
ABSTRACT
Background
Thalassemia, a common autosomal hereditary blood disorder worldwide, mainly contains α- and β-thalassemia. The α-globin gene triplicates allele is harmless for carriers, but aggravates the phenotype of β-thalassemia. Therefore, it is particularly crucial to accurately detect the structural variants of α-globin gene clusters.
Case report
We reported a 28-year-old man, the proband, with microcytic hypochromic anemia. From pedigree analysis, his mother and sister had hypochromic microcytosis, and his father was normal. Genetic testing of thalassemia identified a novel α-globin gene triplicate named αααanti4.2del726bp (NC_000016.10:g.170769_174300dupinsAAAAAA) by third-generation sequencing (TGS) in the proband and his father, which was further validated by multiplex ligation-dependent probe amplification (MLPA) and Sanger sequencing. The genotypes of the proband’s mother and sister were both -α3.7/αα compounded with heterozygous HBB:c.126_129delCTTT. They were categorized as silent α-thalassemia with co-inheritance of β-thalassemia trait. The proband’s genotype additionally had the α-globin gene triplicates compared with his mother and sister, which increased the imbalance between α/β-globin, so the proband had more severe hematological parameters. The proband’s wife was diagnosed as HBA2:c.427T > C heterozygosis, and his daughter had the novel α-globin gene triplicates compounded with HBA2:c.427T > C, therefore the girl might be asymptomatic.
Conclusion
The identification of the novel α-globin gene triplicates provides more insight for the research of thalassemia variants and indicates that TGS has significant advantages on genetic testing of thalassemia for the reliability, accuracy and comprehensiveness.
Introduction
Thalassemia is a common autosomal hereditary blood disorder worldwide, and its clinical severity ranges from asymptomatic to lethal hemolytic anemia [Citation1,Citation2]. At the molecular level, thalassemia is mainly caused by the defects in two gene clusters encoding α- and β-globin genes, which leads to the α- and β-thalassemia, respectively [Citation3]. The clinical severity of thalassemia is typically correlated with an imbalance or decrease of α/β chain synthesis [Citation4].
There are two HBA genes including HBA2 and HBA1 on each allele of chromosome 16. There are three high homology regions (X-box, Y-box and Z-box) in the αglobin gene cluster, and HBA2 and HBA1 are located in Z-boxes [Citation5]. Non-allelic homologous recombination (NAHR) between the homology regions can result deletions and repetitions of αglobin genes. The NAHR between the two Z-boxes causes a -α3.7 allele and a αααanti3.7 allele. The reciprocal recombination of the two X-boxes results in a -α4.2 chromosome and a αααanti4.2 chromosome [Citation6]. Furthermore, the Hong Kongαα (HKαα) allele is the result of a rearrangement crossover of the α-globin gene cluster containing both -α3.7 and αααanti4.2 crossover junctions, and the anti-HKαα allele is the reciprocal product containing both the -α4.2 and αααanti3.7 crossover junctions [Citation7]. Moreover, the more complex rearrangements with four or five a-globin genes have also been discovered [Citation8].
Multiplex ligation-dependent probe amplification (MLPA) is considered as conventional detected methods of structural variants for α-thalassemia. However, MLPA is not a suitable method for large-scale carrier testing because it is labor-consuming. Also, the breakpoints of the deletions or duplications could not be identified. In addition, if the variants are relatively complex or the deletions and duplications concur, it is difficult to estimate the gene rearrangement and genotype.
Recently, third-generation sequencing (TGS) based on single-molecule real-time (SMRT) technology is emerging as a comprehensive approach in carrier screening for thalassemia [Citation9–16]. TGS could not only detect the variants more accurately compared with conventional methods [Citation11,Citation12] but also define novel genotypes and rearrangements of the α-globin gene cluster [Citation17,Citation18]. By TGS-based thalassemia genetic testing, this report described novel alpha-globin gene triplicates containing two entire HBA2 and one entire HBA1.
Case description
The proband was a 28-year-old man from Yunan province, People’s Republic of China (PRC). He and his wife came to our hospital for routine carrier screening of thalassemia. From the routine hematological results, the proband had a microcytic hypochromic anemia with low hemoglobin (HGB) (<131 g/L), low mean corpuscular volume (MCV) (<80 fl) and low mean corpuscular hemoglobin (MCH) (<27 pg). His wife’s hematological results were normal except for a marginally decreased level of MCV (80.9 fl) and an Hb CS peak (1.1%). Then genetic testing of thalassemia was performed for both the couple. Peripheral blood samples from the proband’s family members were also collected for pedigree analysis genetic diagnosis.
This study involved six members of three generations in the prodand’s family (). The hematological analyzer (Mindray, Shenzhen, China) was used for routine hematological examinations. The hemoglobin electrophoresis was performed and quantified by automatic capillary electrophoresis (Sebia, Paris, France). From the hematological data (), the proband’s father (I-1) was normal, but the proband (II-2) as well as his mother (I-2) and sister (II-1) had reduced MCV, reduced MCH and elevated HbA2. They might have β-thalassemia and the proband was the most severe. The proband’s wife (II-3) might have α-thalassemia because of low HbA2 (2%).
Figure 1. Family tree. The square indicates male, the circle indicates female. The proband is labeled with an arrow. Blue border indicates HBA gene allele, red border indicates HBB gene allele. The blanks indicate HBA-WT or HBB-WT.
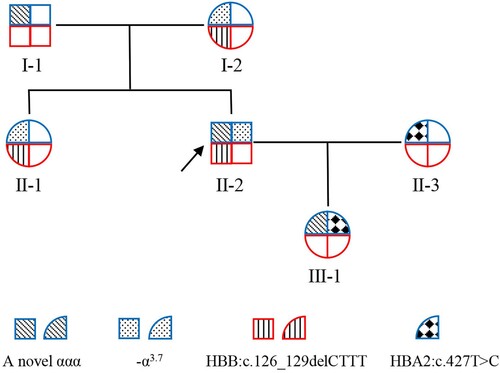
Table 1. Hematological data of the family.
Routine hematological examinations and hemoglobin electrophoresis were the first step of thalassemia screening, and the genetic testing was used for diagnosis. TGS based on SMRT technology was performed on the Sequel II platform(Pacific Biosciences, Menlo Park, USA) as previously described [Citation11,Citation15]. The proband’s wife was diagnosed as HBA2:c.427T>C heterozygous [Citation11] (A), so she was categorized as α-thalassemia trait. A novel α-globin gene triplicate (NC_000016.10:g.170769_174300dupinsAAAAAA), containing two entire HBA2 and one entire HBA1, was identified by TGS in the proband. The genotype of the proband was NC_000016.10:g.170769_174300dupinsAAAAAA/-α3.7 compounded with heterozygous HBB:c.126_129delCTTT which was corresponded to β-thalassemia trait [Citation12] (A). Pedigree analysis based on TGS date showed that he inherited the α-globin gene triplicates allele from his father, and the -α3.7 allele and HBB:c.126_129delCTTT from his mother. The proband’s sister inherited the -α3.7 and HBB:c.126_129delCTTT both from the mother (A). The proband’s genotype had an extra HBA2 compared with his mother and sister, which increased the imbalance between α/β-globin, so the proband had more severe hematological parameters. The proband’s daughter inherited the α-globin gene triplicates allele from the proband and HBA2:c.427T>C from the wife (A). The girl had one allele with an extra HBA2 and one allele with an impaired HBA2, therefore the girl might be asymptomatic.
Figure 2. Results of genetic test. (A) Display of TGS sequences by Integrative Genomics Viewer (IGV). Each colored area represents one chromosome, but no correlations between HBA and HBB gene alleles. HBA2:c.427T > C is labeled with a black frame. The located positions are aligned to genome build hg38. (B) Results of MLPA of the father, mother and the proband. (C) Breakpoint sequences of NC_000016.10:g.170769_173000dupinsAAAAAA duplicate region. Reference sequences encompassing the breakpoints are shown above and below the sequence of the novel haplotype. The microrepetitive sequences are underlined. (D) Agarose gel electrophoresis results of the family numbers carried the novel haplotype. The PCR targeted the characteristic sequences of the X1/X2 fusion fragment. The sample with known αααanti4.2 was used as positive control (with X1/X2 fusion fragment) and WT was used as negative control (αα/αα). (E) Verification of the breakpoints for the novel haplotype by Sanger sequencing.

MLPA and Sanger sequencing were performed to validate the novel α-globin gene triplicates. MLPA was performed by SALSA MLPA P140 HBA probemix (MRC-Holland, Amsterdam, Netherlands), which contained 45 MLPA probes (34 probes for the alpha-globin gene cluster and its flanking regions and 11 reference probes). MLPA result showed that the probe signs from HBA2-up to HBA1-up were increased (nearly 1.5 fold), which implied that the proband’s father carried a duplication involving HBA2 gene. The proband’s mother had a heterozygous deletion at least from HBA2-intr.2 to HBA1-up, because the probe signs were decreased in these regions (0.5 fold). The proband’s MLPA result showed that the probe signs were combinations of the repetition and the deletion (B).
MPLA validated the duplication, and the breakpoints of the duplication were further analyzed by Sanger sequencing. Analysis of the sequences obtained by TGS showed that the duplicated regions originated from X1-box and X2-box (C). The primers (5’-GAGACAGAGTCTCGCTGTGTCGTC-3’ and 5’-TTTTGGGGGGATCATGATGGAAACAT-3’) were designed to amplify the X1/X2 fusion fragment, and the result showed a 3.1-kb band in electrophoresis for individuals I-1, II-3 as well as III-1, and a 3.8-kb band for αααanti4.2 carrier but non-band for the normal people (D). The primer sequences for Sanger sequencing were 5’-GGTTCTCTCTGTCCCGGAATGTGC-3’. Sanger sequencing validated the breakpoints of the novel duplication identified by TGS (E).
Discussion and conclusion
Many clinical data indicate that the carriers of α-globin gene copy number variants (CNVs), such as αααanti3.7, αααanti4.2 and αHKαα, usually have normal hematological phenotype. But they may suffer from moderate to severe anemia when they also carry the β-thalassemia allele [Citation7,Citation11,Citation12,Citation19]. So it is important to accurately detect the structural variants of α-globin gene clusters.
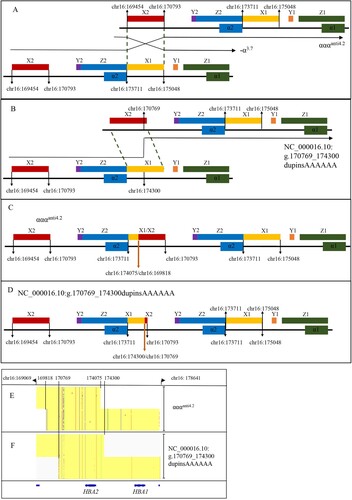
This report identified a novel α-globin gene triplicate (NC_000016.10:g.170769_174300dupinsAAAAAA) by TGS, which was further validated by MLPA and Sanger sequencing. The breakpoints of the duplication were located on X-boxes by analyzing of the sequences (C). The recombination of the two X-boxes results in a -α4.2 chromosome and a αααanti4.2 chromosome [Citation6] as shown in the (A). But the breakpoints of the novel α-globin gene triplicates were different from the αααanti4.2 (NC_000016.10:g.169818_174075) (https://www.ithanet.eu/db/ithagenes?ithaID = 301) (C), which caused a lack of 726 bp in the novel α-globin gene triplicates compared with αααanti4.2 (D and E–F). Furthermore, comparing the upstream sequences and downstream sequences of reference sequences encompassing the breakpoints, a microrepetitive region (6 nucleotide) in the X-boxes was discovered (C). So we speculated that the possible formation mechanism of the novel α-globin gene triplicates might be the non-allelic homologous recombination (NAHR) at the microrepetitive region between X1-box and X2-box of two αα alleles (B). The gene arrangements of the αααanti4.2 (NC_000016.10:g.169818_174075) and the novel α-globin gene triplicates (NC_000016.10:g.170769_174300dupinsAAAAAA) (C–D) explained the gap of 726 bp between them. So, we named the novel α-globin gene triplicates as αααanti4.2del726bp. Moreover, the nearby reference sequences (chr16:170503-chr16:170784) of the downstream breakpoint on the X2-box are the AluSp element sequences. So we speculated the Alu elements might be involved in the breakpoints formation [Citation20]. There may be different mechanisms, and further exploration is needed.
Figure 3. The possible rearrangement mechanism for the novel haplotype (NC_000016.10:g.170769_174300dupinsAAAAAA). The located positions are aligned to genome build hg38. (A) The rearrangement mechanism for the αααanti4.2. (B) The possible rearrangement mechanism for the novel haplotype. (C) The gene arrangement for αααanti4.2. The bold orange arrow shows the most common breakpoints. (D) The gene arrangement for the novel haplotype. The bold orange arrow shows the breakpoints. (E–F) The comparison between αααanti4.2 and NC_000016.10:g.170769_174300dupinsAAAAAA displayed by Integrative Genomics Viewer (IGV).
The report also pointed out the clinical hematologic phenotype of this novel α-globin triplicates allele. The father (I-1), as the novel α-globin triplicates carrier, showed normal routine hematological parameters and hemoglobin electrophoresis parameters. The genotype of the proband’s mother and sister were both -α3.7/αα compounded with heterozygous HBB:c.126_129delCTTT. They were categorized as silent α-thalassemia with co-inheritance of β- thalassemia trait. The proband was the severest in the family for the extra α-globin gene triplicates compared with his mother and sister, which increased the imbalance between α/β-globin. The above results implied that this novel α-globin triplicates might have the same clinical affection as αααanti3.7 and αααanti4.2 [Citation11,Citation12].
In this report, the conventional methods were used step-wise to validate the novel α-globin gene triplicates identified by TGS, but it is difficult to estimate the accurate haplotype only by MLPA. First, it is impossible to clearly detect the breakpoints of the duplication, particularly for novel haplotypes. Second, it is difficult to determine the genotype when the deletions and duplications concurred. However, TGS accurately identified the breakpoints of duplication and the rearrangement of the novel α-globin triplicates. Therefore, TGS based on SMRT had shown significant advantages on thalassemia carrier screening.
In conclusion, this report defined a novel α-globin triplicate and described the phenotype and speculated the possible formation mechanism, which provides more insight for the study of thalassemia variants. Furthermore, accurate molecular diagnosis is particularly crucial. This report proved the allelic coverage, reliability, accuracy and comprehensiveness of TGS for carrier screening and diagnosis of thalassemia.
Author contributions
YC discovered the cases, collected and analyzed the data. TX analyzed the data and wrote the first draft of the manuscript, MM analyzed the data and provided the technical support, JY conducted genetic testing and analyzed the data. YL analyzed the data, DD designed the study, analyzed the data and reviewed the manuscript.
Ethics statement
The study was approved by the Clinical Ethics Committee of The First People’s Hospital of Yunnan Province. All procedures were performed in accordance with the Declaration of Helsinki and international and national guidelines for human studies. Informed written consent was obtained from all the subjects or their legal guardians.
Acknowledgements
The authors would like to thank the patients for their participation in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Additional information
Funding
References
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487. doi:10.2471/blt.06.036673
- Williams TN, Weatherall DJ. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb Perspect Med. 2012;2(9):a011692, doi:10.1101/cshperspect.a011692
- Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379(9813):373–383. doi:10.1016/S0140-6736(11)60283-3
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167. doi:10.1016/S0140-6736(17)31822-6
- Long J. Rapid diagnosis of common deletional alpha-thalassemia in the Chinese population by qPCR based on identical primer homologous fragments. Clin Chim Acta. 2016;456:93–99. doi:10.1016/j.cca.2016.02.023
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13, doi:10.1186/1750-1172-5-13
- Shang X, Li Q, Cai R, et al. Molecular characterization and clinical presentation of HKalphaalpha and anti-HKalphaalpha alleles in southern Chinese subjects. Clin Genet. 2013;83(5):472–476. doi:10.1111/cge.12021
- Farashi S, Vakili S, Faramarzi Garous N, et al. Copy number variations of six and seven alpha-globin genes in a family with intermedia and major thalassemia phenotypes. Expert Rev Mol. 2015;8(5):693–698. doi:10.1586/17474086.2015.1075385
- Huang R, Liu Y, Xu J, et al. Back-to-back comparison of third-generation sequencing and next-generation sequencing in carrier screening of thalassemia. Arch Pathol Lab Med. 2023. doi:10.5858/arpa.2022-0168-OA
- Jiang F, Mao AP, Liu YY, et al. Detection of rare thalassemia mutations using long-read single-molecule real-time sequencing. Gene. 2022;825:146438, doi:10.1016/j.gene.2022.146438
- Liang Q, Gu W, Chen P, et al. A more universal approach to comprehensive analysis of thalassemia alleles (CATSA). J Mol Diagn. 2021;23(9):1195–1204. doi:10.1016/j.jmoldx.2021.06.008
- Liang Q, He J, Li Q, et al. Evaluating the clinical utility of a long-read sequencing-based approach in prenatal diagnosis of thalassemia. Clin Chem. 2023. doi:10.1093/clinchem/hvac200
- Luo S, Chen X, Zeng D, et al. The value of single-molecule real-time technology in the diagnosis of rare thalassemia variants and analysis of phenotype-genotype correlation. J Hum Genet. 2022;67(4):183–195. doi:10.1038/s10038-021-00983-1
- Peng C, Zhang H, Ren J, et al. Analysis of rare thalassemia genetic variants based on third-generation sequencing. Sci Rep. 2022;12(1):9907, doi:10.1038/s41598-022-14038-8
- Xu L, Mao A, Liu H, et al. Long-molecule sequencing: A new approach for identification of clinically significant DNA variants in alpha-thalassemia and beta-thalassemia carriers. J Mol Diagn. 2020;22(8):1087–1095. doi:10.1016/j.jmoldx.2020.05.004
- Zhuang J, Chen C, Fu W, et al. Third-generation sequencing as a new comprehensive technology for identifying rare alpha- and beta-globin gene variants in thalassemia alleles in the Chinese population. Arch Pathol Lab Med. 2023;147(2):208–214. doi:10.5858/arpa.2021-0510-OA
- Long J, Sun L, Gong F, et al. Third-generation sequencing: A novel tool detects complex variants in the alpha-thalassemia gene. Gene. 2022;822:146332, doi:10.1016/j.gene.2022.146332
- Ning S, Luo Y, Liang Y, et al. A novel rearrangement of the alpha-globin gene cluster containing both the -alpha(3.7) and alphaalphaalphaalpha(anti4.2) crossover junctions in a Chinese family. Clin Chim Acta. 2022;535:7–12. doi:10.1016/j.cca.2022.07.020
- Steinberg-Shemer O, Ulirsch JC, Noy-Lotan S, et al. Whole-exome sequencing identifies an alpha-globin cluster triplication resulting in increased clinical severity of beta-thalassemia. Cold Spring Harb Mol Case Stud. 2017;3(6). doi:10.1101/mcs.a001941
- Jourdy Y, Chatron N, Fretigny M, et al. Comprehensive analysis of F8 large deletions: characterization of full breakpoint junctions and description of a possible DNA breakage hotspot in intron 6. J Thromb Haemost. 2022;20(10):2293–2305. doi:10.1111/jth.15835