
ABSTRACT
Objectives
Although cytarabine (AraC) has greatly contributed to improving the prognosis of patients with acute myeloid leukemia (AML), many patients developed drug resistance and eventually succumbed to AML. Thus, resistance to AraC is a major obstacle to improve the efficacy of chemotherapy in AML. Hence, this study aimed to demonstrate that artesunate (ART) can reliably induce cell death in vitro and block AraC resistance.
Methods
AML cell lines resistant to AraC were first constructed by repeated dosing for 5 months. Further, we analyzed whether ART intervention affected the sensitivity of AraC-resistant cells to AraC by cell function experiments, mainly including CCK-8 to assess cell viability, flow cytometry to examine apoptosis, and Western blotting to measure the Janus kinase (JAK)/signal transducers and activators of transcription 3 (STAT3) pathway protein expression. Furthermore, whether JAK/STAT3 pathway knockdown has a blocking effect on the efficacy of ART was also assessed.
Results
Co-treatment of ART and AraC increased the sensitivity of AML cells to AraC. Also, it effectively reversed the resistance of AML cells to AraC that is shown by the significantly reduced proliferation and increased apoptosis rates. ART intervention suppressed the activation of the JAK/STAT3 signaling pathway in AraC-resistant AML cells, suggesting that the function of ART in reversing AraC resistance is indeed dependent on the JAK/STAT3 signaling pathway.
Conclusions
In summary, ART enhanced the sensitivity of AML/AraC-resistant cells to AraC by modulating the JAK/STAT3 pathway.
1. Introduction
Acute myeloid leukemia (AML) is a common and highly heterogeneous malignant clonal disorder of hematopoietic stem cells [Citation1]. In AML, myeloid progenitors fail to differentiate and mature, resulting in malignant clonal proliferation and apoptotic arrest, and thereby suppressing the normal hematopoiesis in the bone marrow and other hematopoietic compartments [Citation2]. From 1990 to 2017, the incidence and mortality rates of AML gradually increased worldwide, wherein males and older people had a higher risk of developing AML [Citation3]. The current treatment for AML broadly encompasses four broad categories: nucleoside analog therapy, epigenetic therapy, molecular-based therapy, and novel therapies using quinolone derivatives, monoclonal antibodies, or immune checkpoint inhibitors [Citation4]. Most of these approaches are still in clinical trials, and chemotherapy using nucleoside analogs is still the conventional therapy currently used to treat AML. Cytarabine (AraC) is a cytidine analog widely used for the treatment of various tumors, including leukemia and lymphoma [Citation5,Citation6]. However, the outcomes for the current AML treatment remain unsatisfactory, and even young adults treated with repeated high-dose cytarabine therapy or stem cell transplantation achieve only 40%–50% complete remission [Citation7]. Treating AML is difficult due to the resistance developed by patients to antileukemic agents; however, no successful treatment has been established for patients with relapsed or primary resistant AML [Citation8]. The low induction remission rate, poor response to chemotherapy, and short survival time with high case fatality rate in relapsed and primary resistant AML are difficult problems encountered during the clinical treatment of AML. Therefore, exploring new therapeutic strategies that can enable patients with AML achieve true complete remission and long-term disease-free survival is critically required.
Artemisinin (ARS) and its derivatives have long been widely used as first-line antimalarial drugs worldwide [Citation9]. Recent studies reported that ARS and their derivatives not only have good antimalarial effects but also have more prominent antitumor performance [Citation10]. Efferth et al. [Citation11] revealed that ART, a derivative of ARS, has good antitumor activity and is widely effective against various tumor cells. Zhao et al. [Citation12] suggested that ART significantly suppressed the proliferation and metabolic aspects of cisplatin-sensitive and cisplatin-resistant bladder cancer cells and, thus, may have the potential to treat advanced and treatment-resistant bladder cancer. Jiang et al. [Citation13] discovered that ART treatment induced apoptosis and autophagy in colon cancer cells, illustrating that it can be a potentially clinically useful anticancer drug for human colon cancer. Wang et al. [Citation14] experimentally reported that ART could reverse the drug resistance of esophageal cancer by regulating the ABCG2 expression in esophageal cancer cells, suggesting that ART may serve as a reversal agent to reverse the multidrug resistance of tumor cells. ART appears to be broadly effective in various cancers with low toxicity and side effects; its successful development as a reversal agent for cancer cell resistance would be highly promising. A recent study demonstrating that ART induced apoptosis in myeloid leukemia KG1a cells and inhibited cell proliferation and stemness in a dose-dependent manner provided new insights into the artesunate (ART)’s anticancer potential mechanism in leukemia [Citation15]. However, whether ART is effective for AraC resistance in AML remains to be further explored.
In this study, AraC induction was used to construct human AML drug-resistant cell lines that enabled us to focus on the effects of ART on drug resistance of drug-resistant cell lines and its possible mechanisms.
2. Experimental
2.1. Construction of cell culture and drug resistance strain
To investigate the ART toxicity on normal cells, human hematopoietic stem cells (HSPC-1; #CL-0547, Procell, Wuhan, China) were treated with a series of ART concentrations (0.1, 0.5, 1, 2, 5, and 10 μM) for 48 h before the cell viability detection.
The human AML cell lines HL-60 (#CL-0110, Procell, Wuhan, China) and THP-1 (Beyotime, Shanghai, China) were cultured in Dulbecco’s Modified Eagle Medium containing 10% fetal bovine serum (ThermoFisher, USA) in a 5% CO2 saturated humidified cell incubator at 37°C. AraC-resistant cell lines were established using the drug concentration escalation approach. Briefly, HL-60 and THP-1 cells (60%–80% confluent) in the logarithmic growth phase were taken and a gradient of 1, 2, 4, 8, and 16 μmol/L AraC (Sigma-Aldrich, USA; cat.no PHR1787) was gradually added to the culture medium for stimulation, and each concentration was replaced by the next higher concentration after 4 weeks of continuous culture until 16 μmol/L AraC was added for 4 weeks. Subsequently, the cells were collected to evaluate the IC50 of AraC against parental cells of HL-60 and THP-1 and resistant cells using the Cell Counting Kit-8 (CCK-8) reagent and to calculate the drug resistance index [Citation16]. Subsequently, HL-60/AraC and THP-1/AraC cells were divided into four groups based on the experimental design, including the control, AraC, ART, and AraC + ART. For the control group, cells were cultured normally without any treatment. For the AraC group, cells were treated with AraC at IC50 concentrations against HL-60 and THP-1 parental cells, respectively, for 48 h. For the ART group, 0.05 μM of ART (Sigma-Aldrich, USA; cat.no PHR2573) was added to the medium to incubate the cells for 48 h. For the AraC + ART group, 0.05 μM of ART and IC50 concentrations of AraC were both added to the medium to incubate the cells for 48 h. Furthermore, cells were divided into four groups: si-NC, si-NC + ART, si-JAK2, and si-JAK2 + ART. For si-NC and si-JAK2 groups, si-NC or si-JAK was transfected into HL-60/AraC and THP-1/AraC cells cultured for 48 h. For the si-NC + ART and siJAK2 + ART groups, si-NC or si-JAK2 was transfected into HL-60/AraC and THP-1/AraC cells for 48 h, accompanied by a culture medium supplemented with μM of ART.
2.2. Cell transfection
Three different siRNAs against JAK2 (si-JAK2#1, si-JAK2#2, and si-JAK2#3) were designed by Qiagen (Qiagen, Germany) to avoid off-target effects. In brief, cells were seeded into six-well plates at a density of 5 × 105 cells/well. The siRNAs or si-NC was transfected into cells using Lipofectamine®3000 transfection reagent based on the manufacturer’s protocols. JAK2 expression was then examined 48 h post-transfection. Information regarding siRNA sequences used in this study was as follows: si-JAK2#1 (5′-AACATATTATGATACACAGGT-3′; target position: 829–851), si-JAK2#2 (5′-TGACAAAGTCATCAAGAAGAG-3′; target position: 1,023–1,045), and si-JAK2#3 (5′-AATTAATGACACGAAAGACAA-3′; target position: 1,682–1,704).
2.3. Cell viability assay
The HL-60 and THP-1 cells after different treatment groups were made into single-cell suspensions and seeded in 96-well plates (2000 cells/well), respectively, with three replicate wells set for each time point. About 10 μL of the CCK-8 reagent (Sigma-Aldrich, USA; cat.no 96992) was added to the wells. These were examined at 72 h, and incubation was continued at 37°C for 3 h. The absorbance at 450 nm was determined in a microplate reader (bio rad, USA). Graphpad prism 7.0 software was used to draw graphs and histograms of HL-60 and THP-1 cell viability.
2.4. Cell apoptosis assay
The HL-60 and THP-1 cells from each group post-transfection were plated at a density of 1 × 104 cells/well were seeded in 24-well plates and cultured for 48 h. After the cells were collected, cell apoptosis was checked by flow cytometry (Attune NxT, ThermoFisher, USA) following the instructions of Annexin V-FITC/PI kit (Pricella, Wuhan, China).
2.5. Western blotting
Cell lysates were used to extract the total cellular protein after different treatments. The proteins on gels were transferred to nitrocellulose membranes by sodium dodecyl-sulfate polyacrylamide gel electrophoresis. Nitrocellulose membranes were blocked with Tris-buffered saline with 0.1% Tween® 20 detergent containing 5% nonfat dry milk for 2 h. Subsequently, primary antibodies (p-JAK2, JAK2, p-STAT3, STAT3, and β-actin) were added and incubated overnight at 4°C with shaking. Secondary antibodies goat anti-rabbit immunoglobulin G were then added for incubation at room temperature for 2 h. Enhanced chemiluminescence luminescent solution was added to the nitrocellulose membrane to react for 1–3 min and exposed in a dark room. The relative expression levels of proteins were analyzed using Image pro-plus 5.0 software, and β-actin was used as a loading control. Information about antibodies is listed in Table S1.
2.6. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
The total RNA of HL-60 and THP-1 cells was extracted using Trizol reagent (Invitrogen, USA). About 300 ng of RNAs was reverse transcribed using the miScript II RT Kit (Qiagen, Germany) based on the manufacturer’s instructions. The JAK2 gene expression in THP-1/AraC cells was assessed using the miScript PCR starter real-time PCR kit (Qiagen, Germany). The primers are presented in Table S2. Glyceraldehyde 3-phosphate dehydrogenase was used as an internal reference, and the relative expression levels were calculated using the 2-ΔΔCT method.
2.7. Statistical analysis
Experimental data were statistically analyzed using Graphpad prism 7.0 software. All experiments were triplicate independent experiments. One-way analysis of variance was used to compare the three groups and above, and t-test was used between the two groups, with P < 0.05 indicating a statistical difference.
3. Results
3.1. Establishing the AraC-resistant AML cells
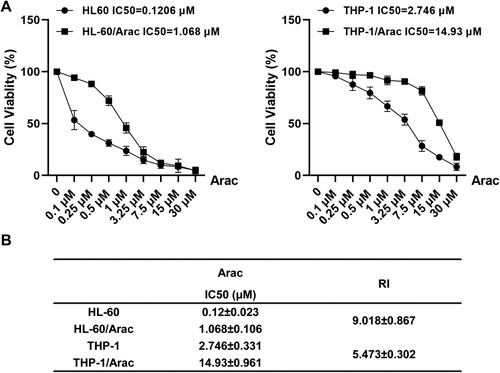
In this study, HL-60 and THP-1 cell lines resistant to AraC were first developed by repeated dosing over 5 months. Subsequently, based on the CCK8 assay, the IC50 of AraC on HL-60 and HL-60/AraC cells was 0.1206 and 1.068 μM, whereas those on THP-1 and THP-1/AraC cells was 2.746 and 14.93 μmol/L, respectively ((A)). The HL-60/AraC resistance index (RI) = 9.018 and THP-1/AraC resistance index RI = 5.473 ((B)) were calculated based on the drug RI formula: cellular RI = IC50 (AML cells/AraC) / IC50 (AML cells).
Figure 1. Establishing the AraC-resistant acute myeloid leukemia cells. A: AraC-resistant cell lines were established by increasing AraC concentrations and the viability rate of HL-60 and HL-60/AraC or THP-1 and THP-1/AraC after the treatment with different AraC doses. B: RI of HL-60/AraC and THP-1/AraC cells to AraC. RI = IC50 (AML cells/AraC) / IC50 (AML cells). Data: mean ± SEM. n = 3.
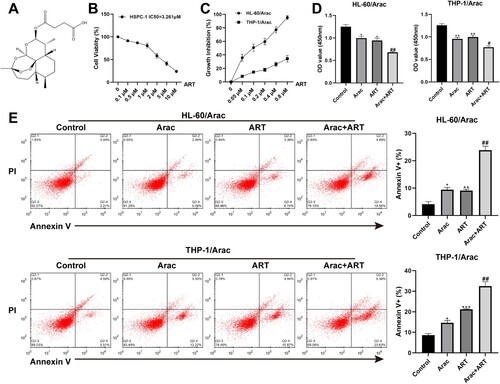
3.2. ART effectively reverses AraC resistance in AraC-resistant AML cells
The HL-60/AraC and THP-1/AraC-resistant cell lines were intervened with a series ART concentrations. Then cell viability was checked by CCK-8 to select the appropriate ART concentration for subsequent experiments. (A) displays the structural formula for ART. As shown in (B), the cell viability of HSPC-1 was >80% when the ART concentration was <1 μM, whereas the IC50 of AraC on HSPC-1 was 3.261 μM, suggesting a low ART toxicity on normal cells. Results in (C) displayed that the cell growth inhibition rates of HL-60/AraC and THP-1/AraC were increased after the ART intervention in HL-60/AraC- and THP-1/AraC-resistant cell lines at different concentrations, and 0.05 μM was selected for subsequent study based on the lowest effective dose method. Next, the effects of ART on AraC resistance of HL-60/AraC and THP-1/AraC were observed after treatment with AraC and ART alone or together, respectively. The results of the cell proliferation and apoptosis assays illustrated that the HL-60/AraC and THP-1/AraC cell proliferation and apoptosis were conspicuously altered when treated with ART or AraC alone, as indicated by the decreased proliferation capacity and the increased apoptosis rate. Interestingly, co-treatment of ART + AraC also increased the sensitivity of cells to AraC and effectively reversed the resistance of AML cells to AraC, as reflected by materially decreased proliferation and increased apoptosis rates ((D–E)). Moreover, the effects of ART on the non-resistant AML cell lines were also investigated. The inhibition of ART on the HL-60 and THP-1 cells was similar to that on HL-60/AraC and THP-1/AraC cells (Figure S1(A)). Notably, suppression of HL-60 and THP-1 cells could not be strengthened after combining AraC with a low dose of ART (Figure S1(B)).
Figure 2. ART effectively reverses AraC resistance in AraC-resistant AML cells. A: The chemical structure formula of ART. B: CCK-8 assay measurement of the cell viability of the HSPC-1 cells intervened with a series of ART concentrations (0.1, 0.5, 1, 2, 5, and 10 μM). C: CCK-8 assay measurement of the cell activity of the HL-60/AraC and THP-1/AraC cells intervened with a series of ART concentrations (0.05, 0.1, 0.2, 0.4, and 0.8 μM). D: CCK-8 assay measurement of cell viability of the HL-60/AraC and THP-1/AraC cell lines intervened with AraC (IC50) and ART (0.05 μM) alone or together. E: Flow cytometry measurement of the cell apoptotic rate in the HL-60/AraC and THP-1/AraC cell lines intervened with AraC (IC50) and ART (0.05 μM) alone or together. Data: mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs Control group. #P < 0.05, ##P < 0.01 vs AraC group. n = 3.
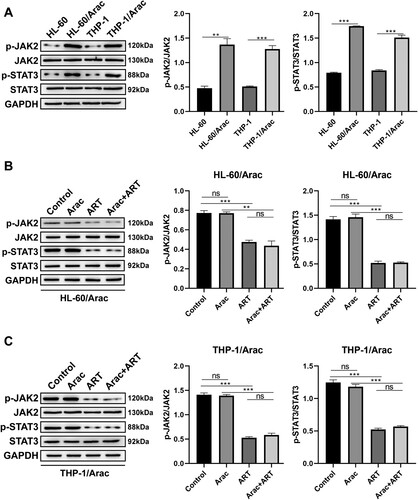
3.3. ART suppresses JAK/STAT3 signaling in AraC-resistant AML cells
Previous related literature reported that the JAK/STAT3 pathway is associated with proliferation, migration, invasion, and drug resistance in various tumors [Citation17–19], and a recent study based on network pharmacology and molecular docking revealed that STAT3 may be an effective target of ART in AML [Citation20]. The study reported that the phosphorylation of JAK2 and STAT3 was significantly higher in the AraC-resistant AML cell lines than that in the parental cell lines ((A)), suggesting that JAK/STAT3 pathway activation is the driver of AraC resistance. This study envisions that ART can affect AraC resistance in AML by modulating the JAK/STAT3 pathway and validating this invasion. Western blot results demonstrated that the ART treatment of HL-60/AraC and THP-1/AraC cells significantly downregulated p-JAK2 and p-STAT3 protein expression levels but not JAK2 and STAT3 total protein levels in the presence or absence of AraC treatment ((B and C)). The ART intervention also reportedly suppressed the activation of the JAK/STAT3 signaling pathway in AraC-resistant AML cells.
Figure 3. ART suppresses JAK/STAT3 signaling in AraC-resistant AML cells. A: Western blotting measurement of p-JAK2, p-STAT3, JAK2, and STAT3 protein levels in AraC-sensitive and AraC-resistant AML cells. B: Western blotting measurement of p-JAK, p-STAT3, JAK, and STAT3 protein levels in the HL-60/AraC cells treated with AraC (IC50) and ART (0.05 μM) alone or together. C: Western blotting measurement of p-JAK2, p-STAT3, JAK2, and STAT3 protein levels in the THP-1/AraC cells treated with AraC (IC50) and ART (0.05 μM) alone or together. Data: mean ± SEM. **P < 0.01, ***P < 0.001. n = 3.
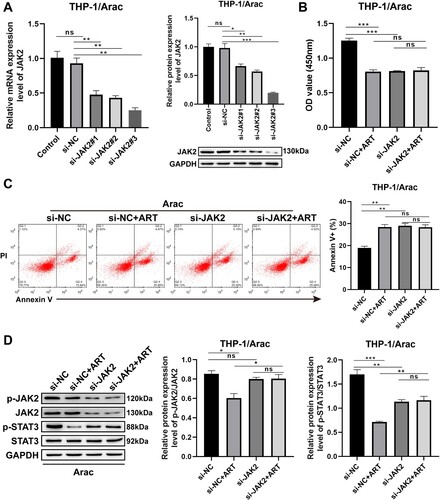
3.4. The function of ART on reversing AraC resistance relies on the JAK/STAT3 signaling
Next, whether preventing the activation of the JAK/STAT3 pathway had a blocking effect on the efficacy of ART was further assessed. Three kinds of siRNAs against JAK2 were used in this study. (A) shows that the knockdown efficiency of si-JAK2#3 was the most significant that downregulated the protein and mRNA levels of JAK2 by more than threefold ((A)). Moreover, cell behavioral results displayed that AraC resistance of THP-1/AraC cells largely disappeared after the ART intervention in JAK2 knockdown THP-1/AraC cells, as indicated by no significant changes in cell proliferation ability and apoptosis rate ((B and C)). Besides, the same examinations were performed in HL-60/AraC cells (Figure S2). The si-JAK2#3 was chosen for the functional assays of HL-60/AraC cells as it showed the highest efficiency for JAK2 silencing (Figure S2(A)). Similar to the results on THP-1/AraC cells, the knockdown of JAK2 diminished the role of ART in reversing AraC resistance of HL-60/AraC cells, as revealed by the absence of significant changes on the cell proliferation ability and apoptosis rate of JAK2 silencing HL-60/AraC cells (Figure S2(B and C)). Furthermore, Western blotting results indicated that none of the changes in the expression levels of JAK/STAT3-pathway-related proteins, including p-JAK2 and p-STAT3, in THP-1/AraC and HL-60/AraC cells treated with ART based on JAK2 knockdown were statistically significant ((D) and S2(D)). These aforementioned results indicate that the function of ART in reversing AraC resistance is indeed dependent on the JAK/STAT3 signaling pathway.
Figure 4. Function of ART on reversing AraC resistance in THP-1/AraC cells relies on JAK/STAT3 signaling. A: RT-qPCR (left) and Western blotting (right) assessment of the mRNA and protein levels of JAK2 in THP-1/AraC cells transfected with si-JAK2#1, si-JAK2#2, and si-JAK2#3. B: CCK-8 assessment of the proliferation ability of THP-1/AraC cells transfected with si-JAK2#3 and/or ART intervention. C: Flow cytometry measurement of the cell apoptotic rate of THP-1/AraC cells transfected with si-JAK2#3 and/or ART intervention. D: Western blotting measurement of p-JAK2, p-STAT3, JAK2, and STAT3 protein levels in the THP-1/AraC cells transfected with si-JAK2#3 and/or ART intervention. Data: mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. n = 3.
4. Discussion
AML, a class of malignant clonal and irreversible expansion neoplasms of myeloid blasts, is highly heterogeneous. Its heterogeneity includes abnormalities in the clinical features, morphological and immunological functions, karyotype, and inheritance [Citation21]. Chromosomal translocations, multigenic mutations, and others often accompany AML. Mutual cooperation among these mutations results in proliferation, increased cell viability and impaired differentiation capacity, a major mechanism contributing to the AML development. The complex karyotype and variable mutated genes in AML make it extremely vulnerable to chemotherapeutic drugs that also pose a big obstacle to currently hinder the remission and complete cure in patients with AML [Citation22]. Drug resistance refers to the tolerance of microorganisms, parasites, and tumor cells to the action of chemotherapeutic drugs. Once drug resistance has developed, the chemotherapeutic effect of the drug has significantly declined [Citation23]. Currently, the cure rate of patients with tumor is affected by drug resistance to varying degrees. daunorubicin, AraC, and doxorubicin have been widely used clinically as common drugs for the treatment of AML, with AraC-based regimens being the currently accepted classic first-line treatment for AML [Citation24]. AraC, a glycoside compound formed from cytosine with arabinose, is a pyrimidine-based antimetabolite mainly acting in the proliferative phase of cells. AraC was found to act as a competitive inhibitor of DNA polymerase and was able to interfere with cell proliferation by inhibiting cellular DNA biosynthesis [Citation25]. Although the combination of AraC with other broad-spectrum chemotherapeutics has shown remarkable antitumor efficacy, especially for the treatment of AML, patients clinically diagnosed with AML are highly susceptible to relapse due to the emergence of resistance to the therapeutic agent AraC after complete remission that greatly hampers the subsequent treatment of drug-resistant patients and severely affects the patient’s quality of survival.
In recent years, several studies have found that ARS and its derivatives, such as dihydroartemisinin, ART, and artemether, have good pharmaceutical activities. They have also shown some efficacy as an anti-infective, antiviral, and anti-schistosomal agent as well as in the treatment of autoimmune diseases [Citation26]. Since the first report of a possible anticancer effect of ARS in 1993, investigators have conducted numerous studies on the potential antitumor effects of ARS and its derivatives [Citation11]. ART, a water-soluble semi-synthetic ARS derivative, is a class of antimalarial drugs with high efficacy and low toxicity. Recent studies have found that ART may function as a completely new class of antineoplastic agents. Some studies demonstrated that ART can induce tumor cell death through the production of reactive oxygen species [Citation27]. Zhang et al. [Citation28] suggested that ART could kill tumor cells by utilizing iron ions in tumor cells to produce free radicals. Additionally, ART can induce lysosomal reactive oxygen species-mediated mitochondrial apoptosis, causing tumor cell DNA damage by inhibiting tumor growth. Previous studies have proposed that ART with improved pharmacokinetic properties may provide therapeutic benefits in combination with conventional therapeutic strategies in AML [Citation29]. Remarkably, ART can reverse drug resistance in addition to its ability to inhibit tumor cell growth. ART also inhibited the stem-cell-like properties of non-small-cell lung cancer cells and inhibited tumor growth in xenografts bearing gefitinib-resistant tumors [Citation30]. Zhang et al. [Citation31] revealed that enhanced targeting of cytarabine to AML by ART through B-cell lymphoma two-interacting mediator of cell-death-mediated apoptosis and Mcl-1 reduction as a bridge to the combination of venetoclax and cytarabine. In this experiment, the human AML drug-resistant HL-60/AraC and THP-1/AraC cell lines were first constructed, and the alteration of drug resistance in drug-resistant cells was detected by CCK-8 and other experiments after the ART intervention with different concentrations for 48 h. Meanwhile, our study established a series of ART concentrations on normal cells to test ART cytotoxicity. Our data revealed that ART exerts relatively low toxicity to HSPC-1 cells when the concentration is <1 μM. To confirm that ART can make the effects on AML cells at the concentration that does not produce an inhibitory effect on normal cells, our study subsequently explored the effects of ART on AraC-resistant AML cells based on low concentrations (0.05–0.8 μM). Furthermore, differences and changes in the behaviors of drug-resistant cells and the protein expression of drug-resistance-related pathways after drug intervention were detected using CCK-8, flow cytometry, real-time PCR, and Western blotting to explore that the ART mechanism and pathway might be used as a reversal agent of drug resistance in cancer. The results revealed that ART treatment increased the sensitivity of cells to AraC and effectively reversed the resistance of AML cells to AraC, as reflected by a significant reduction in proliferation and an increased apoptotic rate. Meanwhile, studies on the effects of ART on the non-resistant AML cell lines showed that AraC suppression on HL-60 and THP-1 cells could not be strengthened after being combined with a low dose of ART. These data suggested that the role of ART in improving the sensitivity of AraC-resistant AML cells to AraC was not attributed to the synergistic effect. The JAK/STAT3 signaling pathway is a more evolutionarily conserved cascade that plays an important role in intercellular signal transduction [Citation32]. The JAK/STAT3 signaling pathway is confirmed as a common pathway involved in the proliferation, invasion, and drug resistance mechanisms of cancer [Citation33]. Several studies have demonstrated that aberrant activation of the JAK/STAT3 signaling pathway can cause phenotypic changes and malignant transformation of AML cells that is beneficial for promoting AML development, progression, invasion, and metastasis while inhibiting AML cell apoptosis [Citation34–37]. We have learned through literature reports that the JAK/STAT3 pathway has an important role in tumorigenesis and drug resistance formation, especially its phosphorylation status. Inhibition of ataxia telangiectasia mutation reverses the epithelial-to-mesenchymal transition and decreases the metastatic potential of cisplatin-resistant lung cancer cells by preventing the activation of the JAK/STAT3 signaling pathway [Citation38]. Ham et al. [Citation39] demonstrated that curcumin, as a suitable natural product, could overcome 5-FU chemotherapy resistance by inhibiting cancer-associated fibroblast-induced activation of the JAK/STAT3 signaling pathway in gastric cancer. Our study showed that the JAK/STAT3 pathway was obviously activated in AML cells after the occurrence of AraC resistance, suggesting that the JAK/STAT3 signal was involved in the AraC resistance of AML cells. Incorporating AraC into the DNA during the process of DNA synthesis was speculated to eventually cause DNA damage, thereby evoking an inflammatory response that causes STAT3 activation [Citation40]. Notably, in a recent study based on network pharmacology and molecular docking, STAT3 has been predicted as an effective target of ART for the treatment of AML [Citation20]. Interestingly, our present study discovered that the levels of phosphorylated proteins of the JAK/STAT3 pathway in AraC-resistant AML cells were inhibited by ART intervention. Besides, the results further indicated no significant changes in the cell proliferation ability and apoptosis rate of both THP-1/AraC and HL-60/AraC cells treated with ART based on JAK2 knockdown during AraC exposure. The aforementioned results indicate that the ART function in reversing AraC resistance is indeed dependent on the JAK/STAT3 signaling pathway. Collectively, our data showed that the combination of ART and AraC appears to synergistically decrease the proliferation and increase apoptosis in AraC-resistant AML cells, providing a rationale for continued exploration of ART as an adjunct treatment modality for patients with AML. Although some studies have investigated this topic, animal experiments were missing. This will be one of our subsequent research focuses.
In summary, this study elucidates that activation of the JAK/STAT3 pathway is one of the causes of AraC resistance in AML cells and suggests strategies for administering ART interventions to overcome AraC resistance and enhance its antitumor activity. The aforementioned results provide an experimental basis for clinical studies of ART and a beneficial therapeutic strategy for the treatment of patients with limited acquired AraC resistance.
Research ethics and patient consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Supplemental Material
Download MS Word (16.4 KB)Supplemental Material
Download MS Word (19.9 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Pelcovits A, Niroula R. Acute myeloid leukemia: a review. R I Med J (2013). 2020;103(3):38–40.
- Saultz JN, Garzon R. Acute myeloid leukemia: a concise review. J Clin Med. 2016;5(3). doi:10.3390/jcm5030033
- Yi M, Li A, Zhou L, et al. The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017: estimates based on the global burden of disease study 2017. J Hematol Oncol. 2020;13(1):72. doi:10.1186/s13045-020-00908-z
- Kantarjian H, Kadia T, DiNardo C, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 2021;11(2):41. doi:10.1038/s41408-021-00425-3
- Strickland SA, Vey N. Diagnosis and treatment of therapy-related acute myeloid leukemia. Crit Rev Oncol Hematol. 2022;171:103607. doi:10.1016/j.critrevonc.2022.103607
- Smallwood K, Harper A, Blackwood L. Lomustine, methotrexate and cytarabine chemotherapy as a rescue treatment for feline lymphoma. J Feline Med Surg. 2021;23(8):722–729. doi:10.1177/1098612X20972066
- Rowe JM. Optimal induction and post-remission therapy for AML in first remission. Hematology Am Soc Hematol Educ Program. 2009: 396–405. doi:10.1182/asheducation-2009.1.396
- Short NJ, Konopleva M, Kadia TM, et al. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 2020;10(4):506–525. doi:10.1158/2159-8290.CD-19-1011
- Ma N, Zhang Z, Liao F, et al. The birth of artemisinin. Pharmacol Ther. 2020;216:107658. doi:10.1016/j.pharmthera.2020.107686
- Kiani BH, Kayani WK, Khayam AU, et al. Artemisinin and its derivatives: a promising cancer therapy. Mol Biol Rep. 2020;47(8):6321–6336. doi:10.1007/s11033-020-05669-z
- Efferth T. From ancient herb to modern drug: artemisia annua and artemisinin for cancer therapy. Semin Cancer Biol. 2017;46:65–83. doi:10.1016/j.semcancer.2017.02.009
- Zhao F, Vakhrusheva O, Markowitsch SD, et al. Artesunate impairs growth in cisplatin-resistant bladder cancer cells by cell cycle arrest, apoptosis and autophagy induction. Cells. 2020;9(12). doi:10.3390/cells9122643
- Jiang F, Zhou JY, Zhang D, et al. Artesunate induces apoptosis and autophagy in HCT116 colon cancer cells, and autophagy inhibition enhances the artesunate-induced apoptosis. Int J Mol Med. 2018;42(3):1295–1304.
- Wang L, Liu L, Chen Y, et al. Correlation between adenosine triphosphate (ATP)-binding cassette transporter G2 (ABCG2) and drug resistance of esophageal cancer and reversal of drug resistance by artesunate. Pathol Res Pract. 2018;214(9):1467–1473. doi:10.1016/j.prp.2018.08.001
- Li Y, Feng L, Li Y, et al. Artesunate possesses anti-leukemia properties that can be enhanced by arsenic trioxide. Leuk Lymphoma. 2014;55(6):1366–1372. doi:10.3109/10428194.2013.829573
- Feng Z, Chen Q. Raised CD40L expression attenuates drug resistance in Adriamycin-resistant THP-1 cells. Exp Ther Med. 2020;19(3):2188–2194.
- Teng Y, Ross JL, Cowell JK. The involvement of JAK-STAT3 in cell motility, invasion, and metastasis. Jakstat. 2014;3(1):e28086.
- Wang Y, Lu HL, Liu YD, et al. Cryptotanshinone sensitizes antitumor effect of paclitaxel on tongue squamous cell carcinoma growth by inhibiting the JAK/STAT3 signaling pathway. Biomed Pharmacother. 2017;95:1388–1396. doi:10.1016/j.biopha.2017.09.062
- Gritsina G, Xiao F, O'Brien SW, et al. Targeted blockade of JAK/STAT3 signaling inhibits ovarian carcinoma growth. Mol Cancer Ther. 2015;14(4):1035–1047. doi:10.1158/1535-7163.MCT-14-0800
- Tao Y, Li W, Yang J, et al. Exploring underlying mechanism of artesunate in treatment of acute myeloid leukemia using network pharmacology and molecular docking. Clin Transl Oncol. 2023;25(8):2427–2437. doi:10.1007/s12094-023-03125-5
- Stubbins RJ, Francis A, Kuchenbauer F, et al. Management of acute myeloid leukemia: a review for general practitioners in oncology. Curr Oncol. 2022;29(9):6245–6259. doi:10.3390/curroncol29090491
- McNeer NA, Philip J, Geiger H, et al. Genetic mechanisms of primary chemotherapy resistance in pediatric acute myeloid leukemia. Leukemia. 2019;33(8):1934–1943. doi:10.1038/s41375-019-0402-3
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi:10.1146/annurev.med.53.082901.103929
- Chang YC, Lo WJ, Huang YT, et al. Deferasirox has strong anti-leukemia activity but may antagonize theanti-leukemia effect of doxorubicin. Leuk Lymphoma. 2017;58(9):1–12. doi:10.1080/10428194.2017.1280604
- Chhikara BS, Parang K. Development of cytarabine prodrugs and delivery systems for leukemia treatment. Expert Opin Drug Delivery. 2010;7(12):1399–1414. doi:10.1517/17425247.2010.527330
- Shi Q, Xia F, Wang Q, et al. Discovery and repurposing of artemisinin. Front Med. 2022;16(1):1–9. doi:10.1007/s11684-021-0898-6
- Huang Z, Gan S, Zhuang X, et al. Artesunate inhibits the cell growth in colorectal cancer by promoting ROS-dependent cell senescence and autophagy. Cells. 2022;11(16):2472. doi: 10.3390/cells11162472
- Zhang Q, Yi H, Yao H, et al. Artemisinin derivatives inhibit non-small cell lung cancer cells through induction of ROS-dependent apoptosis/ferroptosis. J Cancer. 2021;12(13):4075–4085. doi:10.7150/jca.57054
- Drenberg CD, Buaboonnam J, Orwick SJ, et al. Evaluation of artemisinins for the treatment of acute myeloid leukemia. Cancer Chemother Pharmacol. 2016;77(6):1231–1243. doi:10.1007/s00280-016-3038-2
- Cao D, Chen D, Xia JN, et al. Artesunate promoted anti-tumor immunity and overcame EGFR-TKI resistance in non-small-cell lung cancer by enhancing oncogenic TAZ degradation. Biomed Pharmacother. 2022;155:113705. doi:10.1016/j.biopha.2022.113705
- Zhang J, Wang Y, Yin C, et al. Artesunate improves venetoclax plus cytarabine AML cell targeting by regulating the Noxa/Bim/Mcl-1/p-Chk1 axis. Cell Death Dis. 2022;13(4):379. doi:10.1038/s41419-022-04810-z
- Agashe RP, Lippman SM, Kurzrock R. JAK: not just another kinase. Mol Cancer Ther. 2022;21(12):1757–1764. doi:10.1158/1535-7163.MCT-22-0323
- Johnson DE, O'Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–248. doi:10.1038/nrclinonc.2018.8
- Moser B, Edtmayer S, Witalisz-Siepracka A, et al. The ups and downs of STAT inhibition in acute myeloid leukemia. Biomedicines. 2021;9(8). doi:10.3390/biomedicines9081051
- Almajali B, Johan MF, Al-Wajeeh AS, et al. Gene expression profiling and protein analysis reveal suppression of the C-Myc oncogene and inhibition JAK/STAT and PI3 K/AKT/mTOR signaling by thymoquinone in acute myeloid leukemia cells. Pharmaceuticals (Basel). 2022;15(3):307. doi: 10.3390/ph15030307
- Sha C, Jia G, Jingjing Z, et al. miR-486 is involved in the pathogenesis of acute myeloid leukemia by regulating JAK-STAT signaling. Naunyn-Schmiedeberg's Arch Pharmacol. 2021;394(1):177–187. doi:10.1007/s00210-020-01892-4
- Wang L, Zhang H, Lei D. microRNA-146a promotes growth of acute leukemia cells by downregulating ciliary neurotrophic factor receptor and activating JAK2/STAT3 signaling. Yonsei Med J. 2019;60(10):924–934. doi:10.3349/ymj.2019.60.11.1093
- Shen M, Xu Z, Xu W, et al. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 2019;38(1):149. doi:10.1186/s13046-019-1161-8
- Ham IH, Wang L, Lee D, et al. Curcumin inhibits the cancer-associated fibroblast-derived chemoresistance of gastric cancer through the suppression of the JAK/STAT3 signaling pathway. Int J Oncol. 2022;61(1):85. doi: 10.3892/ijo.2022.5375
- Sabaawy HE, Ryan BM, Khiabanian H, et al. JAK/STAT of all trades: linking inflammation with cancer development, tumor progression and therapy resistance. Carcinogenesis. 2021;42(12):1411–1419. doi:10.1093/carcin/bgab075