
ABSTRACT
Hyperammonemia is a rare and often fatal complication following the conditioning therapy in autologous and allogeneic stem cell transplant recipients. It is characterized by anorexia, vomiting, lethargy and coma without any other apparent cause. The diagnosis is often delayed because symptoms can be subtle and ammonia is usually not included among the routine analyzes. Previous reports have not identified the molecular mechanisms behind hyperammonemia in stem cell transplant recipients. Urea cycle disorders (UCDs) are inborn errors of metabolism leading to hyperammonemia that usually presents in early childhood, whereas first presentation in adults is less common. Here we describe an adult woman with hyperammonemia following autologous stem cell transplantation for multiple myeloma. No apparent cause of hyperammonemia was identified, including portosystemic shunting, liver dysfunction or recent hyperammonemia-inducing chemotherapy. Hyperammonemia, normal blood glucose as well as anion gap and a previous history of two male newborns that died early after birth, prompted biochemical and genetic investigations for a UCD. A heterozygous variant in the X-linked gene encoding ornithine transcarbamylase (OTC) was identified and was regarded as a cause of UCD. The patient improved after treatment with nitrogen scavengers and high caloric intake according to a UCD protocol. This case report suggests that UCD should be considered as a possible cause of hyperammonemia following stem cell transplantation.
Introduction
Hyperammonemia following stem cell transplantation is a rare complication in the early post-transplant period and is characterized by hyperammonemia, with no apparent evidence of liver cell damage and no other obvious cause. Only a limited number of case reports have been published as summarized in [Citation1–9]. The complication is associated with a mortality of at least 40% ().
Table 1. Summary of published case reports of hyperammonemia following stem cell transplantation.
Hyperammonemia has also been described following conventional chemotherapy, especially in patients with multiple myeloma (), and after solid organ transplantation, most often lung transplantation [Citation10]. The most common clinical manifestations are hyperventilation, seizures, altered mental status and coma. All reported cases required intensive care treatment and many were fatal. Several pathogenic mechanisms have been suggested, including microvascular abnormalities causing intrahepatic shunting. However, no underlying causes have been identified except for in lung transplant recipients, where a possible association with mycoplasma and ureaplasma infections has been suggested [Citation11].
Case report
A 65-year-old previously healthy Caucasian woman was diagnosed with multiple myeloma (monoclonal IgA kappa, international staging score III) presenting with anemia, hypercalcemia and osteolytic bone disease. She received induction treatment with bortezomib, lenalidomide and dexamethasone followed by stem cell mobilization with cyclophosphamide plus granulocyte colony-stimulating factor and thereafter stem cell harvesting without any major complications. She proceeded to autologous stem cell transplantation with melphalan 200 mg/m2 as the pretransplant conditioning therapy. During the first post-transplant week the patient experienced neutropenic fever accompanied by anorexia and hyperventilation. She was transferred to the intensive care unit five days after stem cell reinfusion. Candida albicans was found in blood cultures eight days post-transplant. Blood gas analysis showed respiratory alkalosis. At this time, she was markedly tired and obtunded without any apparent cause, and required artificial mechanical ventilation. She had near normal glucose levels (6.5 mmol/L, reference range 4,0 - 6,0 mmol/L) and normal serum levels for liver enzymes and bilirubin (8 μmol/L; reference range < 19 μmol/L) and a normal anion gap. Her creatinine was elevated (127 μmol/L, reference range 45 - 90 μmol/L), and arterial blood ammonia levels were increased to 240 μmol/L (normal range < 45 μmol/L). Cerebral computer tomography was normal, abdominal ultrasound examination was also normal without signs of portal venous shunting. Her hyperammonemia persisted despite standard treatment with laxatives, intraluminal antibiotics, volume repletion, and protein restriction. She therefore received additional treatment with rifaximin, lactulose, protein-free oral and parenteral nutrition and continuous hemofiltration resulting in normalized ammonia. The patient was gradually weaned off ventilator and was discharged from the intensive care unit.
Since no apparent cause of hyperammonemia was identified, late-onset of an inborn error of metabolism (IEM) was suspected. The patient had previously had no typical symptoms of an IEM, other than a history of headaches that did not require medical work-up or interventions. However, the patient had two previous pregnancies with male offspring born apparently well at full term, but both became critically ill 24–48 h after birth with trembling, seizures, and subsequent death. Autopsy showed cerebral edema in both cases, tissue samples have not been retrieved at a later stage an thus genetic investigations were not performed.
Metabolic work-up performed in urine and plasma obtained during stable clinical phase revealed repeatedly increased urinary uracil (16 and 24 µmol/mmol creatinine, reference range <13.1 µmol/mmol creatinine) and decreased plasma citrulline (13 and 14 µmol/L, reference range 16–46 µmol/L), while plasma glutamine and the excretion of orotic acid were normal.
Genetic testing using a gene panel for IEM identified a heterozygous variant in the X-linked gene encoding ornithine transcarbamylase (OTC), OTC c.[77+5G>A];[=] (NM_000531.5). The variant was confirmed with Sanger sequencing, but parents were not available for genetic testing. An attempt was also made to verify a splice change by mRNA analysis of blood samples, but – as expected – this was not successful since the OTC-gene is primarily expressed in the liver. This intronic +5 variant most likely affects the donor splice site in exon 1, and has previously been reported once in a newborn with OTC deficiency [Citation12].
The patient was treated according to the recommendations for OTC deficiency with a low-protein diet and avoidance of catabolism [Citation13]. After an attempt of reinitiating normal nutritional support, she experienced an increase in ammonia levels to a maximum of 132 μmol/L and worsening of symptoms, but this improved after the high caloric protein-reduced diet was re-initiated.
Discussion
Ammonia is a neurotoxin that exerts its effects through several and complex mechanisms [Citation13,Citation14] including accumulation of brain glutamate as well as neurotransmitters including N-methyl-D-aspartate (NMDA) receptor agonists, ATP depletion, and altered astrocyte water and K+ balance. Ammonia detoxification leads to glutamine accumulation in astrocytes, which results in cerebral edema due to an increase in osmotic stress with astrocyte swelling and dysfunction. Effective removal of ammonia is required to avoid neurotoxic damage [Citation13,Citation14]. Ammonia concentrations in the portal blood are usually 5–10 times higher than in the systemic circulation due to intestinal bacterial degradation of amino acids and urea, in addition to the hepatic metabolism of amino acids and other nitrogen-containing substances. Excessive ammonia is removed in the mitochondrial matrix and the cytosol of hepatocytes through the urea cycle, where ammonia is converted to non-toxic urea (), which is excreted in urine [Citation13]. Defects in the genes encoding urea cycle enzymes result in impaired removal of ammonia and are referred to as primary UCDs. Primary UCDs also comprise N-acetylglutamate synthase deficiency, as N-acetylglutamate (NAG) is an activator of CPS1 and deficiency of the mitochondrial ornithine transporters and citrin.
Figure 1. An overview of ammonia metabolism and effects on the brain. Excessive ammonia is removed through cytosolic and mitochondrial steps in hepatocytes. Step 1 requires carbamoyl phosphate synthetase (CPS1), step 2 Ornithine transcarbamylase (OTC), step 3 argininosuccinate synthetase converts citrulline, step 4 argininosuccinate, argininosuccinate lyase and step 5 arginase. Ammonia toxicity is complex and incompletely understood. Ammonia exerts its effects through disruption of neurotransmitter by depletion of glutamate, a precursor of GABA and other neurotransmitters (A and C) and blockade of the citric acid cycle (TCA) and there by oxidative phosphorylation (B). Hyperammonemia also leads to astrocyte swelling.
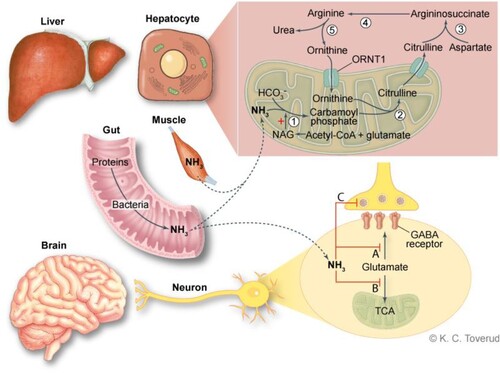
During the fetal period excess ammonia is removed by the mother, and UCDs usually manifest a few days after birth when the infant is fed with protein-containing milk, or when endogenous protein is released due to the normal neonatal catabolic state. Typical manifestations are nausea, neurological symptoms, encephalopathy, and hyperventilation. OTC deficiency is the only X-linked UCD, whereas defects in the other urea cycle enzymes are inherited in an autosomal recessive manner. OTC deficiency was previously thought to only occur in men, but heterozygous females can also present with symptoms depending on skewed X-inactivation in liver cells [Citation15]. Patients with partial defects or female carriers of pathogenic OTC variants may have an atypical presentation with only mild symptoms (e.g. headache) and may develop a fulminant hyperammonemic crisis with severe or lethal outcome after the newborn period if exposed to triggers such as excessive protein intake, catabolic situations, and administration of certain medications [Citation16]. Possible triggers in our patient may have been recent stress related to transplantation, a severe infection and malnutrition.
Urinary orotic acid, a biomarker for OTC deficiency, and plasma glutamine were normal in our patient when the patient was stable. Increased plasma glutamine is an indicator of elevated ammonia, and could be normalized during a metabolic stable phase. Previous studies have shown normal excretion of orotic acid in adult onset patients [Citation17], and thus normal levels of orotic acid should not exclude the diagnosis of OTC deficiency.
To the best of our knowledge, this is the first report of late-onset UCD as the cause of severe hyperammonemia following stem cell transplant in an adult patient. Laemmle et al. [Citation18] described fatal hyperammonemia in a 2-year-old boy following high-dose chemotherapy and autologous stem cell transplant for neuroblastoma. Biochemical patterns of their patient were consistent with CPS1 deficiency, but no mutations in CPS1 or OTC were identified. However, the authors showed that carboplatin and etoposide caused a dose-dependent decrease in CPS1 protein expression, and they concluded that chemotherapy could have induced a UCD-like state.
Conclusion
Hyperammonemia following allogenic stem cell transplantation is rare, but when it occurs, it is associated with high mortality. The diagnosis can easily be delayed or overlooked since ammonia is not a routine analysis; other laboratory analyzes as well as organ functions may be normal, and ammonia testing requires specific precautions (i.e. airtight chilled tubes with ammonia-free sodium heparin, analysis within 30 min). Furthermore, diagnostic work-up requires a multidisciplinary approach to exclude other causes of hyperammonemia and to establish a prompt and accurate diagnosis as a basis for adequate treatment.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Uygun V, Karasu G, Daloglu H, et al. Idiopathic hyperammonemia after hematopoietic stem cell transplantation: A case report. Pediatr Transplant. 2015;19(4):E104–E105.
- Frere P, Canivet JL, Gennigens C, et al. Hyperammonemia after high-dose chemotherapy and stem cell transplantation. Bone Marrow Transplant. 2000;26(3):343–345. doi:10.1038/sj.bmt.1702485
- Graetz R, Meyer R, Shehab K, et al. Successful resolution of hyperammonemia following hematopoietic cell transplantation with directed treatment of Ureaplasma parvum infection. Transpl Infect Dis. 2018;20(2):e12839), doi:10.1111/tid.12839
- Ho AY, Mijovic A, Pagliuca A, et al. Idiopathic hyperammonaemia syndrome following allogeneic peripheral blood progenitor cell transplantation (allo-PBPCT). Bone Marrow Transplant. 1997;20(11):1007–1008. doi:10.1038/sj.bmt.1701003
- Laemmle A, Hahn D, Hu L, et al. Fatal hyperammonemia and carbamoyl phosphate synthetase 1 (CPS1) deficiency following high-dose chemotherapy and autologous hematopoietic stem cell transplantation. Mol Genet Metab. 2015;114(3):438–444. doi:10.1016/j.ymgme.2015.01.002
- Mitchell RB, Wagner JE, Karp JE, et al. Syndrome of idiopathic hyperammonemia after high-dose chemotherapy: review of nine cases. Am J Med. 1988;85(5):662–667. doi:10.1016/S0002-9343(88)80239-0
- Snyder MJ, Bradford WD, Kishnani PS, et al. Idiopathic hyperammonemia following an unrelated cord blood transplant for mucopolysaccharidosis I. Pediatr Dev Pathol. 2003;6(1):78–83. doi:10.1007/s10024-001-0271-3
- Tse N, Cederbaum S, Glaspy JA. Hyperammonemia following allogeneic bone marrow transplantation. Am J Hematol. 1991;38(2):140–141. doi:10.1002/ajh.2830380213
- Davies SM, Szabo E, Wagner JE, et al. Idiopathic hyperammonemia: a frequently lethal complication of bone marrow transplantation. Bone Marrow Transplant. 1996;17(6):1119–1125.
- Seethapathy H, Fenves AZ. Pathophysiology and management of hyperammonemia in organ transplant patients. Am J Kidney Dis. 2019;74(3):390–398. doi:10.1053/j.ajkd.2019.03.419
- Paparoupa M, Barten MJ, de Heer J, et al. Hyperammonemia by Ureaplasma urealyticum pneumonia after lung transplantation. Respir Med Case Rep. 2020;30:101080.
- Tuchman M, Morizono H, Rajagopal BS, et al. Identification of ‘private’ mutations in patients with ornithine transcarbamylase deficiency. J Inherit Metab Dis. 1997;20(4):525–527. doi:10.1023/A:1005301513465
- Haberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis. 2019;42(6):1192–1230. doi:10.1002/jimd.12100
- Auron A, Brophy PD. Hyperammonemia in review: pathophysiology, diagnosis, and treatment. Pediatr Nephrol. 2012;27(2):207–222. doi:10.1007/s00467-011-1838-5
- Musalkova D, Sticova E, Reboun M, et al. Variable X-chromosome inactivation and enlargement of pericentral glutamine synthetase zones in the liver of heterozygous females with OTC deficiency. Virchows Arch. 2018 Jun;472(6):1029–1039. doi:10.1007/s00428-018-2345-x
- Pridmore CL, Clarke JT, Blaser S. Ornithine transcarbamylase deficiency in females: an often overlooked cause of treatable encephalopathy. J Child Neurol. 1995;10(5):369–374. doi:10.1177/088307389501000506
- Brassier A, Gobin S, Arnoux JB, et al. Long-term outcomes in Ornithine Transcarbamylase deficiency: a series of 90 patients. Orphanet J Rare Dis. 2015;10(1):58), doi:10.1186/s13023-015-0266-1
- Laemmle A, Gallagher RC, Keogh A, et al. Frequency and pathophysiology of acute liver failure in ornithine transcarbamylase deficiency (OTCD). PLoS One. 2016;11(4):e0153358.