
ABSTRACT
Background
β-thalassemia is a common inherited hemolytic disorder caused by mutations in the HBB gene. Genetic analysis of 2 new beta-thalassemia patients with deletion mutations in the HBB gene and their family members.
Methods
Their clinical presentation and blood phenotypic tests were analyzed. We detected the approximate degree of deletion of these two new HBB gene deletion mutants and analyzed their specific deletion locations by multiplex ligation-dependent probe amplification (MLPA), reverse breakpoint polymerase chain reaction (GAP-PCR), and sanger DNA sequencing.
Results
Two new deletion mutants of the HBB gene were identified. First, a 49% decrease in the expression of the third exon of the HBB gene was detected by MLPA testing, and then proband 1 and her mother were found to have HBB: exon3del and proband 2 and her mother to have HBB: c.−81A > C by GAP-PCR and sanger sequencing.
Conclusion
When the blood phenotype and clinical manifestations do not match the genotype, the presence of new mutants should be considered, and attention should be paid to further testing to avoid missing the diagnosis, which can help in clinical diagnosis and treatment, prenatal diagnosis and genetic counseling.
KEYWORDS:
1. Introduction
Thalassemia is a common monogenic genetic disorder and is often divided into two main groups, α-thalassemia and β-thalassemia. β-thalassemia is due to partial or complete inhibition of β-globin chain synthesis, resulting in an imbalance in the ratio of alpha and beta chains. Nearly one hundred alleles of β-thalassemia have been reported to have been identified. However, only about 40 species account for 90% or more of global β-thalassemia [Citation1]. The main types of mutations in β-thalassemia are single base substitutions, insertions or deletions resulting in subsequent base pair shift mutations. In China, reverse dot hybridization or flow fluorescence methods are generally used for detection and genetic diagnosis. 17 kits are available for the detection of β-thalassemia that provide comprehensive testing for the mutations: −28、−29、−30、−32、CD14-15、CD17、CD26、CD27-28、CD31、CD41-42、CD43、CD71-72、Int M、IVS-I-1、IVS-I-5、IVS-II-654 and Cap [Citation2]. Clinically, β-thalassemia is characterized by ineffective erythropoiesis, chronic hypoxia, anemia, and iron overload [Citation3,Citation4]. Severity of β-thalassemia depends on inherited alleles. Defects in the β-globin gene result in either absent (β0) or reduced (β+) β-globin chain synthesis. Sagar et al. found that β0 homozygotes were more severe compared to β0 heterozygotes or β0β+ heterozygotes [Citation5]. The severity of disease expression is mainly related to the degree of excess of precipitated α-globin chain in erythrocyte precursors [Citation6]. β-thalassemia major is most often detected shortly after birth and its treatment depends on long-term blood transfusions or hematopoietic stem cell transplants [Citation7].
Because the spectrum of mutations in thalassemia is geographically specific and particularly prevalent in subtropical regions, some rare mutations are very easy to miss [Citation8]. Lack of awareness of these rare pathogenic mutations can largely affect the clinical management, prenatal diagnosis and genetic counseling of thalassemia. Therefore, we need to refine knowledge on the different thalassemia gene mutations in a population.
2. Materials and methods
2.1. Subject
Proband 1, male, 15 years old, from Gui gang City, Guangxi Zhuang Autonomous Region. The family complained that the child appeared pale and yellow at the age of one year, and was diagnosed with thalassemia at the local hospital and started blood transfusion therapy. Until about 8 years of age his anemia worsened, blood transfusions became frequent, and iron removal therapy was started. He came to the First Affiliated Hospital of Guangxi Medical University in December 2016 and had the common 17 thalassemia gene test done and it came back at 41–42 M/N. CD41-42 i.e. HBB c.124_127delTTCT, which is the Traditional name for the HBB gene mutation [Citation9]. The heart iron and liver iron MRIs of thalassemia showed no deposition of cardiac iron, heavy liver iron deposition, and general examination of hepatomegaly and splenomegaly. In April 2022, he came to the clinic for bone marrow transplantation consultation, so we investigated his HSCT donor, his sibling, a 13-year-old male, showed abnormal hemoglobin A2 (Hb A2) and hemoglobin F (Hb F) by hemoglobin electrophoresis and the thalassemia gene of 41-2M/N. Therefore, further investigation of the blood phenotypes of the members of his family line, the father of Proband 1 and the mother of Proband 1. in . the family tree of this family is shown in .
Figure 1. Family lineage chart.
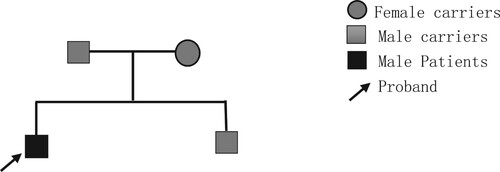
Table 1. Hematological phenotypic analysis of Proband 1 and its family lineage.
Proband 2, female, 1 year and 4 months, from Guangxi. The family complained that the child was found to be pale 10 months after birth, and she was found to have low hemoglobin with a fluctuating range of 60-105 g/L in the routine blood tests at the local hospital several times, so she went to the First Affiliated Hospital of Guangxi Medical University for medical treatment, and the examination of thalassemia gene showed 41-42M/N, which was not consistent with the clinical presentation. Therefore, further investigation of the blood phenotypes of the members of her family line, the father of Proband 2 and the mother of Proband 2. in . The family lineage map of this family is shown in .
Figure 2. Family lineage chart.
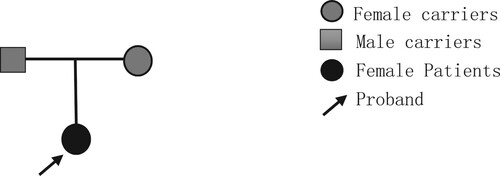
Table 2. Hematological phenotypic analysis of Proband 2 and its family lineage.
The above two families are not related by blood.
The research was approved by the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (ethical approval number: NO.2022-KY-(068)), and the research subjects and guardians signed an informed consent form.
2.2. Methods
2.2.1. Sample collection
A total of 2-3 ml of EDTA-anticoagulated peripheral blood was collected from Proband 1, father of Proband 1, mother of Proband 1, and sibling of Proband 1, and 2-3 ml of EDTA-anticoagulated peripheral blood was collected from Proband 2, father of Proband 2, and mother of Proband 2, respectively, and stored at 4°C. The informed consent form is signed by the individual at the time of blood sample collection.
2.2.2. DNA isolation
Genomic DNA was extracted using the Whole Blood Genomic DNA Extraction Kit (Tiangen Beijing Co., Ltd.). The NanoDrop ONE UV spectrophotometer (Thermo Fisher) determined DNA sample purity of 1.8-2.0 at 50-200 ng/μL and stored at −20°C.
2.2.3. MLPA assay
The P102-D1 HBB kit (MRC-HOLLAND, The Netherlands) was used. According to the instructions, the first step is Left (LPO) and Right Probe Oligo (RPO) bind to their target DNA. The second step is hybridized probe oligos are ligated by ligase enzyme. The third step is amplification. The last step is that PCR products are separated by length and Coffalyser.Net performs a quality check and calculates probe ratios. A probe ratio of 1.0 signifies a normal diploid copy number; a probe ratio of 0.5 a heterozygous deletion. They were entrusted to Nanning Qibo Biological Company for detection.
2.2.4. GAP-PCR and sanger sequencing
Based on the results of MLPA assay and the reference sequence of HBB gene in NCBI database (HBB, NG_00007), primers were designed to detect the promoter region, first exon, second exon and third exon of HBB gene, and the selected primers are shown in . The PCR reaction body contains 2×Rapid Taq Master Mix (Vazyme) 25μL, 2μL each of forward and reverse primers(10μM), 0.1∼1μg of genomic DNA, and ddH2O to make up 50μL of the reaction system. The PCR reaction conditions were pre-denaturation at 95°C for 3 min, denaturation at 95°C for 15 sec, annealing at 55°C for 15 sec, extension at 72°C for 5 sec, 30 cycles, and extension at 72°C for 5 min. The PCR products were separated by 1.5% agarose gel electrophoresis containing Gelred, and the amplification products were determined by fragment size. The PCR products were sent to Shanghai Sangon Biotech for Sanger sequencing.
Table 3. Primer sequences and amplification fragments of HBB gene.
3. Results
3.1. MLPA assay results
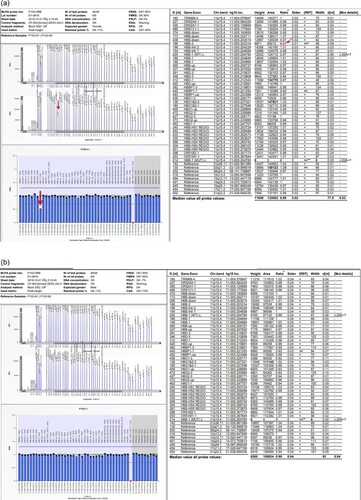
The sample from Proband 1's mother showed a heterozygous deletion at the 166nt probe, which is located in the third exon of the HBB gene, and a 49% decrease in expression there. These result with controls were shown in .
Figure 3. MLPA assay map of the mother of Proband 1 and controls (normal individuals without thalassemia). a. MLPA assay map of the mother of Proband 1 b. MLPA assay map of the mother of controls (normal individuals without thalassemia). The Proband 1's mother showed a 49% decrease in expression at the 166nt probe compared to the control, suggesting a heterozygote deletion. All are pointed out with arrows in the figure.
3.2. GAP-PCR and sanger sequencing results
GAP-PCR and gene sequencing results showed that Proband 1 and his father and sibling had the HBB gene c.124_127delTTCT, and Proband 1 and his mother had the HBB gene exon3del, in and . His father and mother's siblings were all beta-thalassemia carriers. Because of the long deletion fragment in the third exon of both Proband 1 and its mother, the deletion segments were found by sanger sequencing to be
Figure 4. GAP-PCR results of Proband 1 and its family lineage: I. DNA Marker. II.a. From left to right, the second exon PCR results for Proband 1, Proband 1's father, and Proband's sibling. b. Proband 1’s mother exon 3 PCR results. c. Proband 1 exon 3 PCR results. Because Proband 1 is a double heterozygous mutation, the bar has multiple bars.
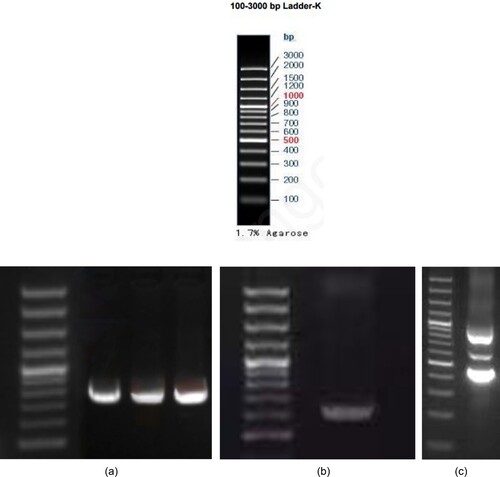
Figure 5. Sequencing map of Proband 1 and its family lineage. : a. a sequencing map of the second exon of Proband 1, c.124_127delTTCT. b. a sequencing map of the second exon of Proband 1’s sibling, c.124_127delTTCT. c. a sequencing map of the second exon of Proband 1’s father, c.124_127delTTCT.d. a sequencing map of the third exon of Proband 1, exon3del. e. a sequencing map of the third exon of Proband 1’s mother, exon3del.
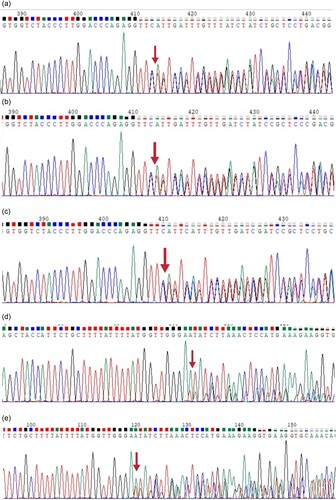
‘CTCCTGGGCAACGTGCTGGTCTGTGTGCTGGCCCATCACTTTGGCAAAGAATTCACCCCACCAGTGCAGGCTGCCTATCAGAAAGTGGTGGCTGGTGTGGCTAATGCCCTGGCCCACAAGTATCACTAAGCTCGCTTTCTTGCTGTCCAATTTCTATTAAAGGTTCCTTTGTTCCCTAAGTCCAACTACTAAACTGGGGGATATTATGAAGGGCCTTGAGCATCTGGATTCTGCCTAATAAAAAACATTTATTTTCATTGC’. The breakpoints are located in the upstream and downstream intron regions of the third exon of HBB.
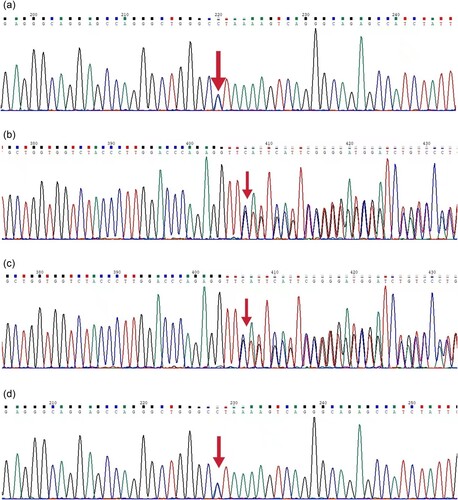
Proband 2 and his father had the HBB gene c.124_127delTTCT, and Proband 2 and his mother had the 5′ UTR region of the HBB gene c.−81A > C, in and . Proband 2, a double heterozygous mutation in the HBB gene c.−81A > C and c.124_127delTTCT resulting in beta chain synthesis deletion, is a beta thalassemia major whose parents are beta thalassemia carriers.
Figure 6. GAP-PCR results of Proband 2 and its family lineage. I. : I: a. Proband 2 promoter region and exon 1 PCR results. b. Proband 2’s mother region and exon 1 PCR results. c. Proband 2 exon 2 PCR results. d. Proband 2’s father exon 2 PCR results. (DNA Marker same as I)
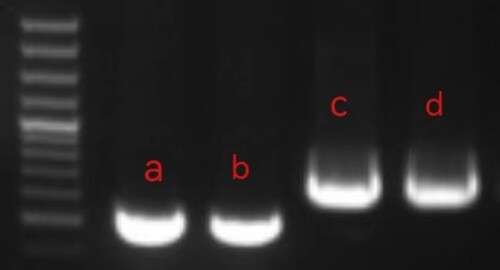
Figure 7. Sequencing map of Proband 2 and its family lineage. . a. a sequencing map of the promoter region of Proband 2 with C/A at position 220 as c.−81A > C. b. a sequencing map of the second exon of Proband 2, c.124_127delTTCT.c. a sequencing map of the second exon of Proband 2’s father, c.124_127delTTCT. d. a sequencing map of the promoter region of Proband 2’s mother with C/A at position 220 as c.−81A > C.
4. Discussion
Proband 1 received blood transfusions every few months from the time he was found to be anemic until he was 8 years old when his anemia worsened and he began to receive frequent transfusions at monthly intervals. However, his thalassemia genetic test results in several hospitals showed HBB 41-42M/N, his father and his sibling also turned out to be HBB 41-42M/N, and his mother was shown not to have thalassemia. Analysis of the clinical manifestations and hematological indices of proband 1 and family members revealed that proband 1 was a manifestation of β-thalassemia major, and the remaining three were all β-thalassemia gene mutation carriers. Since the thalassemia gene mutation was not detected in the mother of proband 1 in several hospitals, it was considered to be a large segmental deletion, which was difficult to detect by one-generation sequencing. In this research, MLPA assay was first performed on the samples from the proband 1's mother. The P102-D1 HBB kit (MRC-HOLLAND, The Netherlands) was used, and the results of the assay showed that a 49% decrease in the expression of HBB was found at the third exon of HBB, 166nt probe, which is a heterozygous deletion, but there was only one such probe, so it was not possible to determine whether this was an experimental error or a third exon deletion. Therefore, multiple pairs of HBB third exon primers were designed to measure the heterozygous deletion of the third exon of the mother of the proband 1 by continuously stretching the GAP-PCR range to find out the specific missing fragment. Proband 1 mother had a large deletion of the third exon of HBB with a breakpoint located in the intron site upstream and downstream of the third exon of HBB. The range is too large to be detected by common genetic sequencing, which could explain the failure to detect thalassemia in the mother of proband 1 in several hospitals. The genetic inheritance of the rare deletion type β- thalassemia in proband 1 came from her mother by GAP-PCR and sanger sequencing. The results can be well interpreted by hemoglobin electrophoresis results, routine blood results and clinical manifestations. Proband 1 was a β-thalassemia major who started long-term blood transfusion at the age of one year. Proband 1's sibling and his mother and father were β-thalassemia carriers with only mild anemia that did not require special treatment, but had abnormal hemoglobin electrophoresis.
Proband 2 is a double heterozygote for the HBB genes c.−81A > C and c.124_127delTTCT. Her clinical presentation and genotype were consistent with beta-thalassemia major. c.−81A > C is located in the 5′UTR region of the HBB gene, which is a non-coding amino acid, but the 5’ UTR region contains several regulatory elements such as upstream ORFs, internal ribosomal entry sites, enhancers and even structural components involved in the regulation of mRNA stability, pre-mRNA splicing and even translation initiation. Defects in these regions of the 5’ UTR can lead to disease. Her mother was a carrier of the HBB gene variant c.−81A > C, and her blood showed routine changes consistent with those seen in common β-depleted carriers, i.e. small red blood cells and mild anemia. Her father is a carrier of HBB gene c.124_127delTTCT. The HBB gene c.−81A > C was inherited from the mother and c.124_127delTTCT was inherited from the father in Proband 2.
The above findings are reported in two unrelated families in the Guangxi Zhuang Autonomous Region of China. There have been many HBB point mutations reported, such as the (β+) −31 (A > G) and (β⁰) Codon 17 (A > T) [Citation10]. However, the two new mutations in the HBB gene identified in this research have not yet been reported, which is crucial for the research of thalassemia gene mutations.
β-thalassemia is a hereditary hemolytic disease [Citation9]. Mild thalassemia does not require treatment; intermediate thalassemia does not require blood transfusion, but in case of infection, stress, surgery, etc., appropriate transfusion of concentrated red blood cells can be given; thalassemia major is often accompanied by long-term blood transfusion and iron removal treatment [Citation11]. Hematopoietic stem cell transplantation (HSCT) is also available and is currently the only powerful treatment for β-thalassemia, but the pre- and post-operative risks are unpredictable and post-transplant rejection (GVHD) and infection are still great risk factors [Citation4,Citation12–14]. Therefore, it is now accepted worldwide that the preferred countermeasure is prenatal screening and that early termination of pregnancy can effectively prevent the development of thalassemia major [Citation15,Citation16]. The current range of thalassemia genetic tests provided by the major kits covers 95%−98% of β-thalassemia-causing genetic defects in the Chinese population, but rare or unknown mutations are missed at a rate of about 2% [Citation17]. When it is difficult to be detected by one-generation sequencing, MLPA assay can be chosen to roughly localize the mutation location first, and then find out the specific mutation site by GAP-PCR and gene sequencing. This research enriches the gene mutation spectrum of human β-thalassemia, which is beneficial for genetic diagnosis, clinical treatment, family genetic counseling and prenatal screening of thalassemia in the worldwide population, and provides more experimental data and methods for prevention, diagnosis and treatment of thalassemia.
Author contributions
Yaxuan Cao and Jianming Luo conceived the idea. Yaxuan Cao collected and analyzed the data. Yaxuan Cao drafted the manuscript. Jianming Luo reviewed the manuscript. Yaxuan Cao contributed to manuscript revision. All authors contributed to the article and approved the submitted version.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of First Affiliated Hospital of Guangxi Medical University [2022-KY-(068)]. Written informed consent to participate in this research was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Acknowledgments
The authors wish to sincerely thank her supervisor, Professor Jianming Luo.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Thein SL. Genetic basis and genetic modifiers of β-thalassemia and sickle cell disease[J/OL]. Adv Exp Med Biol. 2017;1013:27–57. doi:10.1007/978-1-4939-7299-9_2
- Jiang LX, Fan YH, Yi W, et al. Identification of 2 two rare cases of β0-thalassemia mutation odon41(-C)[J]. Chin J Eugenics Genet. 2018;26(11):14–17.
- Rund D, Rachmilewitz E. Beta-thalassemia[J]. N Engl J Med. 2005;353(11):1135–1146. doi:10.1056/NEJMra050436
- Cappellini MD, Motta I. New therapeutic targets in transfusion-dependent and -independent thalassemia[J/OL]. Hematology Am Soc Hematol Educ Program. 2017;2017(1):278–283. doi:10.1182/asheducation-2017.1.278
- Sagar CS, Kumar R, Sharma DC. 等. DNA damage: beta zero versus beta plus thalassemia[J/OL]. Ann Hum Biol. 2015;42(6):585–588. doi:10.3109/03014460.2014.990921
- Origa R. β-Thalassemia[J/OL]. Genet Med. Official J Am College Med Genet. 2017;19(6):609–619. doi:10.1038/gim.2016.173
- Muncie HL, Campbell J. Alpha and beta thalassemia[J]. Am Fam Physician. 2009;80(4):339–344.
- Chen DY, Sun XF. Progress in the study of the modifying role of iron metabolism on the phenotype of β-thalassemia[J]. Chin J Med Genet. 2021;38(1):27–31.
- Xinh PT, Chuong HQ, Hant T. 等. Spectrum of HBB gene mutations among 696 β–thalassemia patients and carriers in Southern Vietnam[J/OL]. Mol Biol Rep. 2022;49(4):2601–2606. doi:10.1007/s11033-021-07062-w
- Panyasai S, Jaiping K, Pornprasert S. Elevated Hb A₂ levels in a patient with a compound heterozygosity for the (β+) -31 (A > G) and (β0) Codon 17 (A > T) mutations together with a single α-globin gene[J/OL]. Hemoglobin. 2015;39(4):292–295. doi:10.3109/03630269.2015.1047513
- Khandros E, Kwiatkowski JL. Beta thalassemia: monitoring and new treatment approaches[J/OL]. Hematol Oncol Clin North Am. 2019;33(3):339–353. doi:10.1016/j.hoc.2019.01.003
- Shang X, Wu X, Zhang X, et al. Clinical practice guidelines for beta-thalassemia][J/OL]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2020;37(3):243–251. doi:10.3760/cma.j.issn.1003-9406.2020.03.004
- La Nasa G, Vacca A, Littera R, et al. What unrelated hematopoietic stem cell transplantation in thalassemia taught us about transplant immunogenetics[J/OL]. Mediterr J Hematol Infect Dis. 2016;8(1):e2016048. doi:10.4084/MJHID.2016.048
- Michlitsch JG, Walters MC. Recent advances in bone marrow transplantation in hemoglobinopathies[J/OL]. Curr Mol Med. 2008;8(7):675–689. doi:10.2174/156652408786241393
- Horvei P, Mackenzie T, Kharbanda S. Advances in the management of α-thalassemia major: reasons to be optimistic[J/OL]. Hematology Am Soc Hematol Educ Program. 2021;2021(1):592–599. doi:10.1182/hematology.2021000295
- Zhu XJ, Liu YP, Liu RY, et al. Analysis of prenatal genetic diagnosis of fetal thalassemia in homozygous α-thalassemia couples in Huizhou[J]. Chin J Child Health. 2015;23(1):15–17.
- Li XY, Chen SQ, Xu LH, et al. Clinical diagnosis and genetic testing in a case of intermediate β-thalassemia[J]. Chin J Pediatric Hematol Oncol. 2014;19(3):138–142.