
Introduction
Wilson’s disease (WD) is an inherited, autosomal recessive, copper accumulation disorder affecting approximately 30 individuals per million [Citation1]. WD is caused by the dysfunction of a copper-transporting P-type ATPase (ATPase copper transporting beta) that plays a crucial role in copper excretion into the bile. The gene encoding a P-type ATPase, ATP7B, is located on chromosome 13q14.3. Copper has a narrow nontoxic window, where submicromolar changes can be deleterious to cells, and living organisms have evolved a strictly controlled copper homeostatic mechanism. However, multiple gene mutations can impair transporter protein function, leading to copper accumulation in the liver, brain, cornea, and kidney, and then organ dysfunction [Citation2]. Medications commonly used to treat WD include D-penicillamine, a chelating agent that binds to excess copper and facilitates its excretion via the urine, but is known to cause myelosuppression. Trientine may also be used as an alternative to D-penicillamine. Zinc acetate or zinc gluconate can also be administered to reduce the amount of copper absorbed from the diet [Citation3].
The American Association for the Study of Liver Diseases, recommends a comprehensive approach be employed to manage WD, with the objective of mitigating the buildup of copper in the body [Citation4] since oxidative damage is caused when the copper level in the liver outweighs the proteins that typically bind it and may result in chronic active hepatitis, fibrosis, and cirrhosis [Citation5].
In acute promyelocytic leukemia (APL), the addition of all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) has significantly improved outcomes [Citation6]. However, the interplay between impaired transporter function and treatment with ATRA/ATO in the setting of baseline organ dysfunction can be very challenging. Additionally, ATP7B has been linked with resistance to platinum-based chemotherapy [Citation7]. Here, we report the first case of APL in a young 22-year-old female patient with WD.
Case description
A 22-year-old woman with WD since the age of 5 years on D-penicillamine, with no renal or neurological manifestations presented with a one-week history of abdominal pain and fatigue that interfered with her daily activities. She had no documented portal hypertension.
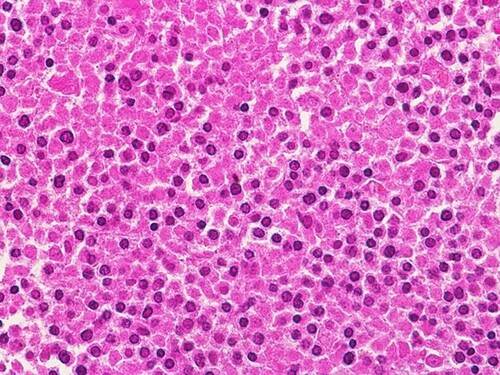
Workup revealed a hemoglobin level of 5.6 g/dL, a white blood cell count of 77000/μL, and a platelet count of 12000/μL. A peripheral blood smear revealed 12% blasts with morphological features compatible with APL () and bone marrow trephine biopsy showed heavy infiltration by promylocytes (). Flow cytometry showed cells expressing CD13, CD33, CD38, CD64, CD58, and MPO but negative for CD34, CD117, CD14, CD15, and HLA-DR. Fluorescence in situ hybridization of the bone marrow aspirate was positive for PML::RARA rearrangement. Karyotype analysis revealed t(15;17). Molecular evaluation of the PML::RARA fusion transcript was performed using polymerase chain reaction (RT–PCR). Computed tomography of the abdomen and pelvis showed features of chronic liver disease, splenomegaly with multiple splenic infarctions, small ascites, and a ruptured hemorrhagic ovarian cyst.
Figure 1. Peripheral blood showing abnormal promyelocytes with hyper granular cytoplasm with an arrow showing group of promylocytes
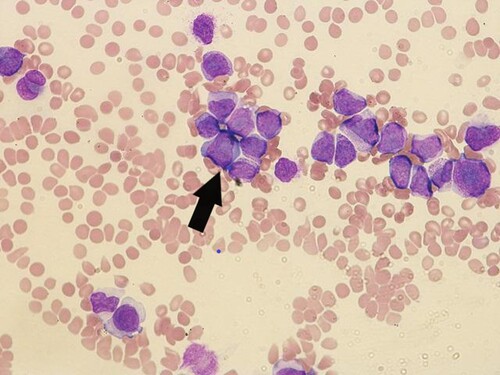
Figure 2. Bone marrow trephine biopsy at base line showing infiltration by promyelocytes
Since the patient was considered high risk APL, as per our center protocol, induction with ATRA, ATO, and idarubicin using the Australasian Leukemia and Lymphoma Group APML4 (ALLG APML4) [Citation8] protocol was initiated, with a 50% dose reduction in idarubicin.
On day 1 of her treatment, she developed a cholestatic liver injury with increasing total bilirubin (mainly direct), reaching the highest level on day 22 at 149 mmol/L, with mild transaminitis (AST, 82 U/L; ALT, 92 U/L), which improved with ATRA for a few days.
Work up for her liver disease was consistent with compensated liver cirrhosis, CHILD A, MELD sore was 10, with preserved liver synthetic capacity and with portal hypertension. At the time of presentation, she had normal ALT level, slightly raised AST 55U/L and high ALP 280 U/L. A Fibro scan was performed during the treatment course, consistent with a fibrosis score of 4 (F4). Serum Ceruloplasmin was 0.50 mg/dl and 24 urine Cu was more than 100 mcg/day.
During her treatment course on day 3, she had mild differentiation syndrome, managed with dexamethasone and diuretics, and ocular hemorrhage, for which she received supportive care.
On day 12 of treatment, her laboratory results showed high anion gap metabolic acidosis (PH = 7.27, HCO3 = 11 mmol/L anion gap = 19) and a blood glucose level of 288 mg/dL, with strongly positive urine ketones. Evaluation of type 1 diabetes mellitus using glutamic acid decarboxylase antibodies, anti-islet cells, and anti-insulin antibodies yielded negative results, and the C-peptide level was normal.
On day 20 of treatment, D-penicillamine was put on hold during the diagnosis of leukemia and she was started on Trientine.
The post-induction bone marrow (day 33) was consistent with complete morphological and cytogenetic responses with less than 5% of blasts as per ELN response criteria 2019 [Citation9].
On day 35 the patient was started on consolidation therapy with subsequent 50% and 33% dose reductions of ATO and ATRA, respectively, due to drug-induced hepatocellular injury (AST and ALT levels up to 135 and 171 U/L, respectively).
The post-consolidation bone marrow showed no evidence of disease, with undetectable PCR results. Following consolidation therapy, the patient was started on maintenance ATRA 20 mg BID for 2 weeks every 3 months, with methotrexate 25% dose reduction of 7.5 mg/m2 weekly and mercaptopurine 50% reduction of approximately 40 mg/m2 daily.
We managed to treat this patient with dose adjustments based on her liver enzyme levels during each phase of treatment, with no major events or irreversible side effects. The patient completed a year of maintenance chemotherapy with no major toxicity or undetectable PML-RARA by PCR and planned to continue maintenance for one more year.
Discussion and literature review
To our knowledge, this is the first case report describing the occurrence of APL in a patient with WD in both the adult and pediatric populations. We discussed in detail the challenges faced in treating APL in a patient with WD in the setting of organ dysfunction in the form of compensated liver cirrhosis and achieving remission by using hepatotoxic medication without inducing further liver injury by using reduced doses of chemotherapy.
Management of such cases is complex, leading to significant delays and, often, discontinuation of therapy. The underlying cirrhotic liver disease presented several potential problems, including fluid retention, hepatic derangement affecting drug metabolism, portal hypertension, variceal bleeding, coagulopathy, and thrombocytopenia due to splenic sequestration. As a result, delivering leukemia-directed therapy can be challenging [Citation10]. Given that the liver metabolizes anthracyclines, dose adjustment was implemented based on total bilirubin and liver enzymes. In preclinical studies, ATRA has been shown to reduce liver fibrosis by reducing transforming growth factor b1, interleukin-6, and type I collagen. Therefore, it has been suggested for the treatment of cirrhotic liver disease [Citation10].
There are several protocols for treating high-risk APL, whether ATRA – ATO or ATRA-chemotherapy. The ALLG APL4 protocol used the ATRA plus ATO-based regimen [Citation9]. There was no significant outcome compared to the historical ATRA chemotherapy, with a 50% reduction in idarubicin, in both low- and high-risk patients. The protocol consisted of an induction with ATRA, ATO, and four doses of idarubicin (6–12 mg/m2, adjusted for age), followed by two consolidation courses of ATRA and ATO, and then by 2 years of maintenance therapy with ATRA and low-dose chemotherapy.
ATP7B further poses a challenge as deletion of the 13q.14.3 region, where ATP7B is located, results in the inactivation of the tumor suppressor gene, retinoblastoma 1 (RB1), which is frequently observed in B-cell chronic leukemia/lymphoma and myeloid neoplasms [Citation2].
Regarding the development of malignancy in patients with WD, two case reports have described patients with WD and acute lymphoblastic leukemia (ALL) [Citation2,Citation11]. The first case was a pediatric patient successfully treated with chemotherapy who was in remission after 2 years of follow-up [Citation2]. The second report described the occurrence of ALL in a 15-year-old girl with a known case of WD after 37 months of D-penicillamine treatment. The patient died after 26 months of acute leukemia with a relapse and secondary infection [Citation11].
Regarding prolonged D-penicillamine exposure and the development of lymphoid malignancies, one case report described the development of chronic lymphoblastic leukemia in a 58-year-old woman who had received 5 years of D-penicillamine treatment for rheumatoid arthritis and was successfully treated with chemotherapy [Citation12].
The copper chelator, D-penicillamine, originally isolated through the hydrolysis of penicillin in 1943, was first prescribed as a treatment for WD in 1956 [Citation13]. However, there is increasing evidence that copper transport mechanisms may play a role in resistance to anti-cancer drugs. Additionally, an animal model study reported that female NZBxNZW F1 hybrid mice receiving D-penicillamine 400 mg/kg body weight five times per week intraperitoneally developed a clinical picture similar to lymphocytic leukemia in humans. Despite the early nature of the findings, D-penicillamine has been linked to this outcome [Citation14]. Another challenge with D-penicillamine in patients undergoing treatment for leukemia is myelosuppression, which includes neutropenia, thrombocytopenia, anemia, agranulocytosis, and rarely, aplastic anemia [Citation13,Citation15–17].
Conclusion
In this report, we describe a young female patient with WD who was successfully treated for high-risk APL using chemotherapy-adjusted doses with manageable liver toxicities. Although D-penicillamine is known to cause immunological effects, as well as myelosuppression and neutropenia, a direct correlation between either D-penicillamine exposure or WD and the development of APL or other hematological malignancies cannot be established based on this case or previous cases reported in the literature. Thus, it is imperative that clinical research is conducted, however, given the rarity of this condition, it may be challenging.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bearn AG. Wilson's disease: hepatolenticular degeneration. Postgrad Med J. 1956;32(372):477–481.
- Maeda S, Matsubara H, Hiejima E, et al. Acute lymphoblastic leukemia in a girl with Wilson's disease. Pediatr Int. 2014;56(4):626–629.
- Moini M, To U, Schilsky ML. Recent advances in Wilson disease. Transl Gastroenterol Hepatol. 2021;6:21.
- Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: executive summary of the 2022 practice guidance on Wilson disease from the American association for the study of liver diseases. Hepatology. 2023;77(4):1428–1455.
- Chaudhry HS, Anilkumar AC. Wilson disease. StatPearls. Treasure Island (FL): StatPearls Publishing; 2023.
- Josephat F, Pruett E. Acute promyelocytic leukemia with t(15;17): a case study. MLO Med Lab Obs. 2012;44(7):22–24.
- Petruzzelli R, Mariniello M, De Cegli R, et al. TFEB regulates ATP7B expression to promote platinum chemoresistance in human ovarian cancer cells. Cells. 2022;11(2).
- Iland HJ, Collins M, Bradstock K, et al. Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: a non-randomised phase 2 trial. Lancet Haematol. 2015;2(9):e357–e366.
- Sanz MA, Fenaux P, Tallman MS, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133(15):1630–1643.
- Ofran Y, Tallman MS, Rowe JM. How I treat acute myeloid leukemia presenting with preexisting comorbidities. Blood. 2016;128(4):488–496.
- Gilman PA, Holtzman NA. Acute lymphoblastic leukemia in a patient receiving penicillamine for Wilson's disease. JAMA. 1982;248(4):467–468.
- Clausen JE, Arndal JC, Gram L, et al. Chronic lymphocytic leukaemia after treatment with penicillamine. Lancet. 1978;2(8081):152.
- Pitman SK, Huynh T, Bjarnason TA, et al. A case report and focused literature review of d-penicillamine and severe neutropenia: A serious toxicity from a seldom-used drug. Clin Case Rep. 2019;7(5):990–994.
- Harris G. Leukaemia-like appearances in mice given penicillamine. Lancet. 1976;2(7999):1356–1357.
- Rau AR, Usha M, Mallya P, et al. Cytopenia and bone marrow dysplasia in a case of Wilson's disease. Indian J Hematol Blood Transfus. 2014;30(Suppl 1):433–436.
- Kay AG. Myelotoxicity of D-penicillamine. Ann Rheum Dis. 1979;38(3):232.
- Richards AJ, Velvin DS, Whitmore DN, et al. Fatal aplastic anaemia and D-penicillamine. Lancet. 1976;307(7960):P646–P647.