
Abstract
Background
Little is known about the changes and mechanisms of intestinal flora in primary immune thrombocytopenia (ITP) patients.
Aim
To explore the structural and functional differences of intestinal flora between ITP patients and healthy controls, and clarify the correlation between intestinal flora and Th1/Th2 imbalance.
Methods
Feces from ITP patients and healthy controls were studied by 16S rRNA and metagenomic techniques at phylum, genus, species or functional levels. Blood samples were collected for the detection of interleukin −2 (IL-2) and IL-4 concentrations.
Results
The following changes in ITP patients were found: a decrease of Bacteroidetes phylum, an increase of Proteobacteria phylum and alterations of ten genera and 1045 species. IL-2 and IL-4 were significantly correlated with six and five genera, respectively. Species of C. freundii, C. rodentium, and C. youngae were negatively correlated with bleeding scores, and S. infantis was positively related to platelet counts. Functionally, the intestinal flora of ITP patients changed mainly in terms of motility, chemotaxis, membrane transport, and metabolism.
Conclusion
The mechanism underlying functional and structural changes of intestinal flora in ITP patients may be related to inflammation and immunity, providing possibilities of probiotics or fecal transplants for ITP.
Introduction
Primary immune thrombocytopenia (ITP) is a kind of hemorrhagic disease mediated by autoimmune abnormalities, accounting for about one third of clinical hemorrhagic diseases [Citation1]. The pathogenesis of ITP is complex and remains unclear though it is known to involve immune imbalance, infection and genetics [Citation2]. The involvement of T cells in the pathogenesis of ITP is well known. One consensus about it is that T helper 1 (Th1) polarization and Th1/Th2 imbalance are important components of the pathogenesis of ITP [Citation3]. Th1 cells mainly secrete cytokines such as interleukin-2 (IL-2), involving in activating cytotoxic responses, inflammation and delayed hypersensitivity. In contrast to the role of Th1 cells, Th2 cells promote antibody production and regulate excessive immune response of the body, secreting cytokines such as IL-4. It was found that serum IL-2, circulating T cells and Th1/Th2 ratio in the spleen were elevated in ITP patients, while IL-4 concentrations were decreased [Citation4].
The differentiation of Th1 and Th2 cells can be regulated by the intestinal flora, a large population of microorganisms colonizing the human digestive tract. Its metabolites such as short-chain fatty acids and sphingolipids can also influence the immune response of the body and thus be involved in the development of inflammatory diseases [Citation5]. In addition, probiotics have been shown to be used in the treatment of inflammatory diseases mediated by Th1 or/and Th2 cell abnormalities [Citation6]. Changes in intestinal flora have also been reported on some autoimmune diseases. In the study of Crohn’s disease, researchers found a decrease in Firmicutes and Actinobacteria phyla, and an increase in Proteobacteria phylum [Citation7]. They believed that the changes might be related to intestinal inflammation and metabolites. Altered intestinal flora was also reported in systemic lupus erythematosus (SLE). Streptococcus and S.anginosus were found to be positively correlated with the activity of SLE, possibly through their pro-inflammatory effects, which might have resulted in increased releasing of inflammatory factors and systemic immunity [Citation8]. In patients with psoriasis, researchers have also found alterations in intestinal flora, specifically Faecalibacterium, Megamonas, Lachnospiraceae, et al, which were presumed to be associated with immune responses in the disease [Citation9].
As a classic autoimmune disease, few papers have studied the intestinal flora of ITP. Helicobacter pylori (Hp) infection is found to be associated with ITP [Citation10], with possible mechanism including molecular simulation [Citation11] and immune regulation [Citation12]. After the elimination of Hp, the ITP patient's platelet counts and cytokine level returned to normal [Citation13]. Liu et al. [Citation14] identified eight and 10 phyla in ITP patients and normal controls, respectively. In the ITP patients, the phylum of Bacteroidetes was elevated while the phylum of Proteobacteria was lowered. Wang et al. [Citation15] showed that the phyla of Actinobacteria, Fusobacteria and Verrucomicrobia increased but no phylum decreased in ITP patients. Although several researchers have also focused on the changes in the microbiome of ITP patients, their results have been inconsistent and the mechanism of intestinal flora in the occurrence and development of ITP is unknown. In this study, we aimed to investigate the alterations of intestinal flora in ITP by 16S rRNA and metagenomics, to explore their correlation with Th1/Th2 imbalance, and to reveal the possible mechanism of intestinal flora in the pathogenesis of ITP.
Methodology
Patients
Forty ITP patients (16 males and 24 females) and 33 healthy controls (13 males and 20 females) were subjected to 16S rRNA sequencing. Sixteen ITP patients (10 females and 6 males) and 16 matched healthy individuals were selected for metagenomic sequencing. All patients were hospitalized in the Hematology Department of the Second Affiliated Hospital of Kunming Medical University and met the diagnostic criteria of Chinese guideline (version 2020) [Citation16]. Healthy controls were selected from the patients’ family members, denying tumors and autoimmune diseases. All subjects were free of antibiotics, antifungal drugs and glucocorticoids for at least one month prior to collection of stool samples. Basic information, including gender, age, dietary preference, and disease characteristics, was shown in . The study was approved by the medical ethics committee of the hospital. All subjects signed informed consent. Clinical trial number was ChiCTR1900027808.
Table 1. Characteristics of patients and controls.
Fecal sample collection and DNA extraction
Fecal samples were collected from all subjects and stored at −80°C. Microbial community genomic DNA was extracted from 0.5g stool samples, using the E.Z.N.A.®Soil DNA Kit (Omega Bio-tek, Norcross, GA, U.S.) according to manufacturer instructions. DNA was stored at −20°C before sequencing. The integrity of DNA was checked on a 1% agarose gel, and DNA concentration and purity were determined using a NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, U.S.A.).
16S rRNA sequencing and processing of sequencing data
The hypervariable region V3-V4 of the bacterial 16S rRNA gene were amplified with primer 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R(5′-GGACTACHVGGGTWTCTAAT-3′). The PCR amplification of 16S rRNA gene was performed according to the following methods: initial denaturation at 95 °C for 3 min, followed by 30 cycles of denaturing at 95 °C for 30 s, annealing at 50 °C for 30 s, extension at 72 °C for 45 s, and single extension at 72 °C for 10 min, ending at 4 °C. PCR products were detected using 2% agarose gel electrophoresis, purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, U.S.A.) and quantified using the Quantus™ Fluorometer (Promega, U.S.A.). Library construction was performed using the NEXTFLEX® Rapid DNA-Seq (Bioo Scientific, Austin, TX, U.S.A.). Sequencing was performed using Illumina's Miseq PE300 platform. Raw 16S rRNA gene sequencing reads were demultiplexed, quality-filtered by fastp (version 0.20.0) [Citation17] and merged by FLASH (version 1.2.7) [Citation18]. Operational taxonomic units (OTUs) formation was performed using the QIIME1, and clustered with 97% similarity cutoff by UPARSE (version 7.1) [Citation19], and chimeric sequences were identified and removed. The taxonomy of each OTU representative sequence was analyzed by RDP Classifier (version 2.2) [Citation20] against the 16S rRNA database (Silva v138) using confidence threshold of 0.7.
Metagenomic sequencing and processing of sequencing data
The DNA was segmented and a fragment of about 400 bp was screened for the construction of libraries. Paired-end sequencing was performed on Illumina NovaSeq (Illumina Inc., San Diego, CA, U.S.A.) at Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The paired-end Illumina reads were trimmed of adaptors, and low-quality reads (length < 50 bp or with a quality value <20 or having N bases) were removed by fastp (version 0.20.0) [Citation17]. Reads were compared to the human DNA sequences by the software BWA (version 0.7.9a) [Citation21]. Contaminating reads with high comparative similarity were removed. Metagenomics data were assembled using MEGAHIT (version 1.1.2) [Citation22] through succinct de Bruijn graphs. Contigs with the length being or over 300 bp were selected as the final assembling result, and then used for further gene prediction and annotation. Open reading frames (ORFs) from each assembled contig were predicted using MetaGene [Citation23]. A non-redundant gene catalog was constructed using CD-HIT (version 4.6.1) [Citation24] with 90% sequence identity and 90% coverage. Using SOAPaligner software (version 2.21) [Citation25], the high-quality reads of each sample were compared to the non-redundant gene set separately (95% identity) to count the gene abundance information. Representative sequences from the non-redundant gene catalog were aligned with the NCBI NR database using Diamond (version 0.8.35) [Citation26] for taxonomic annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation.
Blood sample collection, IL-2 and IL-4 testing
3-5ml of venous whole blood was aseptically extracted from ITP patients and healthy controls and placed in EDTA anticoagulant blood collection vessel. Blood samples were centrifuged at 3000 rpm for 10 min and stored at −80°C. Concentrations of IL-2 and IL-4 in plasma were determined by enzyme-linked immunosorbent assay (ELISA) using Elabscience IL-2/IL-4 kit according to manufacturer’s instructions.
Statistical analysis
The basic information of the participants, such as age, body mass index (BMI), gender, et al. and levels of IL-2/IL-4, was first described by mean and standard deviation and then analyzed by Student’s t-test, Mann–Whitney U test or chi-square test. Diversity analysis of a single sample (alpha diversity) demonstrated the richness and diversity of microbial communities. The Sobs index was used to estimate the number of OTUs contained in the samples, demonstrating the abundance of the community. The higher the Sobs index, the higher the richness of the intestinal flora. The Shannon and Simpson indices were used to reveal the diversity of the community. The higher the Shannon index, the lower the Simpson index, the higher the diversity of the intestinal flora. Student’s t-test were used to compare the alpha diversity indices of two groups. Beta diversity analysis explores similarities or differences in community composition between different groups. In this study, Principal Co-ordination Analysis (PCoA) and Non-metric Multidimensional Scaling (NMDS) based on unweighted_unifrac distance matrix were used to analyze the beta diversity of the two groups of intestinal flora. Differences in intestinal flora between the two groups were obtained by Wilcoxon rank-sum test and corrected by FDR multiple testing. Meanwhile, linear discriminant analysis effective size (LEFSe) was used to identify differential species. Features with significant differences in abundance were detected by Kruskal–Wallis rank-sum test. Linear discriminant analysis (LDA) was used to estimate the magnitude of the effect of each component abundance on variability. The screening criteria for differential species were P < 0.05 and LDA > 2. The relationship between intestinal flora and bleeding-related indicators was detected by Spearman correlation analysis. The correlation between intestinal microflora and inflammatory factors was analyzed by Multivariate Association with Linear Models (MaAslin). The above analysis was done by R software (v3.6.2). P < 0.05 was considered significant.
Results
Intestinal flora was altered in ITP patients
Changes in the alpha and beta diversity of intestinal flora in ITP patients
By metagenomic sequencing, a total of 1,031,874,740 high-quality clean sequences were obtained from ITP patients, with an average of 64,492,171 sequences per sample. 1,010,993,086 high-quality clean sequences were obtained from the control group, with an average of 63,187,068 sequences per sample. A total of 4,294,801 valid sequences were obtained by 16S rRNA sequencing. The length of these sequences mainly ranged from 410 to 440bp, with an average length of (417.44 ± 3.220) bp.
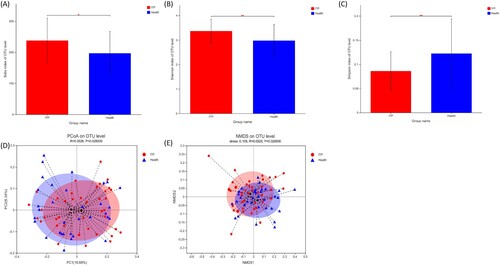
Sobs, Shannon and Simpson indices were calculated to evaluate the alpha diversity of gut microbiome. It was found that the alpha diversity of the ITP group was higher than that of healthy controls, with higher Sobs and Shannon indexes, and lower Simpson index ((a–c)). The beta diversity of intestinal flora was analyzed by PCoA and NMDS. Both analyses showed a statistically significant difference in beta diversity between the two groups ((d–e)). Thus, the intestinal microbiome of ITP patients is disorganized, as both alpha and beta diversity are altered.
Figure 1. Alterations of the alpha and beta diversity in ITP patients. (a–c) were Sobs, Shannon and Simpson indexes, respectively. (d–e) were the beta diversity by PCoA and NMDS, respectively.
Bacteroidetes was decreased and Proteobacteria was increased in ITP patients at the phylum level
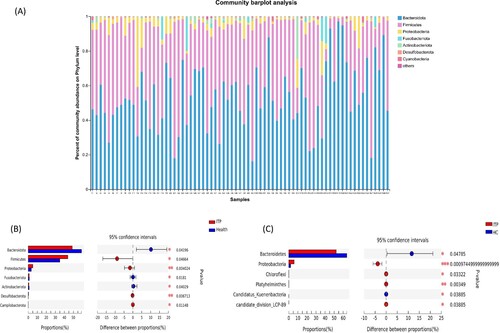
The results of 16S rRNA sequencing showed that the intestinal flora of both ITP patients and healthy controls consisted mainly of three phyla (Bacteroidetes, Firmicutes and Proteobacteria), with the sum of the relative abundance of Bacteroidetes and Firmicutes >90% ((a)). Also, four elevated phyla (Firmicutes, Proteobacteria, Desulfobacterota, Campilobacterota) and three decreased phyla (Bacteroidetes, Fusobacteriota, Actinobacteriota) in ITP patients were found ((b)). By metagenomics, it showed four phyla (Proteobacteria, Chloroflexi, Platyhelminthes, candidate_division_LCP-89) increased and two phyla (Bacteroidetes, Candidatus_Kuenenbacteria) decreased ((c)) in the ITP group. Both sequencing methods demonstrated a decrease in Bacteroidetes and an increase in Proteobacteria in ITP patients.
Figure 2. Composition (a) and differences (b, c) in intestinal flora at the phylum level between the ITP group and the healthy control group. (a) and (b) were the result of 16S rRNA, and C was the results of metagenomics.
Ten genera altered in ITP patients
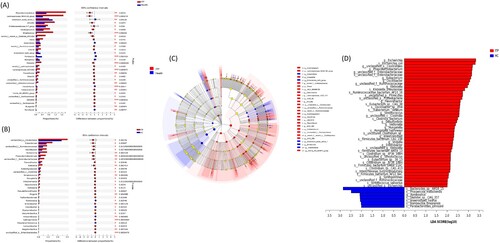
A total of 55 genera were different between ITP and healthy groups at the genus level by 16S rRNA sequencing ((a)). Among them, the relative abundance of 46 genera was significantly higher in the ITP group while nine genera were lower. By metagenomic sequencing, 212 genera were found statistically different between ITP and healthy groups ((b)). Of these, 22 genera were reduced in the ITP group and 190 genera were increased. LEfSe analyses was also performed, showing similar results ((c–d)). Further generalization revealed that two methods consistently identified changes in 10 genera in the ITP group, including increases of Citrobacter, Flavonifractor, Hungatella, Klebsiella, Phascolarctobacterium, Sphingomonas, Streptococcus, g_unclassified_f__Enterobacteriaceae and g_unclassified_f__Ruminococcaceae, and a decrease of Romboutsia.
Figure 3. Differences at the genus level. (a) and (b) showed the top 30 genera with differences by 16S rRNA and metagenomics, respectively. (c) and (d) were the analysis of LEfSe by 16S rRNA and metagenomics, respectively.
Alterations of intestinal flora in ITP patients at the species level
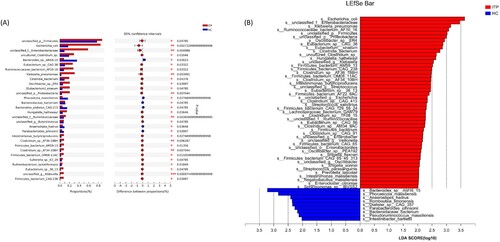
Our study identified 1045 strains that differed between the ITP and healthy controls, including 25 Citrobacter spp., six Flavonifractor spp., two Hungatella spp., 20 Klebsiella spp., 35 Sphingomonas spp., 82 Streptococcus spp., one g_unclassified_f__Enterobacteriaceae spp., six g_unclassified_f__Ruminococcaceae spp., and four Romboutsia spp. ((a)). Consistent results were found by LEfSe analysis, with some species showing enrichment in ITP patients, namely, Streptococcus (S. parasanguinis and S. salivarius, unclassified_g__Streptococcus), Hungatella (H. hathewayi), unclassified_f__Ruminococcaceae (R. bacterium_AF10_16 and unclassified_f__Ruminococcaceae), Klebsiella, (K. pneumoniae and unclassified_g__Klebsiella), and unclassified_f__Enterobacteriaceae (unclassified_f__Enterobacteriaceae). Meanwhile, one bacterium in the genus Romboutsia (R. timonensis) was enriched in the healthy control group ((b)). The differences at the species level were consistent with the results at the genus level.
Figure 4. Differences at the species levels. (a) showed the top 30 species with differences by metagenomics. (a) was the differential species analyzed by LEfSe.
Differences in gut microbiome among newly diagnosed ITP, persistent ITP and chronic ITP
We analyzed the differences of intestinal flora in different stages of ITP (newly diagnosed, persistent and chronic). There were 564, 576 and 1090 OTUs in the New, Persistent and Chronic group, of which 17 (1.47%), 41 (3.55%) and 410 (35.50%) were unique, respectively. The total number of OTUs shared by the three groups was 388, accounting for 33.59% of the total number of OTUs ((a)).
Figure 5. Differences among different stages of ITP. (a) The number of OTUs in three groups. (b) LefSe analysis of intestinal microflora among different stages of ITP.
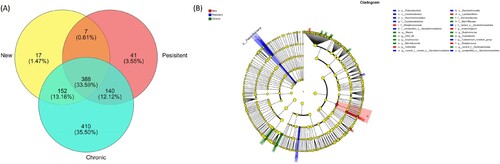
LEfSe was conducted with LDA > 2 as the threshold, and showed that five, ten and nine distinct microbiome communities were enriched in New, Persistent and Chronic group, respectively ((b)). Among them, Lactobacillales, Streptococcaceae, Streptococcus, Veillonella and Anaerostignum were enriched in the newly diagnosed ITP. Saccharimonadales, Patescibacteria, Saccharimonadia, Eubacterium nodatum group, Caulobacteraceae and Caulobacterales were enriched in the persistent ITP. Blautia, Barnesiellaceae, Marinifilaceae, Coprococcus, Butyricimonas, CAG 56 and Coprobacter were enriched in the chronic ITP.
Correlation between intestinal microflora and clinical indices in ITP patients
The relationship of bleeding-related indicators to intestinal flora
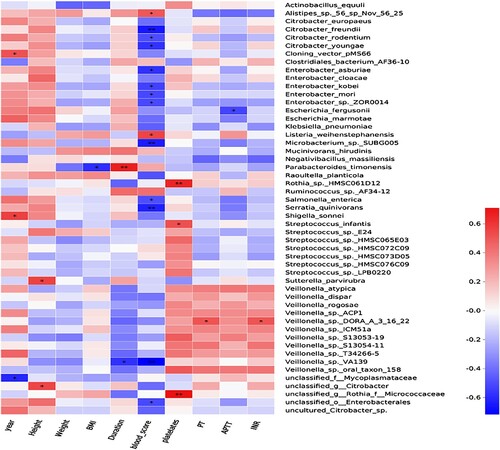
Spearman correlation analysis was conducted to identify the relationship between the patients’ personal characteristics (age, height, weight and BMI) and bleeding-related indicators (bleeding score, disease duration, platelet count, PT, APTT and INR) and intestinal flora. The results showed that P. timonensis was positively while Veillonella sp. VA139 was negatively related to the disease duration. Some species were negatively correlated with the bleeding score, including C. freundii, C. rodentium, C. youngae, E. asburiae, E. kobei, E. mori, Enterobacter sp. ZOR0014, et al. Two species (Alistipes p.56 sp Nov 56 25 and L. weihenstephanensis) were positively correlated with bleeding score. Three species (Rothia sp. HMSC061D12, S. infantis and s__unclassified_g__Rothia_f__Micrococcaceae) were positively correlated with platelet counts ().
Figure 6. Heatmap of the relationship between bleeding-related indicators and intestinal flora in the ITP group. * P < 0.05,** P < 0.01.
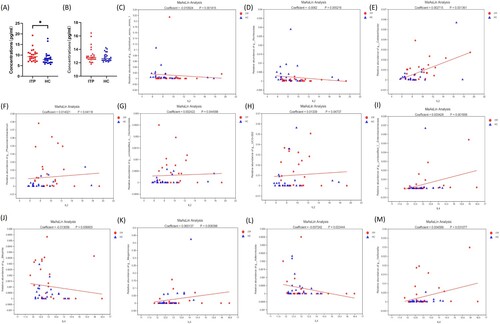
Association between IL-2/IL-4 and intestinal flora in ITP patients
ITP is known to be an autoimmune disease dominated by Th1 polarization and Th1/Th2 imbalance. In order to search for possible mechanisms of microbial changes in ITP patients, we examined the concentration of cytokine IL-2 secreted by Th1 cells and cytokine IL-4 secreted by Th2 cells. In the ITP group, the plasma IL-2 concentration was higher than that in the healthy group (P = 0.0138) ((a)). No difference in plasma IL-4 levels between the two groups was found (P = 0.4329) ((b)). Six genera (Clostridium sensu stricto 1, Romboutsia, Colidextribacter, Phascolarctobacterium, g_unclassified_o_Oscillospirales, and UCG-002) were correlated with plasma IL-2 concentrations, with correlation coefficients of −0.01092, −0.0062, 0.00272, 0.01402, 0.00242 and 0.01339, respectively ((c–h)). Five genera (G_unclassified_f_Enterobacteriaceae, Bilophila, Megamonas, Adlercreutzia and Veillonella) were correlated with IL-4, with correlation coefficients of 0.00343, −0.01306, 0.06014, −0.00724 and 0.0046, respectively ((i–m)).
Figure 7. Relationship between IL-2/4 and intestinal flora in ITP patients. (a) and (b) were the levels of IL-2 and IL-4, respectively. (c–h) were the MaAslin analysis of specific bacteria with IL-2. (i–m) were the MaAslin analysis of specific bacteria with IL-4.
Changes of intestinal flora in ITP patients at the functional level
KEGG pathway enrichment analysis results showed that the intestinal flora differed between the ITP and healthy groups were mainly enriched in three primary and six secondary pathways ((a, b)). Further analysis showed that ABC transporters was the main reason for the enrichment of Membrane transport pathway in ITP. The increases of Aminobenzoate degradation, Nitrotoluene degradation and Benzoate degradation were the main reason for the enrichment of Xenobiotics biodegradation and metabolism in ITP. Increased Bacterial chemotaxis was the main reason for the enrichment of Cell motility in ITP ((c)).
Figure 8. Differences of intestinal flora at the primary (a), secondary (b) and tertiary (c) pathways of KEGG between ITP patients and healthy controls.

Discussion
In this research, two methods were used to study the intestinal flora in ITP patients. It was found that the intestinal flora of ITP patients changed both functionally and structurally. The possible mechanism was further explored, and may involve in the imbalance of Th1/Th2 in ITP.
Our study revealed that the alpha and beta diversity were altered in ITP patients. Further analysis demonstrated a decrease in the Bacteroidetes phylum and an increase in the Proteobacteria phylum in ITP patients, which were partially consistent with other studies [Citation14, Citation15, Citation27]. Bacteroidetes is a group of Gram-negative bacteria containing about 7000 species of bacteria that are widely distributed in different ecological environments. Bacteroidetes can promote the maturation of epithelial cells and repair epithelial tissues, and they encode enzymes that degrade carbohydrates into short-chain fatty acids (SCFAs). SCFAs can carry out β -oxidation in mitochondria and provide energy for the body. Also, SCFAs can interact with G Protein-coupled Receptors (GPR) of intestinal mucosal epithelial cells and play anti-inflammatory roles [Citation28]. Decreased Bacteroidetes was also found in other autoimmune diseases [Citation29]. Therefore, the decrease of Bacteroidetes may contribute to the occurrence of inflammatory diseases. Proteobacteria includes a large number of pathogenic Gram-negative bacteria, such as Citrobacter, Sphingomonas, Enterobacteriaceae and Klebsiella [Citation30, Citation31], which has also been found increased in ITP in our study. The main component of the outer membrane of Proteobacteria is lipopolysaccharide (LPS), which can cause damage to the intestinal mucosal barrier, resulting in intestinal bacterial migration, and then endotoxemia [Citation32].
Apart from the variation at the phylum level, we also identified ten genera and 1045 species that were altered in ITP patients. Combining the study of differential bacterial communities between the two groups and their correlation with clinical indicators, we found that Phascolarctobacterium was positively correlated with IL-2 levels, while Romboutsia was negatively correlated with IL-2 levels. For indicators related to disease severity, C. freundii, C. rodentium, and C. youngae were negatively correlated with bleeding scores, and S. infantis was positively related to platelet counts.
Phascolarctobacterium was increased in ITP patients and was positively correlated with IL-2 levels, whereas Romboutsia, especially R. timonensis, was decreased in ITP patients and was negatively correlated with IL-2 levels. Elevated Phascolarctobacterium has also been found in other diseases [Citation33, Citation34]. In a study on psoriasis, an immune-related disease, intestinal flora was found to be positively associated with IL-2 receptors [Citation9]. Similarly, Romboutsia has been found reduced in other autoimmune diseases, such as primary nephrotic syndrome [Citation35], IBD [Citation5] and pituitary adenoma [Citation36]. Noticeably, one study found that R. timonensis was reduced in children with autism and was the only bacterium associated with the diagnosis [Citation37]. However, the mechanisms remain unclear. Intestinal flora has been shown to affect the balance between Th1 and Th2 cells [Citation38]. In a mouse model, a bacterial strain not only suppressed the allergic response, but also downregulated cytokines secreted by Th cell and increased the number of Th cells [Citation6]. Furthermore, disruption of the symbiotic relationship between microbes and the mucosal immune system can lead to increased activation of Th1 cells, resulting in excessive inflammatory response [Citation39]. By transplanting intestinal bacteria, the Th1/Th2 cell imbalance presented in germ-free mice can be corrected and the formation of lymphoid organs can be induced [Citation40]. Therefore, we speculate that Phascolarctobacterium and Romboutsia may be involved in the differentiation of Th cells and subsequently affect IL-2 levels.
In addition, Citrobacter and Streptococcus were more elevated in ITP patients than in healthy controls, with C. freundii, C. rodentium, and C. youngae negatively correlated with bleeding scores, and S. infantis positively related to platelet counts. The overgrowth of Citrobacter has been shown to be related to intestinal infection and inflammation. C. freundii has been shown to activate the NLRP3 inflammasome [Citation41]. C. rodentium can lead to the formation of adherent and effacing lesions on the surface of the intestinal epithelium, disrupting the intestinal mucosal barrier and leading to intestinal infections [Citation42]. C. youngae can cause intra-abdominal infections in immunosuppressed individuals [Citation43]. Overall, there are fewer studies on Citrobacter, mostly related to infection and inflammation, and its role in ITP remains to be studied.
In this study, S. infantis, a species of Streptococcus, was found to be positively correlated to platelet counts. Liu et al. [Citation14] also found a correlation between intestinal bacteria and platelet counts as well as platelet activation indicators. It was found that intestinal microbes affected platelet function by producing trimethylamine N-oxide, leading to platelet hyperreactivity and enhanced thrombosis potential [Citation44]. However, the mechanism by which Streptococcus affect platelets remains to be investigated. On the other hand, Streptococcus can affect immune system. More elevated Streptococcus was also found in IBD [Citation45] and SLE [Citation8] patients. Streptococcus has a pro-inflammatory effect and some of its species (e.g. S. parasanguinis and S. salivarius) have been shown to affect gene expression in Th cells [Citation46]. In addition, S. infantis has been reported to be associated with periodontitis [Citation47], gestational diabetes mellitus (GDM) [Citation48] and Behcet’s disease [Citation49]. When explaining the relationship between the change in S. infantis and GDM, Gao [Citation48] hypothesized that S. infantis was involved in the imbalance of inflammation and metabolic, leading to insulin resistance and the disease. Therefore, we speculate that Streptococcus may play a key role in the disease severity of ITP by affecting platelet functions and immunomodulatory processes. Since the ability of the sequencing methods to discriminate species is limited, our results and speculation remain to be further investigated.
In addition to the changes in taxonomic levels (phylum, genus and species), we also analyzed the functional enrichment of bacteria with differences. The results showed that the differential bacteria were mainly functionally enriched in motility and chemotaxis, suggesting an altered ability of intestinal bacteria to pass through the gastrointestinal barrier and migrate elsewhere [Citation50]. Enhanced membrane transport, and metabolism of aminobenzoate, nitrotoluene and benzoate were also found in ITP patients, which may represent the virulence profile and metabolic changes of bacteria involved in intestinal inflammation. There are still some limitations in this study. Firstly, the sample size was somewhat limited. Secondly, the participants involved were mainly from rural areas of southwest of China. Ideally, a wider range of people needs to be covered and multi-center cooperation with a large sample size is required.
Conclusion
In conclusion, we found the intestinal flora of ITP patients altered at functional and structural levels. Some bacterial communities were correlated with cytokines and bleeding-related indicators. The possible mechanisms may involve inflammation and immunity. The rationale for using probiotics or fecal transplants to treat ITP was provided.
Fundings
This work was supported in part by grants from the National Natural Science Foundation of China (82060031), the Training Plan of Yunnan Medical Leaders (L-2017005), the Famous Doctor Project of Xing Dian Talent Support Program (RSC2018MY005), the Second Affiliated Hospital of Kunming Medical University National Clinical Medical Research Center for Hematologic Diseases Branch Center (GF2021001). The project was also funded by the China Postdoctoral Science Foundation (2022MD723789) and the Yunnan Provincial Postdoctoral Science Foundation.
Acknowledgements
We thank Professor Luo Na of Lanzhou University for her extensive writing support on this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data and materials during this study are available from the corresponding author on reasonable request. The datasets generated during the current study are available in the Sequence Read Archive (SRA) with BioProject No. PRJNA858054 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA858054) and PRJNA858062 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA858062).
References
- Cooper N, Ghanima W. Immune thrombocytopenia. N Engl J Med. 2019;381(10):945–955. doi:10.1056/NEJMcp1810479
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–2393. doi:10.1182/blood-2008-07-162503
- Zufferey A, Kapur R, Semple JW. Pathogenesis and therapeutic mechanisms in immune thrombocytopenia (ITP). J Clin Med. 2017;6(2). doi:10.3390/jcm6020016
- Audia S, Mahévas M, Samson M, et al. Pathogenesis of immune thrombocytopenia. Autoimmun Rev. 2017;16(6):620–632. doi:10.1016/j.autrev.2017.04.012
- Schirmer M, Garner A, Vlamakis H, et al. Microbial genes and pathways in inflammatory bowel disease. Nat Rev Microbiol. 2019;17(8):497–511. doi:10.1038/s41579-019-0213-6
- Torii A, Torii S, Fujiwara S, et al. Lactobacillus acidophilus strain L-92 regulates the production of Th1 cytokine as well as Th2 cytokines. Allergol Int. 2007;56(3):293–301. doi:10.2332/allergolint.O-06-459
- Qiu X, Zhao X, Cui X, et al. Levels of TB-IGRA may help to differentiate between intestinal tuberculosis and Crohn’s disease in patients with positive results. Therap Adv Gastroenterol. 2020;13:175628482092200. doi:10.1177/1756284820922003
- Li Y, Wang HF, Li X, et al. Disordered intestinal microbes are associated with the activity of Systemic Lupus Erythematosus. Clin Sci (Lond). 2019;133(7):821–838. doi:10.1042/CS20180841
- Zhang X, Shi L, Sun T, et al. Dysbiosis of gut microbiota and its correlation with dysregulation of cytokines in psoriasis patients. BMC Microbiol. 2021;21(1):78. doi:10.1186/s12866-021-02125-1
- Kodama M, Kitadai Y, Ito M, et al. Immune response to CagA protein is associated with improved platelet count after Helicobacter pylori eradication in patients with idiopathic thrombocytopenic purpura. Helicobacter. 2007;12(1):36–42. doi:10.1111/j.1523-5378.2007.00477.x
- Takahashi T, Yujiri T, Shinohara K, et al. Molecular mimicry by Helicobacter pylori CagA protein may be involved in the pathogenesis of H. pylori-associated chronic idiopathic thrombocytopenic purpura. Br J Haematol. 2004;124(1):91–96. doi:10.1046/j.1365-2141.2003.04735.x
- Asahi A, Nishimoto T, Okazaki Y, et al. Helicobacter pylori eradication shifts monocyte Fcgamma receptor balance toward inhibitory FcgammaRIIB in immune thrombocytopenic purpura patients. J Clin Invest. 2008;118(8):2939–2949. doi:10.1172/jci34496
- Frydman GH, Davis N, Beck PL, et al. Helicobacter pylori eradication in patients with immune thrombocytopenic purpura: A review and the role of biogeography. Helicobacter. 2015;20(4):239–251. doi:10.1111/hel.12200
- Liu C, Cheng L, Ji L, et al. Intestinal microbiota dysbiosis play a role in pathogenesis of patients with primary immune thrombocytopenia. Thromb Res. 2020;190:11–19. doi:10.1016/j.thromres.2020.03.012
- Wang Y, Liu F, Zhang G, et al. Gut microbiome alterations and its link to corticosteroid resistance in immune thrombocytopenia. Sci China Life Sci. 2021;64(5):766–783. doi:10.1007/s11427-020-1788-2
- xxx. [Chinese] guideline on the diagnosis and management of adult primary immune thrombocytopenia (version 2020)]. Zhonghua Xue Ye Xue Za Zhi. 2020;41(8):617–623. doi:10.3760/cma.j.issn.0253-2727.2020.08.001
- Chen S, Zhou Y, Chen Y, et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
- Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi:10.1093/bioinformatics/btr507
- Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10(10):996–998. doi:10.1038/nmeth.2604
- Wang Q, Garrity GM, Tiedje JM, et al. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi:10.1128/AEM.00062-07
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi:10.1093/bioinformatics/btp324
- Li D, Liu CM, Luo R, et al. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics. 2015;31(10):1674–1676. doi:10.1093/bioinformatics/btv033
- Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006;34(19):5623–5630. doi:10.1093/nar/gkl723
- Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152. doi:10.1093/bioinformatics/bts565
- Li R, Li Y, Kristiansen K, et al. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–714. doi:10.1093/bioinformatics/btn025
- Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60. doi:10.1038/nmeth.3176
- Zhang X, Gu S, You L, et al. Gut microbiome and metabolome were altered and strongly associated with platelet count in adult patients with primary immune thrombocytopenia. Front Microbiol. 2020;11:1550. doi:10.3389/fmicb.2020.01550
- Jobin C. Gpr109a: The missing link between microbiome and good health? Immunity. 2014;40(1):8–10. doi:10.1016/j.immuni.2013.12.009
- Aomatsu T, Imaeda H, Fujimoto T, et al. Terminal restriction fragment length polymorphism analysis of the Gut microbiota profiles of pediatric patients with inflammatory bowel disease. Digestion. 2012;86(2):129–135. doi:10.1159/000339777
- Litvak Y, Byndloss MX, Tsolis RM, et al. How bacterial pathogens use type III and type IV secretion systems to facilitate their transmission. Curr Opin Microbiol. 2017;35:1–7. doi:10.1016/j.mib.2016.08.007
- Rizzatti G, Lopetuso LR, Gibiino G, et al. Proteobacteria: a common factor in human diseases. Biomed Res Int. 2017;2017:9351507. doi:10.1155/2017/9351507
- Croxen MA, Law RJ, Scholz R, et al. Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol Rev. 2013;26(4):822–880. doi:10.1128/CMR.00022-13
- Iglesias-Vázquez L, Van Ginkel Riba G, Arija V, et al. Composition of Gut microbiota in children with autism spectrum disorder: a systematic review and meta-analysis. Nutrients. 2020;12(3). doi:10.3390/nu12030792
- Hung CC, Chang CC, Huang CW, et al. Gut microbiota in patients with Alzheimer's disease spectrum: a systematic review and meta-analysis. Aging (Albany NY). 2022;14(1):477–496. doi:10.18632/aging.203826
- Kang Y, Feng D, Law HK, et al. Compositional alterations of gut microbiota in children with primary nephrotic syndrome after initial therapy. BMC Nephrol. 2019;20(1):434. doi:10.1186/s12882-019-1615-4
- Lin B, Wang M, Gao R, et al. Characteristics of Gut microbiota in patients with GH-secreting pituitary adenoma. Microbiol Spectr. 2022;10(1):e0042521. doi:10.1128/spectrum.00425-21
- Yap CX, Henders AK, Alvares GA, et al. Autism-related dietary preferences mediate autism-gut microbiome associations. Cell. 2021;184(24):5916–5931.e5917. doi:10.1016/j.cell.2021.10.015
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268–1273. doi:10.1126/science.1223490
- Hand TW, Dos Santos LM, Bouladoux N, et al. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science. 2012;337(6101):1553–1556. doi:10.1126/science.1220961
- Bermudez-Brito M, Borghuis T, Daniel C, et al. L. plantarum WCFS1 enhances Treg frequencies by activating DCs even in absence of sampling of bacteria in the Peyer Patches. Sci Rep. 2018;8(1):1785. doi:10.1038/s41598-018-20243-1
- Liu L, Song L, Deng R, et al. Citrobacter freundii activation of NLRP3 inflammasome via the type VI secretion system. J Infect Dis. 2021;223(12):2174–2185. doi:10.1093/infdis/jiaa692
- Schauer DB, Falkow S. Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect Immun. 1993;61(6):2486–2492. doi:10.1128/iai.61.6.2486-2492.1993
- Chen KJ, Chen TH, Sue YM. Citrobacter youngae and pantoea agglomerans peritonitis in a peritoneal dialysis patient. Perit Dial Int. 2013;33(3):336–337. doi:10.3747/pdi.2012.00151
- Zhu W, Gregory JC, Org E, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165(1):111–124. doi:10.1016/j.cell.2016.02.011
- Pascal V, Pozuelo M, Borruel N, et al. A microbial signature for Crohn's disease. Gut. 2017;66(5):813–822. doi:10.1136/gutjnl-2016-313235
- Li S, Li N, Wang C, et al. Gut microbiota and immune modulatory properties of human breast milk streptococcus salivarius and S. parasanguinis strains. Front Nutr. 2022;9:798403. doi:10.3389/fnut.2022.798403
- Yu XL, Chan Y, Zhuang L, et al. Intra-oral single-site comparisons of periodontal and peri-implant microbiota in health and disease. Clin Oral Implants Res. 2019;30(8):760–776. doi:10.1111/clr.13459
- Wei J, Qing Y, Zhou H, et al. 16S rRNA gene amplicon sequencing of gut microbiota in gestational diabetes mellitus and their correlation with disease risk factors. J Endocrinol Invest. 2022;45(2):279–289. doi:10.1007/s40618-021-01595-4
- Shimizu J, Kubota T, Takada E, et al. Relative abundance of Megamonas hypermegale and Butyrivibrio species decreased in the intestine and its possible association with the T cell aberration by metabolite alteration in patients with Behcet’s disease (210 characters). Clin Rheumatol. 2019;38(5):1437–1445. doi:10.1007/s10067-018-04419-8
- Di Paola M, Cavalieri D, Albanese D, et al. Alteration of fecal microbiota profiles in juvenile idiopathic arthritis. Associations with HLA-B27 allele and disease status. Front Microbiol. 2016;7:1703. doi:10.3389/fmicb.2016.01703