
ABSTRACT
Congenital thrombotic thrombocytopenic purpura (TTP) is a rare autosomal recessive genetic disorder caused by mutations in the ADAMTS13 gene. Approximately 200 mutations of the ADAMTS-13 gene have been identified, although only a few have been characterized through in vitro expression studies. We conducted an investigation on a male congenital TTP patient with reduced plasma levels of ADAMTS13 activity. DNA sequence analysis revealed two mutations on chromosome 9 (1.9q34.2) in the patient’s ADAMTS13 gene. One mutation was a non-synonymous mutation (exon 5: c.A530G: p.Y177C), while the other was a nonsense mutation (exon 21: c.G2651A: p.W884X). Both mutations were found to be heterozygous. The patient’s parents had no history of thrombocytopenia or neurological symptoms. DNA sequence analysis showed the patient’s father was a heterozygote for the nonsense mutation of the ADAMTS13 gene (exon 21: c.G2651A: p.W884X), while the mother was a heterozygote for the non-synonymous mutation of the ADAMTS13 gene (exon 5: c.A530G: p.Y177C). To investigate the mechanism behind ADAMTS13 deficiency in this patient, wild type (WT), ADAMTS13 p.Y177C, and ADAMTS13 p.W884X were transiently expressed in 293-6E cells. Expression studies revealed a significant reduction in enzyme activity and secretion, although the protease was detected within the cells. The 3D structures of the natural and mutated ADAMTS-13 proteins were partially reconstructed using the Phyre2 web server. The mutation that replaces the tyrosine residue at amino acid position 177 with cysteine may result in decreased steric hindrance and a looser structure. This mutation affects the binding of calcium ions and the secretion of the enzyme from intracellular to extracellular compartments.
Introduction
Congenital thrombotic thrombocytopenic purpura (TTP), also known as Upshaw-Schulman syndrome, is a rare autosomal recessive genetic disorder caused by mutations in the ADAMTS13 gene. The annual incidence of this genetic disorder is uncertain, some experts estimate this to be 0.5–2 cases per million; further investigation is needed [Citation1]. It is more common in females and approximately twice as common in males [Citation2]. This condition belongs to the group of thrombotic microangiopathies (TMAs) and is characterized by microangiopathy, hemolytic anemia, decreased platelet aggregation consumption, and organ damage caused by microthrombosis, such as in the kidneys and central nervous system [Citation3].
Clinically, the main manifestations of congenital TTP can be summarized using the pentalogy: thrombocytopenia, microangiopathic hemolytic anemia, neuropsychiatric symptoms, fever, and renal impairment [Citation4]. Laboratory tests can determine the plasma ADAMTS13 activity and perform ADAMTS13 gene sequencing. The diagnostic criteria include an ADAMTS13 activity level below 10% [Citation5]. The pathogenesis of this disorder primarily involves mutations in the coding region of the ADAMTS13 gene, resulting in the inability of ADAMTS13 to synthesize or the production of an abnormal protein structure, leading to enzyme activity loss or reduction [Citation6]. Consequently, large von Willebrand factor (vWF) polymers cannot be inactivated, resulting in enhanced platelet aggregation. The diagnosis of hereditary thrombotic thrombocytopenic purpura relies on symptom identification (especially early recognition), ADAMTS13 activity assessment, and ADAMTS13 gene DNA sequencing.
The ADAMTS13 gene, also known as ADAM metallopeptidase with thrombospondin type 1 motif 13, is approximately 37kb long and consists of 29 exons. It is located on chromosome 9q34.2 [Citation7]. This gene encodes a member of the zinc finger metalloproteinase family, which possesses various domains, including a hydrophobic signal peptide, a propeptide, a metalloproteinase catalytic domain, an integrin-binding domain, multiple thrombin-sensitive protein motifs (TSP), a cysteine-rich domain, a spacer region, and two CUB domains [Citation8]. The structure of ADAMTS13 is closely related to its function. Its primary role is to cleave vWF into two fragments, 170kD and 140kD, preventing platelet aggregation and adhesion. This process is crucial in preventing microvascular thrombosis.
Mutations in the ADAMTS13 gene result in functional impairment, leading to decreased or complete loss of enzyme activity. In an autosomal recessive manner, these mutations can cause congenital TTP. Homozygous or compound heterozygous mutations in the ADAMTS13 gene are responsible for the development of this disorder. Over 200 mutations in the gene have been reported, including homozygous and heterozygous mutations [Citation7, Citation9, Citation10]. These mutations are distributed throughout the coding region of the ADAMTS13 gene. The most common types of mutations are nonsense mutations, followed by missense mutations, frame-shift insertions, or deletions [Citation11].
Salvage treatment involves performing plasma exchange promptly to remove uncleaved large molecule vWF and provide ADAMTS13 enzyme supplementation, aiming to improve the prognosis of patients. Prophylactic treatment requires regular injection of at least 1–2 units of fresh frozen plasma every 3 weeks. Maintaining ADAMTS13 activity levels between 5–10% can help prevent prophylactic episodes. Infusion therapy with ADAMTS13 has shown promising results in certain regions [Citation12].
In this manuscript, we present a case study of congenital TTP and conducted a molecular investigation on two natural ADAMTS13 mutants characterized by homozygous mutations at positions 177 within the metalloprotease domain and 884 between TSP4 and TSP5 domains. To gain insight into the effects of these mutations, we performed in vitro expression studies using 293E cells. We carried out a comprehensive biochemical characterization of these mutant enzymes and utilized software to construct 3D structural models. Our findings shed light on the potential impact of these mutations on ADAMTS13, including calcium binding, conformational changes, and secretion. These results contribute to a better understanding of the functional implications of the mutated sites in ADAMTS13.
Materials and methods
Case report
A 15-year-old boy was admitted to our hospital after suddenly losing consciousness for 5 days. Upon physical examination, he was found to be comatose with impaired mental state, poor light reflex, and no apparent pathological signs. His lips appeared pale, and petechial hemorrhages were observed on the oral mucosa and the skin throughout his body. Three years after birth, he was hospitalized due to fatigue and loss of appetite. Thrombocytopenia and hemolytic anemia were diagnosed following examination. Subsequently, no specific treatment was given, but regular blood routine check-ups showed a platelet count maintained at (30–80) × 109/L. Three years ago, he experienced sudden vomiting without an obvious cause, accompanied by dizziness and discomfort. Needle-shaped petechial hemorrhages were observed on the skin during examination, and the patient received treatment with gamma globulin and methylprednisolone. According to the family history, the patient was born with the umbilical cord wrapped around his neck, and he developed bruised skin after birth. There is no known familial genetic or tumor history in his family.
Initial laboratory tests revealed the following results: white blood cell count of 10.87 (reference range: 3.50 ∼ 9.50) × 109/L, hemoglobin concentration of 95.0 (reference range: 130.0 ∼ 175.0) g/L, platelet count of 10 (reference range: 125 ∼ 350) × 109/L, urea nitrogen level of 14.77 (reference range: 3.10 ∼ 8.00) mmol/L, creatinine level of 213.40 (reference range: 57 ∼ 111) umol/L, alanine aminotransferase level of 20.7 (reference range: 5 ∼ 50) IU/L, aspartate aminotransferase level of 67.0 (reference range: 15 ∼ 45) IU/L, total bilirubin level of 31.36 (reference range: 3.4 ∼ 21.0) umol/L, direct bilirubin level of 9.13 (reference range: 0.0 ∼ 5.0) umol/L, and indirect bilirubin level of 7.81 (reference range: 0 ∼ 18) umol/L. The activated partial thromboplastin time (APTT) was 45 (reference range: 20.00 ∼ 40.00) seconds. Examination of red blood cell morphology revealed approximately 0.9% fragmented erythrocytes. Creatine kinase (CK) level was elevated at 353 (reference range: 26 ∼ 192) IU/L, and lactate dehydrogenase (LDH) level was 6556.30 (reference range: 120 ∼ 250) IU/L. Other laboratory tests showed a hypersensitive troponin I level of 3.080 (reference range: 0.000 ∼ 0.034) ug/L, an amino terminal B-type natriuretic peptide precursor level of 11600 (reference range: 0 ∼ 450) pg/ml, and a myoglobin level of 181.40 (reference range: 0.00 ∼ 61.50) ug/L. Urine analysis revealed 1 + occult blood and 2 + protein. Head and chest CT scans showed no notable abnormalities in the brain, but revealed thickening of the left pleura and a small amount of pericardial effusion. The electrocardiogram (ECG) results were normal. Additionally, cranial magnetic resonance imaging (MRI) with plain scan and contrast enhancement exhibited multiple abnormal signals in the central regions of the bilateral hemispheres and lateral ventricles, suggesting demyelination of the white matter and impaired development of the white matter (Supplemental Figure 1).
The patient was admitted to the hospital presenting with unconsciousness. Initially, diazepam was administered for sedation, followed by endotracheal intubation and catheterization of the right femoral vein. Cefoxitin and high-dose methylprednisolone treatment (1g on days 1–3, 500mg on days 4–5) were initiated. Plasma exchange treatment was performed for 2 days, with 2000 ml of plasma replacement. Following the plasma exchange, the patient’s condition stabilized, although their mental status remained unclear. The platelet count increased to 94 × 109/L, and renal function improved with a decrease in creatinine levels by day 5. The infection index, indicated by CRP levels (which were elevated at 145.15mg/L), suggested a pulmonary infection. Consequently, the patient received maintenance treatment with prednisone (50mg on days 6–9, 45mg on days 10–13, 40mg on days 14–26) and piperacillin-tazobactam as an anti-infective agent. On the 6th day, 1500ml of plasma was administered (800ml in the morning, 700ml in the evening). Although the patient’s consciousness remained unclear, platelet levels returned to normal, leading to extubation and subsequent recovery of consciousness on the 12th day. On day 21, an additional 900ml of plasmapheresis was performed without any reported discomfort or complications. Vital signs remained stable, and subsequent blood routine examinations showed a normal platelet count of 163.0 × 109/L. After discharge, Dexamethasone therapy was discontinued, and regular injections of fresh frozen plasma (400ml once a week) were administered at the hospital. The treatment flow chart for the patient over the course of 25 days is provided in Supplemental Figure 2.
Measurement of ADAMTS13 activity, ADAMTS13 antigen, anti-ADAMTS13 antibodies
Serum ADAMTS13 activity was assessed using surface-enhanced laser desorption/ionization time of flight mass spectrometry (SELDI-TOF). The ADAMTS13 antigen was measured using the MultiskanFC thermo and the ADAMTS-13/vWF-cp ELISA kit (CUSABIO, Catalog Number: CSB-E13487h, Lot number: 201906). The antibody was tested using the Thermo MK3 Enzyme standard instrument and the Human von Willebrand Factor Cleaving Protease ELISA kit (SHANGHAI JINGKANG BIOENGINEERING CO.LTD, Catalog Number: JK139766, Lot number: 201907). The normal value of the antibody was determined based on measurements from 120 normal human specimens.
Measurement of vWF:Ag
Serum vWF antigen was tested by Beckman ACL TOP 700 using Turbidimetric inhibition immunoassay kit (Instrument Laboratory, Catalog Number: 0020002300, Lot number: B30993).
Mutation of ADAMTS-13 gene
All DNA analyses were conducted with the approval of the Ethics Committees of our hospital and the institute responsible for gene analysis. Written informed consent was obtained from his parents. Peripheral blood leukocytes were used to extract DNA, and a total of 700 genes associated with hereditary blood diseases were captured and sequenced using the Kapa HTP Library Prep Kit and Roche Nimblegen SeqCap EZ on an Illumina Hiseq X Ten platform. The primer sequences and amplification parameters can be provided upon request. To exclude the possibility of disease-causing ADAMTS13 mutations being common polymorphisms, individuals from the general population were screened.
Plasmid preparation
DNA sequences encoding the target proteins were custom-designed and synthesized. Subsequently, the entire sequences were subcloned into pTT5 vectors to facilitate expression in 293-6E cells. Detailed cloning strategies can be found in the Appendix section.
Cell culture and transient transfection
293-6E cells were cultured in serum-free FreeStyleTM 293 Expression Medium (Thermo Fisher Scientific). The cells were maintained in Erlenmeyer Flasks (Corning Inc., Acton, MA) at 37°C with 5% CO2 on an orbital shaker (VWR Scientific, Chester, PA). One day prior to transfection, the cells were seeded at an appropriate density in Corning Erlenmeyer Flasks. On the day of transfection, an optimal ratio of DNA and transfection reagent was mixed and added to the flask containing the cells prepared for transfection. Recombinant plasmids encoding the target proteins were transiently transfected into 40 mL suspension cultures of 293-6E cells. Cell density and viability were assessed on day 2, day 4, and day 5. The supernatant and cell pellet samples collected on these days post-transfection were used to evaluate protein expression.
Detection and analysis
Cell culture supernatant and cell pellet samples collected on day 2, day 4, and day 5 post-transfection were subjected to analysis using SDS-PAGE and Western blot techniques. For Western blotting, the primary antibody used was Mouse-anti-his mAb (GenScript), and the secondary antibody employed was Goat Anti-Mouse IgG Antibody (H&L) (HRP).
Molecular dynamics simulations
The FASTA sequence was obtained from the NCBI Protein database with an accession code of NP_620594.1. The Y177C mutation was manually incorporated into the sequence. To reconstruct the three-dimensional (3D) structures of the natural and mutated ADAMTS13 protein, the Phyre2 web server13 was utilized [Citation13]. Since there was no available template encompassing the full length of 1427 amino acids, residues 70–533 were selected for the reconstruction of the Y177C models. The structure of vascular apoptosis-inducing protein-1 (PDB code: 2ERP) was used as the template for this purpose. Additionally, models for the W884X mutation were constructed using residues 615–883 of ADAMTS13, with the structure of human complement component C8 (PDB code: 3OJY) serving as the template.
Results
ADAMTS13 levels in affected subjects
The patient’s plasma showed a serum ADAMTS13 activity of less than 5%, very low ADAMTS13 antigen levels lower than the normal limit, and negative ADAMTS13 antibody, indicating a diagnosis of congenital TTP. The parents of the patient had normal levels of ADAMTS13 antigen and activity, as well as negative ADAMTS13 antibody (A).
Table 1. ADAMTS13 activity and antibody level in plasma of the propositus and his parents (A), ADAMTS13 protein in the conditioned media of 293-6E cells transfected with WT and mutant expression vectors in reducing or non-reducing condition(B).
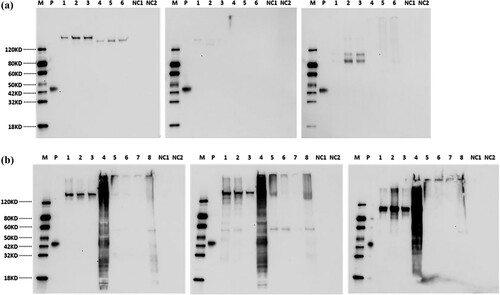
In our study, we successfully detected three variants of ADAMTS13, namely ADAMTS13-WT, ADAMTS13-Y177C, and ADAMTS13-W884X, in 293-6E cells. Both ADAMTS13-WT and ADAMTS13-W884X target proteins were successfully expressed at detectable levels in the cell culture supernatant and cell lysate, as illustrated in . However, for ADAMTS13-Y177C, the target protein was only detected at a detectable level in the cell lysate, not in the supernatant. ADAMTS13-W884X exhibited a smaller molecular weight (∼110kDa) that was detectable in both the cell and supernatant samples on Western blot analysis. The estimated molecular weights under reducing and non-reducing conditions are summarized in B.
Genetic analysis
DNA sequence analysis revealed that the patient carried two heterozygous mutations in the ADAMTS13 gene on chromosome 9 (1.9q34.2). One mutation was a missense mutation, c.530A > G (p.Y177C), inherited from the father. The other mutation was a nonsense mutation, c.2651G > A (p.W884X), inherited from the mother. Based on these findings, the patient was diagnosed with hereditary TTP. The patient’s parents did not exhibit any history of thrombocytopenia or neurological symptoms ( and ). More importantly, we did not identify any other family members of the patient who had congenital TTP. The patient’s DNA sequence analysis identified a non-synonymous mutation and a stopgain mutation in the ADAMTS13 gene located on chromosome 9. The non-synonymous mutation was inherited from the father, while the stopgain mutation was inherited from the mother.
Figure 1. ADAMTS13 mutation in the patient and his parents. DNA sequence chromatograms corresponding to exon 5 and exon 21 are shown from a healthy control, the patient and his parents.
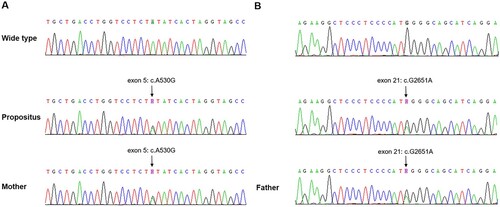
Figure 2. Family pedigree. The circle represents a female and squares represent males. The arrow indicates the index patient. Both the mother and the father of the patient are asymptomatic carriers.
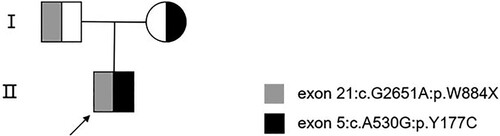
Expression and activity of the exon5: A530G and exon21: G2651A mutant in cell cultures
Western blot analysis was performed on cell culture supernatant and cell lysates derived from 293-6E cells at day 2, day 4, and day 5, under both reducing and non-reducing conditions. A comparison with the WT ADAMTS13 revealed that the p.Y177C mutation (inherited from the patient’s mother) resulted in the secretion of a smaller molecular weight protein compared to the wild-type protein bands. Similarly, the W884X mutation (inherited from the patient’s father) was detected in both the cells and the supernatant, showing a smaller molecular weight compared to the wild-type protein bands (). The presence of the wild-type protein in the conditioned media and cell lysates was confirmed through immunoblotting (A&B). SDS-PAGE analysis of the cell culture supernatant from 293-6E cells transduced with wild type, p.Y177C, and p.W884X mutation plasmids is presented in Supplement Figure 3A&B.
Figure 3. A Western blot analysis of cell culture supernatant from 293-6E cells respectively transducted with wide type, p.Y177C, p.W884X mutation plasmid. Lane M: Protein Marker, Lane P: Multiple-tag (GenScript, Cat.No.M0101) as positive control, Lane 1∼3: Cell culture supernatant from day 2, day 4 and day 5 under a reducing condition, Lane 4∼6: Cell culture supernatant from day 2, day 4 and day 5 under a non-reducing condition, Lane NC1: Negative control under a reducing condition, Lane NC2: Negative control under a non-reducing condition. B Western blot analysis of cell lysate from 293-6E cells respectively transducted with wide type, p.Y177C, p.W884X mutation plasmid. Lane M: Marker, Lane P: Multiple-tag (GenScript, Cat.No.M0101) as positive control, Lane 1∼3: Cell lysate supernatants from day 2, day 4 and day 5 post-transfection under a reducing condition, Lane 4: Cell debris from day 4 post-transfection under a reducing condition, Lane 5∼7: Cell lysate supernatants from day 2, day 4 and day 5 post-transfection under a non-reducing condition, Lane 8: Cell debris from day 4 post-transfection under a non-reducing condition, Lane NC1: Negative control under a reducing condition, Lane NC2: Negative control under a non-reducing condition.
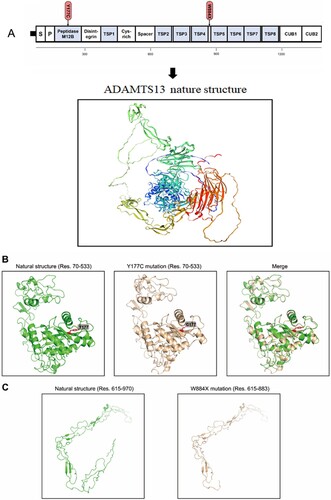
Modeling
The 3D models of normal ADAMTS13 and ADAMTS13 mutation are presented in . Based on the model, there were no significant overall differences observed. The substitution of tyrosine at position 177 with cysteine might have an impact on the steric hindrance of adjacent groups. However, since Y177 is not located in the catalytic active center, the effects on the overall protein activity may be limited. On the other hand, the W884X mutation, resulting in truncation, is likely to cause significant structural changes in the protein. The TSP5-TSP8, CUB1, and CUB2 domains are completely lost, leading to the corresponding loss of their natural functions. Previous reports have indicated that these domains are responsible for the exocytosis of the entire protein [Citation14]. Therefore, ADAMTS13 with the W884X mutation is expected to have impaired exocytosis function.
Figure 4. Domain organization and 3D model of ADAMTS13. (A). Schematic representation of ADAMTS13 protein domain and the location of the ADAMTS13 mutations. (B). Models of the wild type of ADAMTS13 protein and the Y177C mutation (residues 70–533 were modeled). (C) Models of the wild type of ADAMTS13 protein (residues 615–970 were modeled) and the truncated mutation (residues 615–883 were modeled).
Discussion
Upon admission, the patient was diagnosed with TTP based on the presence of thrombocytopenia, microangiopathic hemolysis, neurological symptoms, renal impairment, and fever. Subsequent laboratory tests confirmed the diagnosis as the ADAMTS13 enzyme activity was less than 5% and the ADAMTS13 antibody was negative. In cases of childhood onset, recurrent convulsions, coma, and congenital TTP, genetic testing for non-synonymous heterozygous mutations in the ADAMTS13 gene, including missense mutations, should be considered to support the diagnosis. In this patient, the diagnosis of congenital TTP was further supported by the identification of heterozygous mutations in both parents, suggesting a familial relationship with the patient’s disease gene mutations.
The patient received timely rescue treatment. Immediate fresh frozen plasma exchange was initiated upon clinical diagnosis, followed by regular administration of fresh frozen plasma for replenishment. The patient’s condition improved, with no symptoms observed, and platelet levels remained within the normal range. Although clinical trials using recombinant human ADAMTS13 enzyme for replacement therapy are available in other countries, they are not currently available in China. Therefore, the treatment approach in our case involved removal of vWF polymers through plasmapheresis and the administration of fresh frozen plasma for enzyme replenishment.
The first putative Ca2+-binding site involves the amino acid residues Glu83, Asp173, Cys281, and Asp284, which are highly conserved among ADAMTS and other metalloproteases. They are believed to mediate low-affinity Ca2+ binding. The second putative Ca2+-binding site includes residues Glu164 and Asp166, along with one or more of residues Asn162, Asp165, and Asp168. Mutations at this site do not affect the Ca2+-dependent activity of ADAMTS13. The third putative site is predicted to encompass residues Asp187 and Glu212, together with Asp182 or Glu184. Mutations at this site significantly reduce Ca2+-induced ADAMTS13 activity, suggesting that these residues play a crucial role in high-affinity Ca2+ binding and proteolytic activity [Citation15].
Lancellotti S reported that Asp173, which is located near the calcium cluster at sites 1 and 2, when mutated, reduces the interface area between the metalloprotease and disintegrin-like domain, resulting in improper folding and reduced secretion efficiency of ADAMTS13 [Citation16]. In our study, the mutant plasmid derived from the patient showed no protein secretion in the supernatant, while the cell exhibited a normal protein count. We speculate that this mutation may impact protein secretion from intracellular to extracellular compartments. The substitution of Tyr177 by Cys involves the aromatic R group of tyrosine being replaced by a cysteine with reduced steric hindrance. This change does not cause significant fluctuations in the overall protein structure but leads to fewer covalent bonds. Consequently, it may loosen the calcium ion binding region, affecting the binding and secretion of the enzyme from intracellular to extracellular compartments.
The other mutation, p.W884X, was inherited from the patient’s father and is located in exon 21, specifically in the region between TSP4 and TSP5. ADAMTS13 consists of eight TSR domains, and there are three linker regions between TSR2/TSR3, TSR4/TSR5, and TSR8/CUB1 domains [Citation17]. One of the frequently observed mutations in congenital TTP patients is Arg1060Trp in TSR7. Although this mutation does not affect protease activity, it impairs protein secretion, resulting in ADAMTS13 deficiency [Citation18]. Studies have shown that C-terminal truncated forms of ADAMTS13, lacking TSR2-7 and the CUB1-2 domains, can still cleave vWF [Citation19]. In the case of the p.W884X mutation, the overall structure and function of the TSR 5–8 and CUB1, CUB2 domains are lost. This mutation causes premature termination of protein translation, resulting in the loss of approximately half of the amino acid sequence. This significant structural change in the protein may lead to premature protein degradation. In vitro experiments with the ADAMTS13-W884X plasmid showed concentrated bands with lower molecular weight (110kd) in the Western blot analysis. Unfortunately, the p.W884X mutation was only found in the humanized model and not in the mouse model. Therefore, we were unable to further investigate ADAMTS13 protein expression in the mouse model to validate the findings.
In conclusion, genetic testing serves as a crucial tool not only for diagnosing hereditary thrombotic thrombocytopenic purpura but also for assessing asymptomatic family members who carry a risk of developing the disease. In this case, we successfully diagnosed the patient using second-generation sequencing, promptly initiated plasma exchange for rescue treatment, and implemented regular low-dose plasma replacement therapy every 2–3 weeks for prevention. As a result, the patient experienced complete remission and achieved a favorable prognosis. We conducted a comprehensive study on this patient and their family affected by congenital TTP, identifying two novel mutations in the ADAMTS13 gene. Through in vitro experiments, we verified the impact of these mutations on the pathogenesis of the disease and analyzed the underlying mechanisms. These findings contribute to our understanding of hereditary thrombotic thrombocytopenic purpura. Looking ahead, gene therapy offers a promising avenue for the future treatment of hereditary thrombotic thrombocytopenic purpura. It holds the potential to correct specific ADAMTS13 gene mutations or introduce the ADAMTS13 enzyme gene, providing a novel therapeutic approach for this condition.
Author contributions
Each author is expected to have made substantial contributions to the conception or design of the work. Conceptualization, Li Zhou; Methodology, Xinhui Zhang, Shanglong Feng and Li Zhou; Data Curation, Zhitao Wang, Xueqin Lu and Peng Peng; Patient management, Huiru Huang and Li Zhou; Writing – Original Draft Preparation, Zhitao Wang, Xinhui Zhang and Xueqin Lu; Writing – Review & Editing, Li Zhou.
Acknowledgements
Thanks to everyone on the team for their hard work. And we thank the good care to the patient that Qian Fan, Guangyu Sun, Kaidi Song, Lei Zhang and nurses in the department of hematology and ICU provided.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Kremer Hovinga JA, George JN. Hereditary thrombotic thrombocytopenic purpura. N Engl J Med. 2019;381(17):1653–1662.
- Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–494. doi:10.1038/35097008
- Dainese C, Valeri F, Pizzo E, et al. Adamts13 autoantibodies and burden of care in immune thrombotic thrombocytopenic purpura: new evidence and future implications. Clin Appl Thromb Hemost. 2022;28:10760296221125785.
- Abou-Ismail MY, Kapoor S, Citla Sridhar D, et al. Thrombotic microangiopathies: an illustrated review. Res Pract Thromb Haemost. 2022;6(3):e12708, doi:10.1002/rth2.12708
- Zheng XL, Vesely SK, Cataland SR, et al. ISTH guidelines for the diagnosis of thrombotic thrombocytopenic purpura. J Thromb Haemost. 2020;18(10):2486–2495. doi:10.1111/jth.15006
- Lancellotti S, De Cristofaro R. Progress in molecular biology and translational science. Prog Mol Biol Transl Sci. 2011;99:105–144. doi:10.1016/B978-0-12-385504-6.00003-8
- Rashid A, Mushtaq N, Mansoori H. Congenital thrombotic thrombocytopenic purpura with a novel ADAMTS13 gene mutation. Cureus. 2020;12(12):e12053.
- Graça NA, Ercig B, Pereira LCV, et al. Modifying ADAMTS13 to modulate binding of pathogenic autoantibodies of patients with acquired thrombotic thrombocytopenic purpura. Haematologica. 2020;105(11):2619–2630.
- Choi HS, Cheong HI, Kim NK, et al. ADAMTS13 gene mutations in children with hemolytic uremic syndrome. Yonsei Med J. 2011;52(3):530–534. doi:10.3349/ymj.2011.52.3.530
- Yang L, Li X, Zhu X, et al. Novel ADAMTS13 mutation in a family with three recurrent neonatal deaths: a case report and literature review. Transl Pediatr. 2022;11(5):766–773. doi:10.21037/tp-22-114
- Dekimpe C, Roose E, Sakai K, et al. Toward gene therapy for congenital thrombotic thrombocytopenic purpura. J Thromb Haemost. 2023;21(5):1090–1099. doi:10.1016/j.jtha.2022.12.018
- Loirat C, Girma JP, Desconclois C, et al. Thrombotic thrombocytopenic purpura related to severe ADAMTS13 deficiency in children. Pediatr Nephrol. 2009;24(1):19–29. doi:10.1007/s00467-008-0863-5
- Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi:10.1038/nprot.2015.053
- Pimanda JE, Maekawa A, Wind T, et al. Congenital thrombotic thrombocytopenic purpura in association with a mutation in the second CUB domain of ADAMTS13. Blood. 2004;103(2):627–629. doi:10.1182/blood-2003-04-1346
- Lancellotti S, Peyvandi F, Pagliari MT, et al. The D173G mutation in ADAMTS-13 causes a severe form of congenital thrombotic thrombocytopenic purpura. Thromb Haemost. 2016;115(1):51–62. doi:10.1160/TH15-02-0119
- Gardner MD, Chion CK, De Groot R, et al. A functional calcium-binding site in the metalloprotease domain of ADAMTS13. Blood. 2009;113(5):1149–1157. doi:10.1182/blood-2008-03-144683
- Deforche L, Roose E, Vandenbulcke A, et al. Linker regions and flexibility around the metalloprotease domain account for conformational activation of ADAMTS-13. J Thromb Haemost. 2015;13(11):2063–2075. doi:10.1111/jth.13149
- Camilleri RS, Cohen H, Mackie IJ, et al. Prevalence of the ADAMTS-13 missense mutation R1060W in late onset adult thrombotic thrombocytopenic purpura. J Thromb Haemost. 2008;6(2):331–338. doi:10.1111/j.1538-7836.2008.02846.x
- Davidesko S, Pikovsky O, Al-Athamen K, et al. von Willebrand factor antigen: a biomarker for severe pregnancy complications in women with hereditary thrombotic thrombocytopenic purpura? J Thromb Haemost. 2023;21(6):1623–1629. doi:10.1016/j.jtha.2023.02.022