
ABSTRACT
Objective
The 4.2 kb deletion (-α4.2/) is a common a+-thalassemia with a carrier rate, followed by the South-East Asian deletion (–SEA) and the 3.7 kb deletion (-α3.7/). There are few reports about 4.2 kb deletion sub-types. Herein, we present a patient with double heterozygous -α4.2Ⅰ/-α4.2Ⅱwho was identified using third-generation sequencing (TGS).
Methods
Hematology and hemoglobin fraction analysis were carried out by complete blood count (CBC) and capillary electrophoresis (CE). Gap-PCR was used to detect the common deletional α-thalassemia, and multiple ligation-dependent probe amplification (MLPA) was performed to screen the large deletion. Sanger sequencing identified the variant. The different deletions were confirmed by TGS.
Results
CBC showed the patient with microcytic hypochromic anemia, and CE indicated the presence of a Hb variant. Gap-PCR and MLPA detected 4.2 kb deletion homozygotes (-α4.2/-α4.2). The Hb variant was confirmed as Hb Q-Thailand by Sanger sequencing. The patient was identified as compound heterozygous of 4.2 kb deletion and Hb Q-Thailand (-α4.2/-α4.2-Q-Thailand, -α4.2Ⅰ/-α4.2Ⅱ) using TGS.
Conclusions
Hb Q-Thailand (-α4.2-Q-Thailand/) complex 4.2 kb deletion heterozygote (-α4.2/) is easily misdiagnosed as 4.2 kb homozygous using Gap-PCR and MLPA. The TGS enables the identification of the two different 4.2 kb deletion sub-types.
Introduction
α-thalassemia is the most common inherited monogenic disease worldwide [Citation1,Citation2]. According to the presence or absence of globin chain function, it can be classified into α+-thalassemia and α0-thalassemia. In China, the most common form of α0-thalassemia is the SEA deletion (–SEA), and that of α+-thalassemia is the 3.7 kb deletion (-α3.7) [Citation3]. 4.2 kb deletion is a+-thalassemia with a carrier rate second only to 3.7 kb deletion. The 3.7 kb deletion is created by an unequal crossing over or crossing to the right of the Z-box of the α-globin gene cluster, resulting in one chromosome with one α-globin gene (-α3.7) and another with a triplet of α-globin genes (ααα3.7). Similarly, crossovers between misaligned X-box will give rise to -α4.2 and ααα4.2 [Citation4]. Either -α3.7 or -α4.2, when combined with –SEA, can cause phenotypic variable Hb H disease (–SEA/-α3.7 or –SEA/-α4.2). Hb H disease is highly prevalent in southern China, placing an economic burden on families and forming a public health problem. There are some reports in the literature that a 3.7 kb deletion can be classified as sub-type Ⅰ (-α3.7Ⅰ), sub-type Ⅱ (-α3.7Ⅱ), and sub-type Ⅲ (-α3.7Ⅲ) depending on the deletion breakpoints [Citation5–7]. However, there is only one report on the 4.2 kb deletion sub-type [Citation8]. In this study, we report the rare -α4.2Ⅰ and -α4.2Ⅱsub-types in a Chinese patient identified by third-generation sequencing (TGS).
Case report
A 32-year-old female was referred to our hospital for a routine pregnancy examination. According to the Guangxi policy, she was recommended to be screened for thalassemia; if the screening result was positive, she was required to perform a genetic analysis. After informed consent was given and signed, the patient’s blood sample was collected. In the hemoglobin (Hb) analysis by capillary electrophoresis (CE) (Capillarys 2 Flex Piercing; Sebia, Lisses, Paris, France), the Hb F was found to be 42.2% (reference: 0–5.0%), whereas Hb A2 (reference: 2.4–3.5%) and Hb A (reference: 92.5–97.5%) were 1.3% and 55.5%, respectively; additionally, there was an abnormal peak with a 1% value migrated in zone 1 ((A)). A complete blood count (CBC) showed Hb 12.3 g/dL (reference: 12.0–16.0 g/L), mean corpuscular volume (MCV) 63.8 fL (reference: 80.0–99.0 fL), and mean corpuscular hemoglobin (MCH) 20.3 pg (reference: 27.0–35.0 pg) (Sysmex XT 1800i; Sysmex Corporation, Kobe, Japan). Her screening results suggested the possibility of thalassemia.
Figure 1. Hb analysis by CE showed an abnormal peak migrating to zone F (A). Results of agarose electrophoresis of Gap-PCR amplification products (B): 1. DL2000 (from top to bottom: 2.0, 1.7, 1.4, 1.2, 0.9 kb), 2. -α4.2/αα, 3. -α4.2/-α4.2, 4. Normal. A mutation of GAC>CAC in a heterozygous state was observed at codon 74 of the HBA1 gene using Sanger sequencing, corresponding to Hb Q-Thailand (C).
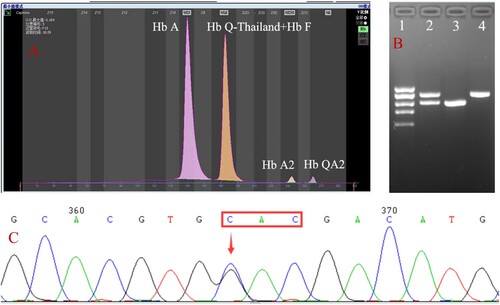
Common deletions and non-deletions of α-thalassemia in the Chinese population were detected with Gap-PCR and PCR-reverse dot blot with genetic analysis kits (Yaneng Biosciences, Guangdong, China). The Gap-PCR results revealed a homozygous -α4.2 deletion but did not match the CE results ((B)). DNA sequencing was further performed based on CE results suggesting the suspected presence of Hb variants. A mutation of GAC>CAC in a heterozygous state was observed at codon 74 of the HBA1 gene, resulting in a histidine replacement of the aspartic acid ((C)). This mutation corresponds to Hb Q-Thailand. In addition, by sequence comparison, we also found some base inconsistencies near the breakpoint. It indicated that there might be two kinds of deletions.
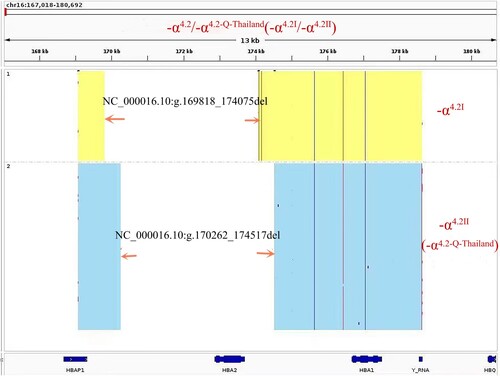
To confirm our speculation, further analysis was performed by multiplex ligation-dependent probe amplification (MLPA) and TGS (the same as single molecule real-time sequencing, SMRT). A homozygous deletion was found between probes 391 and 190 using the SALSA MLPA kit (HBA P140; MRC-Holland, Amsterdam, Netherlands), which involved the HBA1 and HBA2 genes, corresponding to -α4.2 (). This result was consistent with the Gap-PCR result. In order to identify the position of the break-point, TGS was done, and the results revealed the presence of two different lengths of fragment deletions; one was NC_000016.10:g.169818_174075del and the other was NC_000016.10:g.170262_174517del ().
Figure 2. MLPA analysis of the patient. The column chart (red dots) indicated a deletion region extending from probe 391 to 190. The red line diagrams represent the location of type 1 (-α4.2Ⅰ) and type 2 (-α4.2Ⅱ) deletions, respectively.
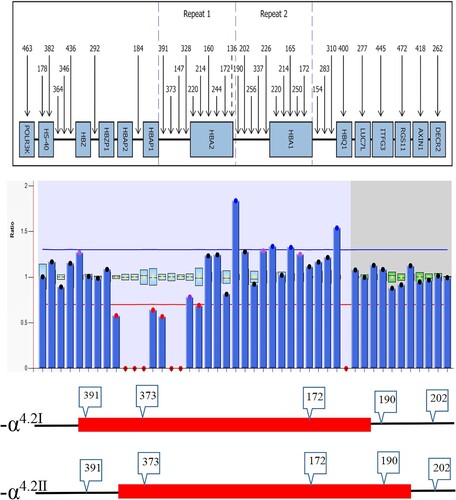
Figure 3. TGS identified two different 4.2 kb deletions in the patient, namely -α4.2Ⅰ and -α4.2Ⅱ.
Discussion
The literature that mentions subtype -α4.2 is relatively limited, and -α4.2 can be divided into type 1 (-α4.2Ⅰ) and type 2 (-α4.2Ⅱ), according to Chang et al [Citation8]. Type 1 is a common type of 4.2 kb deletion, and type 2 is linkage to Hb Q-Thailand (-α4.2-Q-Thailand) (also known as Hb G-Taichung, Hb Mahidol, Hb Asabara, and Hb Kurashiki-Ⅰ). In clinical laboratories in China, Gap-PCR is the most common technique to detect a 4.2 kb deletion. Since the kit primers cover both -α4.2Ⅰ and -α4.2Ⅱ upstream and downstream of the breakpoint, it is difficult to distinguish between -α4.2Ⅰ and -α4.2Ⅱ from the amplified 4.2 kb deletion. This may be the reason why we have obtained little information regarding the prevalence of sub-type 4.2 kb deletions in China. In this study, the patient was diagnosed as having a 4.2 kb homozygous deletion. However, DNA sequencing was performed based on the electrophoresis results, suggesting the presence of Hb Q-Thailand. As far as we know, Hb Q-Thailand is always linked with a 4.2 kb deletion. Until then, we had not noticed the sub-type 4.2 kb deletions and therefore did not know which sub-type of 4.2 kb deletions was linked to the Hb Q-Thailand.
In many regions of southern China, especially primary hospitals, screening for thalassemia is conducted with only a complete blood count (CBC) and rarely with Hb analysis. As previously reported, 35.7% of patients with Hb Q-Thailand may be misdiagnosed using only CBC screening [Citation9]. In addition, only electrophoretic screening can indicate the presence of Hb Q-Thailand. So, some cases with Hb Q-Thailand may be undiagnosed using the CBC and routine Gap-PCR methods. Hb Q-Thailand and its combinations with heterozygous 4.2 kb deletion (-α4.2/) will be diagnosed as homozygous 4.2 kb deletion (-α4.2/-α4.2). In the present study, it was corrected to Hb Q-Thailand combined with a 4.2 kb deletion (-α4.2/-α4.2-Q-Thailand) based on the electropherogram.
TGS is a recently developed and advanced technique for identifying several complex and rare thalassemias [Citation10,Citation11]. It has been reported that it has also been used to detect the 3.7 kb deletion sub-type (-α3.7Ⅲ) [Citation5]. In our study, TGS showed two large segment deletions, NC_000016.10:g.169818_174075del and NC_000016.10:g.170262_174517del, corresponding to -α4.2Ⅰ and -α4.2Ⅱ. This also demonstrated that this patient was not homozygous for a 4.2 kb deletion but a double heterozygote for both sub-types of 4.2 kb deletion. Although the -α4.2Ⅰ and -α4.2Ⅱ heterozygotes present as an absence or mild hypochromic polyglobulia microcytic RBC, they can lead to Hb H disease when combined with α0-thalassemia. Some patients with Hb H disease may have to be treated with blood transfusions. Clinicians should exercise caution when interpreting 4.2 kb deletion results from Gap-PCR and recommend a complementary electrophoretic analysis if no electrophoretic screening results are available.
It has always been assumed that Hb Q-Thailand is linked to -α4.2. Recently, it has been reported that Hb Q-Thailand can be inherited without linkage to -α4.2, so it is necessary to accurately identify Hb Q-Thailand, -α4.2Ⅰ and -α4.2Ⅱ [Citation12]. TGS can detect not only deletions but also variants of the globins’ genes, demonstrating the advantages of a more precise and comprehensive technology that promises to identify these rare and complex mutations.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data in this study are shown in the figures and tables.
Additional information
Funding
References
- Tesio N, Bauer DE. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am. 2023;37(2):273–299. DOI:10.1016/j.hoc.2022.12.001
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167. DOI:10.1016/S0140-6736(17)31822-6
- Xiong F, Sun MN, Zhang XH, et al. Molecular epidemiological survey of haemoglobinopathies in the Guangxi Zhuang autonomous region of southern China. Clin Genet. 2010;78(2):139–148. DOI:10.1111/j.1399-0004.2010.01430.x
- Charoenwijitkul T, Singha K, Fucharoen G, et al. Molecular characteristics of α+-thalassemia (3.7 kb deletion) in Southeast Asia: molecular subtypes, haplotypic heterogeneity, multiple founder effects and laboratory diagnostics. Clin Biochem. 2019;71:31–37. DOI:10.1016/j.clinbiochem.2019.06.005
- Bao X, Wang J, Qin D, et al. The -α3.7III subtype of α+-thalassemia was identified in China. Hematology. 2022;27(1):826–830. DOI:10.1080/16078454.2022.2101913
- Bowie LJ, Reddy PL, Nowak EM. alpha-thalassemia subtyping and the detection of silent mutations by high-resolution fragment analysis and DNA sequencing. Mol Diagn. 1998;3(1):43–53. DOI:10.1016/S1084-8592(98)80026-X
- Chen SQ, Li HY, Chen Z, et al. Detection and analysis of the sub-types of -alpha3.7 in Chinese. Yi Chuan. 2003;25(6):649–651.
- Chang JG, Liu TC, Chiou SS, et al. Rapid detection of –α4.2 deletion of α-thalassemia-2 by polymerase chain reaction. Ann Hematol. 1994;69:205–209. DOI:10.1007/BF02215955
- Li YQ, Chen ZZ, Liang L. Analysis of the phenotype-genotype relationship of hemoglobin Q-Thailand in Guangxi. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2016;33(2):164–168.
- Jiang F, Mao AP, Liu YY, et al. Detection of rare thalassemia mutations using long-read single-molecule real-time sequencing. Gene. 2022;825:146438. DOI:10.1016/j.gene.2022.146438
- Li YQ, Liang L, Guo WL, et al. Identification of a novel 107 kb deletion in the alpha-globin gene cluster using third-generation sequencing. Clin Biochem. 2023;113:36–39. DOI:10.1016/j.clinbiochem.2022.12.010
- Qin D, Wang J, Yao C, et al. Hb Q-Thailand heterozygosity unlinked with the (-α4.2/) α+-thalassemia deletion allele identified by long-read SMRT sequencing: hematological and molecular analyses. Hematology. 2023;28:2184118. DOI:10.1080/16078454.2023.2184118