hand, downregulation of p19INK4d reduces GATA1 protein levels and impairs human terminal erythroid differentiation, which is adjusted by the p-ERK-HSP70-GATA1 pathway, where PEBP1 serves as a connection between p19INK4d and the p-ERK-HSP70-GATA1 pathway [Citation45].
Transcriptional regulation of KLF1 in erythropoiesis
KLF1 is a protein that is abundant in erythroid cells and contains three C2H2 zinc fingers at its C-terminus. It connects its DNA consensus sequence (5′ccmcrcccn) mainly in the distal regulatory region, and is expressed in both primitive and nucleated erythrocytes within the hematopoietic compartment [Citation46]. Its primary role in erythropoiesis is the activation of gene transcription, and it inhibits the differentiation of megakaryocytes while promoting early differentiation of RBCs [Citation47].
KLF1 plays an important part in the final stages of the cell cycle and chromatin condensation, which stimulate RBC output. In the absence of KLF1, RBC denuclearization is impaired, CDKN2C and CDKN1A levels are reduced, and RBC proliferation is raised. Although KLF1 is predominantly a transcriptional activator, it can also act as a transcriptional inhibitor [Citation16]. Coactivator-coinhibitory interactions under KLF1 acetylation dynamically regulate downstream transcriptional effects [Citation47].
Transcriptional regulation of erythropoiesis by other factors
TGF-beta is a crucial factor in cell differentiation and has significant effects on hematopoiesis [Citation48]. TGFβ1 is involved in cell survival, proliferation, and differentiation. Megakaryocytes are the primary source of TGFβ1 in the bone marrow, but they can also be synthesized by immune cells, macrophages, and marrow stromal cells. TGFβ1 inhibits early progenitor cell proliferation and stimulates erythroid terminal differentiation through Smad2/3-Smad4 or tiff-1g signaling. Dysregulation of TGFβ1 may lead to myelofibrosis, while blocking it may result in the proliferation of erythroid progenitors [Citation49]. Additionally, members of the TGFβ family promote the degradation of HIF1α by inhibiting VHL, an E3 ubiquitin ligase, which enhances hypoxia responses [Citation50].
Unusual and common erythropoietic disorders
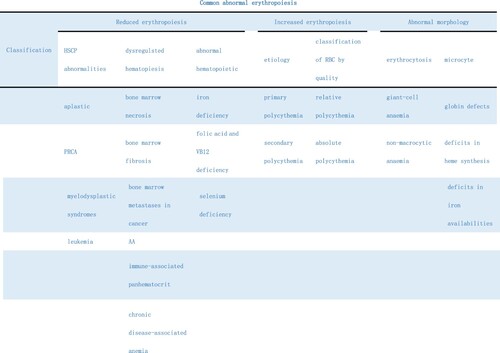
Anemia is a condition characterized by a hemoglobin level below 13 g/dL in adult males and below 12 g/dL in non-pregnant females [Citation51], and can result from decreased erythropoiesis, excessive red blood cell destruction, or blood loss [Citation52]. Reduced erythropoiesis can be sorted into several categories, including anemia caused by abnormalities in hematopoietic stem cells (e.g. aplastic anemia, PRCA, myelodysplastic syndrome, leukemia), abnormalities in hematopoietic regulation (e.g. myeloid necrosis, myelofibrosis, tumor disease, myeloid metastasis, bone marrow stromal-cell-involved diseases), hyperlymphocyte dysfunction (e.g. AA and immune-associated pancytopenia), abnormalities in hematopoietic regulatory factors (e.g. chronic disease-associated anemia), hyperapoptosis (e.g. paroxysmal nocturnal hemoglobinuria), or deficiency of hematopoietic materials (e.g. iron-deficiency anemia, deficiency of iron, leucine, and vitamin B12, and megaloblastic anemia) [Citation53]. Additionally, low levels of selenium, which is involved in redox reactions by binding to the selenium family of cysteine-containing proteins and has an antioxidant effect, can lead to reduced erythroid cell number and differentiation under stress, resulting in an ineffective response. It is important not to overlook selenium deficiency in cases of reduced erythropoiesis [Citation54].
Reduced erythropoiesis
Anemia due to hematopoietic stem cell abnormalities
MDS refers to a cluster of clonal diseases characterized by morphological dysplasia of hematopoietic stem cells, hematopoietic failure, and peripheral cell loss, leading to decreased erythropoiesis. MDS cells, usually derived from pluripotent hematopoietic stem cells, are malignant tumors that typically cannot be cured except through allogeneic stem cell transplantation (SCT) [Citation55]. About 50% of MDS patients possess somatic mutations in the spliceosome gene, with SF3B1 being the most frequent one [Citation56], and SF3B1 mutations primarily linked to abnormal 3′ mRNA splicing. In vitro and in vivo, SF3B1K700E cells showed raised sensitivity to the spliceosome regulator E7107, and the usage of spliceosome regulators may prove effective in treating SF3B1 mutational hematologic malignancies [Citation57]. Moreover, loss or mutations in MDS and TET2(4q24) have been linked, and TET2 mutations can cause various disorders such as myeloproliferative neoplasms, chronic myelomonocytic leukemia, acute myeloid leukemia, and decreased erythropoiesis [Citation58].
Aplastic anemia (AA) is characterized by marrow failure (BMF) status, which results in cytopenia and ineffective hematopoiesis. The disease is classified as hereditary or acquired, with the latter usually associated with established germline mutations, and occurs in two peaks, one in patients aged 15–25 years and another in those older than 60 years. The prognosis of severe or very severe acquired AA with supportive therapy alone is poor, with mortality rates exceeding 80% after two years [Citation59]. The major factors that lead to AA are direct bone marrow injury, genetic mutations that reduce the DNA repair capacity of hematopoietic stem cells, immune factors, and possibly environmental contamination and viral infections [Citation60]. Hematopoietic stem cell transplantation is the curative treatment for AA, while immunosuppressive treatment (IST) with horse anti thymocyte globulin and cyclosporine A is the first-line treatment for elderly patients and all patients lacking matched sibling donors [Citation61].
Anemia due to abnormal hematopoietic regulation
Chronic disease anemia (ACD), also acknowledged as inflammatory anemia (AI), is the second most common type of anemia worldwide. This type of anemia is primarily induced by hepcidin, an inflammatory cytokine that regulates iron homeostasis by blocking intestinal iron absorption, trapping iron in reticuloendothelial cells, and reducing iron availability for erythropoiesis [Citation62]. The transferring of iron from macrophages to developing erythroid cells also requires transferrin FPN. In the absence of FPN, iron remains trapped in macrophages, leading to impaired iron transferring and erythroid maturation arrest in developing erythroid cells. Inflammatory cytokines also decrease EPO synthesis, impair erythroid progenitor differentiation, and shorten the lifespan of mature erythroid cells [Citation63]. These signals also increase peripheral red cell depletion [Citation50].
Oncogenic anemia is a type of chronic disease anemia associated with the activation of cytokines like interferon-gamma, interleukin-1, and tumor necrosis factor (TNF), which can suppress the output of endogenous EPO, damage iron utilization, and decrease erythroid precursor proliferation [Citation64]. Apoptosis is a significant contributor to the pathogenesis of anemia in chronic kidney disease, and approximately half of the patients with chronic heart failure anemia also have evidence of endogenous apoptosis and/or functional iron deficiency [Citation65].
A new treatment for anemia in chronic kidney disease is the hypoxia-inducible factor proline hydroxylase inhibitor, which blocks the degradation of the transcription factor hypoxia-inducible factor, stimulating erythropoiesis to physiological levels [Citation66].
Anemia with abnormal erythrocyte morphology
Erythrocyte abnormalities can manifest as large, small, or misshapen cells. Normal erythrocytes have a diameter of 7–8 μm and a mean cell volume (MCV) of 80–95 fl, with larger erythrocytes above this range and smaller erythrocytes below. The different subgroups of erythrocyte abnormalities include globin chain defects (such as hemoglobinemia or thalassemia), heme synthesis defects, iron availability defects, or precursor iron acquisition defects [Citation67]. Macrocytic anemia, which is characterized by enlarged erythrocytes, can be further categorized into megaloblastic anemia (caused by defects in RNA and DNA synthesis) and non-megaloblastic anemia [Citation68].
Plasmodium infection not only reduces erythropoiesis but also inhibits the bone marrow’s response to EPO and leads to abnormalities in nuclear ultrastructure, including polynuclei, nuclear fragments, internuclear bridges, and irregular nuclear shapes. Furthermore, parasites have immune escape mechanisms that modify host immune responses by increasing cytokine output, including interleukin-6 (IL-6), IL-12, IL-1, IL-10, tumor necrosis factor-alpha (TNFalpha), and interferon-gamma (IFN-gamma). These inflammatory cytokines can strongly impact erythropoiesis [Citation69].
Thalassemia
Mutations in the hemoglobin synthesis gene lead to reduced or absent production of α or β-globin chains and an imbalance in the ratio of α / non-α-globin chains, resulting in thalassemia.
In beta-thalassemia, mutations in the HBB gene lead to insufficient production of beta-globin. This leads to excessive amounts of unstable alpha-globin chains that aggregate in red blood cell precursors, leading to ineffective production of red blood cells and premature destruction of red blood cells [Citation70].
There are two alpha-globin genes on chromosome 16 – α1 and α2, and alpha-thalassemia can be caused by a deletion or point mutation of one or both alpha-globin genes, resulting in a defect in the alpha-globin chain. When alpha-globin production is impaired, excess β-globin chains form tetramers (HbH) with high oxygen affinity and poor stability, leading to hemolysis [Citation71].
The severity of thalassemia is determined by the degree of imbalance in the alpha – / non-alpha globin ratio. Patients with transfusion-dependent thalassemia (TDT) have severe imbalances and require lifelong transfusions. Patients with non-transfusion-dependent thalassemia (NTDT) have less imbalance and anemia.
The body tries to compensate for anemia by increasing red blood cell production in the bone marrow. However, this enlarged but ineffective erythrocyte production leads to further abnormalities. For example, it increases the intestinal absorption of iron and inhibits the iron-regulating hormone hepcidin. This can lead to an iron overload in the liver and heart. Ineffective erythropoiesis also causes bone marrow dilatation and extramedullary hematopoiesis, leading to bone abnormalities and splenomegaly [Citation72].
Erythrocytosis
Polycythemia is a condition where the erythrocyte count exceeds the normal sex range, and it can be categorized into relative polycythemia induced by a decrease in plasma volume and absolute polycythemia induced by an increase in the quality of red blood cells. Primary polycythemia occurs due to the spontaneous output of erythrocytes, usually from myeloproliferative neoplasms (also known as pseudo polycythemia or PV). On the other hand, secondary polycythemia occurs when the body responds appropriately to elevated serum erythropoietin levels [Citation73], such as in post-renal transplant polycythemia where erythropoiesis is driven by both allografts and native kidneys [Citation74]. Polycythemia includes various types such as thrombosis, idiopathic polycythemia, congenital polycythemia, hypoxic lung disease, and post-transplant polycythemia [Citation75]. The majority of polycythemia vera cases are linked to acquired JAK2 variants. Different types of familial polycythemia are associated with various defects, such as erythropoietin hypersensitivity, defects in the oxygen sensitivity pathway, or an increase in hemoglobin affinity for oxygen [Citation76]. Polycythemia vera may also result from mutations in the congenital hypoxia-inducible factor pathway [Citation77]. It is crucial to distinguish PV from polycythemia due to other causes because early detection and treatment of PV can prevent numerous vasomotor and thrombotic complications. Patients with PV often have an enlarged plasma volume that can mask the polycythemia, making diagnosis difficult if this basic fact is ignored [Citation78]. Polycythemia may not cause any symptoms, or it may present with thrombosis, vasomotor symptoms, or splenomegaly [Citation73].
When erythropoiesis is reduced, the body compensates by inducing extramedullary erythropoiesis, also known as stress erythropoiesis. Unlike the continuous output of erythrocytes in steady-state erythropoiesis, stress-induced erythropoiesis produces a large number of new erythrocytes through synchronous differentiation of progenitor cells. The bone marrow temporarily migrates hematopoietic cells to the spleen to re-establish this process [Citation50]. Early progenitor cells, which are usually in the quiescent phase, are favored by the HSPC component in extramedullary tissue, but proliferation is inhibited [Citation79]. Additionally, anemia stress increases erythrocyte phagocytosis, and splenic macrophages promote CCL2 output, which recruits circulating monocytes and forms new hematopoietic islands in the spleen. Macrophages regulate the circulation of splenic erythroid cells by removing aging and impaired red blood cells, and by interacting with developing proto-red blood cells [Citation80]. Moreover, macrophages interact with erythroblasts through direct adhesion and indirect cells, such as erythrocyte-release positive regulators that inhibit the interaction of specific protein 6 (Gas6), vascular endothelial growth factor A (VEGF-A), and placental growth factor (PlGF) to regulate stress erythropoiesis [Citation81] ().
Figure 5. Classification of Common Abnormal Erythropoietic Diseases. Abnormalities of erythropoiesis may be classified as increased erythropoiesis. Reduced erythropoiesis and abnormal erythrocyte morphology. Reduced erythropoiesis can be divided into hematopoietic stem cell abnormalities, dysregulation of hematopoiesis, and obstruction of hematopoietic material utilization. Hypererythropoiesis may be classified as primary or secondary, or as relative or absolute, depending on the amount of red blood cell mass.
Therapeutic significance
Anemia has a complex multi-factorial etiology. The main causes include nutritional deficiencies (iron, vitamin A, B12, folate), infectious diseases (malaria, HIV, hookworm), inherited hemoglobin disorders (sickle cell, thalassemia), and inflammation. Fetal hemoglobin (HbF) induction remains a promising approach for the treatment of sickle cell disease and beta-thalassemia. Strategies include targeting transcription factors that regulate fetal to adult hemoglobin conversions, such as BCL11A, KLF1, and MYB, or using epigenetic regulators such as HDAC and LSD1 inhibitors. Gene therapy using lentiviral vectors to express the β-globin gene or shRNA targeting BCL11A in hematopoietic stem cells has shown the potential to permanently correct hemoglobinopathy. Gene editing using CRISPR/Cas9 to correct potential mutations is also being explored. Erythropoietic drugs such as PHD inhibitors and activin receptor traps (such as Sotercept) are emerging to treat inflammatory anemia, chronic kidney disease, cancer, and more. Management of iron homeostasis with hepcidin modulators, small hepcidin, or TMPRSS6 inhibitors may help treat iron overload in patients with transfusion-dependent thalassemia. Anti-inflammatory therapies such as selectin inhibitors, anti-CXCR2, or iNKT cell blocking may alleviate vaso-blocking crises and organ damage in sickle cell disease. Enzyme or molecular replacement gene therapy appears to hold promise for treating rare inherited anemia such as pyruvate kinase deficiency and congenital erythropoietic porphyria [Citation53].
For polycythemia vera, maintaining hematocrit targets of <45% in men and <42% in women by phlebotomy is the basis of treatment to prevent thrombosis, which is a major immediate health threat. Targeted therapies such as ruxolitinib and pegylated interferon can be used for symptom control and to reduce splenomegaly, but their role in preventing thrombosis is unclear. Hydroxyurea is effective in reducing cells, but does not affect disease progression, with risks such as myelofibrosis and leukemia transformation. Therefore, its use should be limited. Treatment for pregnant patients includes strict blood pressure control and phlebotomy, but aspirin and anticoagulants are avoided unless at high risk. Pegylated interferon is safe when necessary. For advanced myelofibrosis, ruxolitinib, thalidomide-prednisone, and azacytidine may be selected, but hematopoietic stem cell transplantation is curable when feasible [Citation78].
In conclusion, erythropoiesis is a complex and dynamic process that occurs primarily in the marrow, but can also occur extramedullary under stress conditions. The process is influenced by various factors including bone marrow microenvironment, macrophages, exogenous and endogenous factors, and intrinsic transcriptional regulators such as GATA and KLF. Abnormalities in erythropoiesis can lead to various hematologic disorders such as anemia, erythrocytosis, and polycythemia vera. Studies on erythropoiesis have provided valuable clinical insights, leading to the development of new treatments for hematologic malignancies and anemia, and early recognition and prevention of thrombosis in polycythemia vera.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Nandakumar SK, Ulirsch JC, Sankaran VG. Advances in understanding erythropoiesis: evolving perspectives. Br J Haematol. 2016;173(2):206–218. DOI:10.1111/bjh.13938
- Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med. 2013;3(4):011–601.
- Yumine A, Fraser ST, Sugiyama D. Regulation of the embryonic erythropoietic niche: a future perspective. Blood Res. 2017;52(1):10–17. DOI:10.5045/br.2017.52.1.10
- Yamane T. Cellular basis of embryonic hematopoiesis and its implications in prenatal erythropoiesis. Int J Mol Sci. 2020;21(24):9346. DOI:10.3390/ijms21249346
- Popescu DM, Botting RA, Stephenson E, et al. Decoding human fetal liver haematopoiesis. Nature. 2019;574(7778):365–371. DOI:10.1038/s41586-019-1652-y
- Palis J. Interaction of the macrophage and primitive erythroid lineages in the mammalian embryo. Front Immunol. 2016;7:669.
- Baron MH, Vacaru A, Nieves J. Erythroid development in the mammalian embryo. Blood Cells, Molecules, & Diseases. 2013;51(4):213–219. DOI:10.1016/j.bcmd.2013.07.006
- Chakraborty S, Andrieux G, Kastl P, et al. Erythropoietin-driven dynamic proteome adaptations during erythropoiesis prevent iron overload in the developing embryo. Cell Rep. 2022;40(12):111360. DOI:10.1016/j.celrep.2022.111360
- Hidalgo D, Bejder J, Pop R, et al. Epor stimulates rapid cycling and larger red cells during mouse and human erythropoiesis. Nat Commun. 2021;12(1):7334. DOI:10.1038/s41467-021-27562-4
- Barminko J, Reinholt B, Baron MH. Development and differentiation of the erythroid lineage in mammals. Dev Comp Immunol. 2016;58:18–29. DOI:10.1016/j.dci.2015.12.012
- Eggold JT, Rankin EB. Erythropoiesis, EPO, macrophages, and bone. Bone. 2019;119:36–41. DOI:10.1016/j.bone.2018.03.014
- Zivot A, Lipton JM, Narla A, et al. Erythropoiesis: insights into pathophysiology and treatments in 2017. Mol Med. 2018;24(1):11–11. DOI:10.1186/s10020-018-0011-z
- Iolascon A, Andolfo I, Russo R. Congenital dyserythropoietic anemias. Blood. 2020;136(11):1274–1283. DOI:10.1182/blood.2019000948
- Valent P, Büsche G, Theurl I, et al. Normal and pathological erythropoiesis in adults: from gene regulation to targeted treatment concepts. Haematologica. 2018;103(10):1593–1603. DOI:10.3324/haematol.2018.192518
- Ramos P, Casu C, Gardenghi S, et al. Macrophages support pathological erythropoiesis in polycythemia vera and beta-thalassemia. Nat Med. 2013;19(4):437–445. DOI:10.1038/nm.3126
- Caulier AL, Sankaran VG. Molecular and cellular mechanisms that regulate human erythropoiesis. Blood. 2022;139(16):2450–2459. DOI:10.1182/blood.2021011044
- Le Goff S, Boussaid I, Floquet C, et al. P53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood. 2021;137(1):89–102. DOI:10.1182/blood.2019003439
- Gibson JS, Rees DC. Lipid metabolism in terminal erythropoiesis. Blood. 2018;131(26):2872–2874. DOI:10.1182/blood-2018-05-850255
- Yan H, Ali A, Blanc L, et al. Comprehensive phenotyping of erythropoiesis in human bone marrow: evaluation of normal and ineffective erythropoiesis. Am J Hematol. 2021;96(9):1064–1076. DOI:10.1002/ajh.26247
- Sun L, Yu Y, Niu B, et al. Red blood cells as potential repositories of MicroRNAs in the circulatory system. Front Genet. 2020;11:442. DOI:10.3389/fgene.2020.00442
- Parisi S, Finelli C, Fazio A, et al. Clinical and molecular insights in erythropoiesis regulation of signal transduction pathways in myelodysplastic syndromes and β-thalassemia. Int J Mol Sci. 2021;22(2):827. DOI:10.3390/ijms22020827
- Heil J, Olsavszky V, Busch K, et al. Bone marrow sinusoidal endothelium controls terminal erythroid differentiation and reticulocyte maturation. Nat Commun. 2021;12(1):6963. DOI:10.1038/s41467-021-27161-3
- Tsiftsoglou AS. Erythropoietin (EPO) as a key regulator of erythropoiesis, bone remodeling and endothelial transdifferentiation of multipotent mesenchymal stem cells (MSCs): implications in regenerative medicine. Cells. 2021;10(8):2140. DOI:10.3390/cells10082140
- Shih H, Wu C, Lin S. Physiology and pathophysiology of renal erythropoietin-producing cells. J Formos Med Assoc. 2018;117(11):955–963. DOI:10.1016/j.jfma.2018.03.017
- Oikonomidou PR, Rivella S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018;32(2):130–143. DOI:10.1016/j.blre.2017.10.001
- Kautz L, Nemeth E. Molecular liaisons between erythropoiesis and iron metabolism. Blood. 2014;124(4):479–482. DOI:10.1182/blood-2014-05-516252
- Shah YM, Xie L. Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology. 2014;146(3):630–642. DOI:10.1053/j.gastro.2013.12.031
- Zhang A, Enns CA. Grab and go: transferrin uptake in erythropoiesis. Blood. 2022;140(10):1061–1063. DOI:10.1182/blood.2022017638
- Ginzburg YZ. Chapter two – hepcidin-ferroportin axis in health and disease. In: G Litwack, editor. Vitamins and hormones. Academic Press; 2019. p. 17–45.
- Sinha S, Pereira-Reis J, Guerra A, et al. The role of iron in benign and malignant hematopoiesis. Antioxid Redox Signal. 2021;35(6):415–432. DOI:10.1089/ars.2020.8155
- Chifman J, Laubenbacher R, Torti SV. A systems biology approach to iron metabolism. In: J Chifman, R Laubenbacher, SV Torti, editors. Advances in Experimental Medicine and Biology. New York, NY: Springer New York; 2014. p. 201–225.
- Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020;105(2):260–272. DOI:10.3324/haematol.2019.232124
- Wojtaszek E, Glogowski T, Malyszko J. Iron and chronic kidney disease: still a challenge. Front Med (Lausanne). 2020;7:565135. DOI:10.3389/fmed.2020.565135
- Nemeth E, Ganz T. Hepcidin-ferroportin interaction controls systemic iron homeostasis. Int J Mol Sci. 2021;22(12):6493. DOI:10.3390/ijms22126493
- Morales M, Xue X. Targeting iron metabolism in cancer therapy. Theranostics. 2021;11(11):8412–8429. DOI:10.7150/thno.59092
- Kohgo Y, Niitsu Y, Kondo H, et al. Serum transferrin receptor as a new index of erythropoiesis. Blood. 1987;70(6):1955–1958. DOI:10.1182/blood.V70.6.1955.1955
- Blanchette NL, Manz DH, Torti FM, et al. Modulation of hepcidin to treat iron deregulation: potential clinical applications. Expert Rev Hematol. 2016;9(2):169–186. DOI:10.1586/17474086.2016.1124757
- Zhou ZD, Tan EK. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol Neurodegener. 2017;12(1):75. DOI:10.1186/s13024-017-0218-4
- Ginzburg YZ, Fleming RE. Tfr2 suppression benefits B-thalassemic erythropoiesis. Blood. 2018;132(21):2215–2216. DOI:10.1182/blood-2018-10-876110
- Kim A, Nemeth E. New insights into iron regulation and erythropoiesis. Curr Opin Hematol. 2015;22(3):199–205. DOI:10.1097/MOH.0000000000000132
- Pantopoulos K. Tfr2 links iron metabolism and erythropoiesis. Blood. 2015;125(7):1055–1056. DOI:10.1182/blood-2014-12-617571
- Fujiwara T. GATA transcription factors: basic principles and related human disorders. Tohoku J Exp Med. 2017;242(2):83–91. DOI:10.1620/tjem.242.83
- Romano O, Petiti L, Felix T, et al. GATA factor-mediated gene regulation in human erythropoiesis. iScience. 2020;23(4):101018. DOI:10.1016/j.isci.2020.101018
- Barbarani G, Fugazza C, Strouboulis J, et al. The pleiotropic effects of GATA1 and KLF1 in physiological erythropoiesis and in dyserythropoietic disorders. Front Physiol. 2019;10:91. DOI:10.3389/fphys.2019.00091
- Han X, Zhang J, Peng Y, et al. Unexpected role for p19INK4d in posttranscriptional regulation of GATA1 and modulation of human terminal erythropoiesis. Blood. 2017;129(2):226–237. DOI:10.1182/blood-2016-09-739268
- Gnanapragasam MN, Bieker JJ. Orchestration of late events in erythropoiesis by KLF1/EKLF. Curr Opin Hematol. 2017;24(3):183–190. DOI:10.1097/MOH.0000000000000327
- Kim MY, Yan B, Huang S, et al. Regulating the regulators: the role of histone deacetylase 1 (HDAC1) in erythropoiesis. Int J Mol Sci. 2020;21(22):8460. DOI:10.3390/ijms21228460
- Nakamura K, Smyth MJ. Aberrant erythropoiesis fuels tumor growth. Cell Res. 2018;28(6):611–612. DOI:10.1038/s41422-018-0047-1
- Villeval J, Vainchenker W. Megakaryocytes tame erythropoiesis with TGFβ1. Blood. 2020;136(9):1016–1017. DOI:10.1182/blood.2020006906
- Paulson RF, Ruan B, Hao S, et al. Stress erythropoiesis is a key inflammatory response. Cells. 2020;9(3). DOI:10.3390/cells9030634
- Tomasz G, Ewa W, Jolanta M. Biomarkers of iron metabolism in chronic kidney disease. Int Urol Nephrol. 2021;53(5):935–944. DOI:10.1007/s11255-020-02663-z
- Chaparro CM, Suchdev PS. Anemia epidemiology, pathophysiology, and etiology in low- and middle-income countries. Ann N Y Acad Sci. 2019;1450(1):15–31.
- Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med. 2015;21(3):221–230. DOI:10.1038/nm.3814
- Dulmovits BM, Blanc L. Stress erythropoiesis: selenium to the rescue!. Blood. 2018;131(23):2512–2513. DOI:10.1182/blood-2018-04-844720
- Hellstrom-Lindberg E, Tobiasson M, Greenberg P. Myelodysplastic syndromes: moving towards personalized management. Haematologica. 2020;105(7):1765–1779. DOI:10.3324/haematol.2020.248955
- Malcovati L, Stevenson K, Papaemmanuil E, et al. SF3B1-mutant MDS as a distinct disease subtype: a proposal from the international working group for the prognosis of MDS. Blood. 2020;136(2):157–170. DOI:10.1182/blood.2020004850
- Obeng EA, Chappell R, Seiler M, et al. Physiologic expression of SF3B1K700E causes impaired erythropoiesis, aberrant splicing, and sensitivity to pharmacologic spliceosome modulation. Cancer Cell. 2016;30(3):404–417. DOI:10.1016/j.ccell.2016.08.006
- Wojchowski DM. Ineffective erythropoiesis of TET2 deficiency. Blood. 2018;132(22):2320–2321. DOI:10.1182/blood-2018-10-878645
- Schoettler ML, Nathan DG. The pathophysiology of acquired aplastic anemia: current concepts revisited. Hematol Oncol Clin North Am. 2018;32(4):581–594. DOI:10.1016/j.hoc.2018.03.001
- NS Y. Aplastic anemia. N Engl J Med. 2018;17(379):1643–1656.
- Bacigalupo A, Passweg J. Diagnosis and treatment of acquired aplastic anemia. Hematol Oncol Clin North Am. 2009;23(2):159–170. DOI:10.1016/j.hoc.2009.01.005
- Weiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood. 2019 ;133(1):40–50. DOI:10.1182/blood-2018-06-856500
- Fraenkel PG. Anemia of inflammation: a review. Med Clin N Am. 2017;101(2):285–296. DOI:10.1016/j.mcna.2016.09.005
- Abdel-Razeq H, Hashem H. Recent update in the pathogenesis and treatment of chemotherapy and cancer induced anemia. Crit Rev Oncol Hematol. 2020;145:102837. DOI:10.1016/j.critrevonc.2019.102837
- Cazzola M. Introduction to a How I treat series on anemia. Blood. 2020.
- Hanna RM, Streja E, Kalantar-Zadeh K. Burden of anemia in chronic kidney disease: beyond erythropoietin. Adv Ther. 2021;38(1):52–75. DOI:10.1007/s12325-020-01524-6
- Cappellini MD, Russo R, Andolfo I, et al. Inherited microcytic anemias. Hematology. 2020;2020(1):465–470. DOI:10.1182/hematology.2020000158
- Newhall DA, Oliver R, Lugthart S. Anaemia: a disease or symptom. Neth J Med. 2020;3(78):104–110.
- Dumarchey A, Lavazec C, Verdier F. Erythropoiesis and malaria, a multifaceted interplay. Int J Mol Sci. 2022;23(21):12762. DOI:10.3390/ijms232112762
- Musallam KM, Bou-Fakhredin R, Cappellini MD, et al. 2021 update on clinical trials in β-thalassemia. Am J Hematol. 2021;96(11):1518–1531. DOI:10.1002/ajh.26316
- Munkongdee T, Chen P, Winichagoon P, et al. Update in laboratory diagnosis of thalassemia. Front Mol Biosci. 2020;7:74–74. DOI:10.3389/fmolb.2020.00074
- Motta I, Bou-Fakhredin R, Taher AT, et al. Beta thalassemia: new therapeutic options beyond transfusion and iron chelation. Drugs (New York, NY). 2020;80(11):1053–1063.
- Mithoowani S, Laureano M, Crowther MA, et al. Investigation and management of erythrocytosis. CMAJ. 2020;192(32):913–918. DOI:10.1503/cmaj.191587
- Alzoubi B, Kharel A, Machhi R, et al. Post-transplant erythrocytosis after kidney transplantation: a review. World J Transplant. 2021;11(6):220–230. DOI:10.5500/wjt.v11.i6.220
- McMullin MFF, Mead AJ, Ali S, et al. A guideline for the management of specific situations in polycythaemia vera and secondary erythrocytosis: a British society for haematology guideline. Br J Haematol. 2019;184(2):161–175. DOI:10.1111/bjh.15647
- Gaspersic J, Kristan A, Kunej T, et al. Erythrocytosis: genes and pathways involved in disease development. Blood Transfus. 2021;19(6):518–532.
- Cazzola M, Cazzola M. Introduction to a review series on normal and pathologic erythropoiesis. Blood. 2022;139(16):2413–2414. DOI:10.1182/blood.2022015497
- Spivak JL. How I treat polycythemia vera. Blood. 2019;4(134):345–352.
- Mende N, Bastos HP, Santoro A, et al. Unique molecular and functional features of extramedullary hematopoietic stem and progenitor cell reservoirs in humans. Blood. 2022;139(23):3387–3401. DOI:10.1182/blood.2021013450
- Haldar M. Stressed about erythropoiesis: EBI macrophages. Blood. 2018;132(24):2530–2532. DOI:10.1182/blood-2018-10-882183
- Ulyanova T, Phelps SR, Papayannopoulou T. The macrophage contribution to stress erythropoiesis: when less is enough. Blood. 2016;128(13):1756–1765. DOI:10.1182/blood-2016-05-714527