
ABSTRACT
Backgroud
Li-Fraumeni syndrome is a hereditary tumor syndrome characterized by an elevated risk of malignancy, particularly acute lymphoblastic leukemia (ALL), which can be caused by the heterozygous germline mutation. TP53 gene germline mutation is considered a potential risk factor and crucial prognostic parameter for acute leukemia development and diagnosis, but rarely occurs in adults, and its specific pathogenic significance in acute leukemia is unclear.
Case presentation
We describes a case of a 45-year-old woman diagnosed with ALL. Whole-exome sequencing approach identified one of the TP53 germline mutations from her bone marrow sample with possible pathogenic significance, c.848G>A (p.Arg283His) heterozygous missense mutation located on exon 8, which was further verified in her hair, oral mucous and nail samples. Family pedigree screening revealed that the same TP53 genetic variant was present in the patient's father and non-donor son, whereas not in the donor. Digital PCR observed that this point mutation frequency dropped post-transplantation but remained low during maintenance therapy when the patient was leukemia-free.
Conclusion
This suspected Li-Fraumeni syndrome case report with a likely pathogenic heterozygous TP53 variant expands the cancer genetic spectrum. Screening her family members for mutations facilitates identifying the optimal relative donor and avoids unnecessary treatment by monitoring TP53 germline mutations for minimal residual disease following hematopoietic stem cell transplantation. Its potential roles in hematological malignant tumor development and clinical pathogenic implications necessitate further probing.
Introduction
Acute leukemia (AL) is a hematologic malignancy originating from hematopoietic stem/progenitor cells with complex pathogenesis, which is characterized by the aberrant maturation, excessive proliferation, and apoptosis of cells during differentiation. TP53 gene encodes the tumor protein p53, which is a DNA-binding protein known for its role as a tumor suppressor and is involved in a variety of cellular stress responses, inducing cell cycle arrest, apoptosis, and DNA repair mechanisms. As diagnostic technology advances, especially the clinical application of whole-exome sequencing (WES), TP53 gene mutations have been shown to be closely associated with the development and prognosis of acute leukemia [Citation1–4]. The TP53 gene, which encodes the tumor suppressor protein p53, is the most frequently mutated gene in human cancers [Citation5]. Various biological processes involving distinct TP53 transcripts contribute to the progression of diseases including cancer [Citation6]. TP53 mutations can be classified as either somatic or germline mutations, depending on when and where they arise. Somatic mutations are more common and arise in non-germline cells, whereas germline mutations are predominantly observed in children and arise in the germ cells that give rise to oocytes or sperm cells [Citation7].
Germline mutations originate from sperm or oocytes and exist in all cells throughout the body. Approximately 10% of patients have a combination of both somatic and germline TP53 mutations [Citation8]. As the genetic basis of Li-Fraumeni syndrome, germline TP53 mutation is linked to tumorigenesis, disease progression, and inadequate therapeutic responses [Citation9]. The detection and monitoring of TP53 germline mutations may offer predictive information to aid in the treatment of patients with hematologic malignancies [Citation10–13]. Previous studies [Citation14,Citation15] have indicated significant heterogeneity in the presentation of germline mutations. Consequently, further investigation is required to delineate the clinical significance of TP53 germline mutations.
In this case, we report a 45-year-old female patient diagnosed with acute lymphoblastic leukemia (ALL) carrying a heterozygous missense mutation c.848G>A (p.Arg283His) in the TP53 gene. The patient underwent haploidentical allogeneic hematopoietic stem cell transplantation (haplo-HSCT) with stem cells sourced from her son, who does not carry the mutated gene.
Case presentation
Clinical and laboratory findings
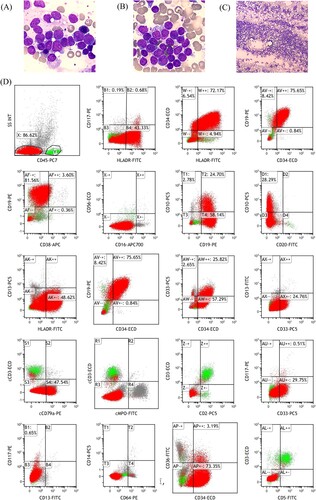
In September 2020, the patient underwent medical examination due to abnormal blood counts. The patient's complete blood count revealed a lower than normal white blood cell count (WBC) of 2.24 × 109/l and a lower absolute neutrophil count (ANC) of 0.86 × 109/l, while the hemoglobin level was 118 g/l and the platelet count was 356 × 109/l. Bone marrow smear suggested ALL diagnosis ((A–C)). Flow cytometry analysis revealed that progenitor B lymphocytes accounted for 86.62% of blasts in the bone marrow, considered as common-B-ALL ((D)).
Figure 1. Bone marrow smear findings at high magnification (A and B) and low magnification (C), as well as preliminary bone marrow flow cytometry results (D) at the patient’s initial consultation. (A–C) The bone marrow was actively proliferating, granulocyte-red ratio of 0.38/1 with an inverted ratio; the lymphocyte lineage accounted for 88.5%, with an extremely high proportion, and cells of all stages were detected, with a predominance of lymphoblasts, accounting for a total of 86%. (D) Flow cytometry of the bone marrow showed a normal lymphocyte population (cluster Y, green). Blasts population (cluster X) occupying approximately 86.62% of the nucleated cells, which expressed CD34, CD19, CD10, cCD79a, HLA-DR, CD33 (partially), and did not express CD117, CD13, CD56, CD5, CD7, CD3, CD2, CD64, CD14, CD38, CD20, CD16, CD11b, cCD3, cMPO.
Genetic analyses
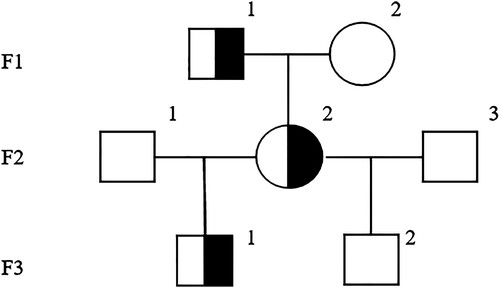
Cytogenetic analysis on her bone marrow sample revealed a karyotype of 46, XX. WES was performed, which identified a common TP53 (NM_000546) c.848G>A (p.Arg283His) heterozygous missense mutation located on exon 8 in the patient's bone marrow sample, with the mutation frequency of 51%. Sanger sequencing was performed to validate the TP53 c.848G>A (p.Arg283His) variant. The mutation frequency of TP53 c.848G>A (p.Arg283His) in the patient's hair, oral mucosa, and nail samples was approximately 50% ((A)). Moreover, family pedigree screening based on peripheral blood and oral mucosa sampling confirmed that the identical TP53 variant was present in the patient's father and another son ((B)), whereas her elder son did not carry the variant. The latter was therefore selected as her donor for hematopoietic stem cell transplantation (HSCT). depicted the genetic profile of such TP53 germline mutation in her family.
Figure 2. Sanger sequencing peaks of TP53 c.848G>A (p.Arg283His) mutation detection in the patient (A) and her relatives (B).
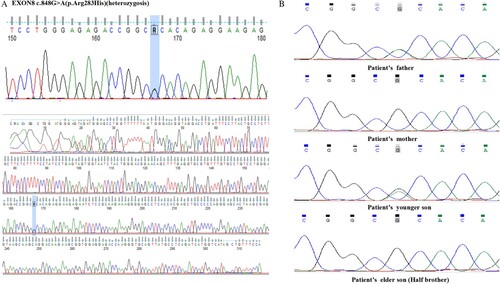
Figure 3. Genetic profile of the patient with TP53 c.848G>A (p.Arg283His) mutations.
Note: In F3, the number ‘1’ and ‘2’ mean the son A and the son B, respectively.
Treatment and follow up
The patient underwent multi-drug chemotherapy and achieved complete remission. Subsequently, she underwent haplo-HSCT from her son following the ‘Beijing protocol’. The conditioning regimen for this patient included: Ara-C 2g·m−2 once a day from day −8 to −7, busulfan (Bu) 0.8mg-kg−1 once a day from day −8 to −5, cyclophosphamide (Cy) 1.8g·m−2 once a day from day −4 to −3, anti-thymocyte globulin (ATG) 7.5mg·m−2 once a day from day −4 to −1. Additionally, Cyclosporine A (CsA) was administered at 1.25 mg-kg−1 twice a day from day −8, mycophenolate mofetil (MMF) at 500 mg twice a day from day −8, and methotrexate (MTX) at 15 mg·m−2 on day +1, followed by 10 mg·m−2 on days +3, +6, and +11 for graft-versus-host disease (GVHD) prophylaxis. The patient underwent haplo-HSCT from her son-donor (HLA 6/12 genotype, blood type matched), receiving 152 ml of donor peripheral blood stem cells (TNC 15 × 108·kg−1, CD34 4 × 106·kg−1, MNC 8.98 × 108·kg−1, CD3 1.33 × 108·kg−1), along with third-party cord blood stem cell transfusion transplantation.
The patient received ‘multi-drug induction regimen’ containing vincristine, idarubicin, cyclophosphamide and prednisone. After induction chemotherapy, the patient achieved complete remission (CR). Following this, she underwent multiple cycles of consolidation chemotherapy, and lumbar puncture-intrathecal chemotherapy was performed for central nervous system leukemia prophylaxis. At eight months after diagnosis, minimal residual disease (MRD) detected by flow cytometry became negative, and one month later, haplo-HSCT was performed.
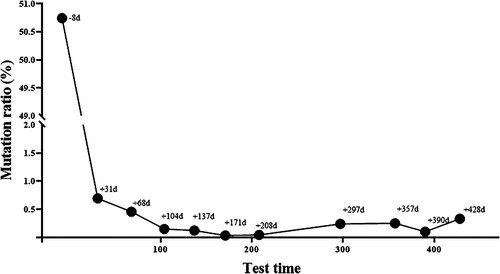
The engraftment of donor hematopoietic stem cells was good, with complete chimerism. Following transplantation, the patient achieved sustained remission and was MRD-negative by flow cytometry. DPCR-based TP53 gene quantification was performed regularly after transplantation. The testing utilized the Droplet Digital PCR (Bio-Rad Laboratories, Hercules, CA, USA) method [Citation16] to quantify gene mutations in the samples, with results expressed as (mutation copies/total effective copies) × 100%. And the percentage of the TP53 gene mutation in the patient decreased rapidly after transplantation and maintained at below 1% (). The patient is presently in a leukemia relapse-free state.
Figure 4. Quantitative monitoring of bone marrow TP53 c.848G>A (p.Arg283His) in the patient after allo-HSCT. Allo-HSCT, allogeneic hematopoietic stem cell transplantation.
Discussion
In this report, we presented a rare case of ALL in a patient carrying the heterozygous missense germline mutation c.848G>A (p.Arg283His) in TP53. To our knowledge, this is the first report of this mutation in a patient with ALL. In addition, this is a successful illustration of haplo-HSCT from a descendant donor who does not carry the identical germline mutation. The TP53 gene is a crucial tumor suppressor gene that plays a critical role in genomic integrity, cell cycle regulation, and apoptosis. Currently, the pathogenic and prognostic implications of TP53 somatic mutations are relatively well defined [Citation5,Citation6]. However, the clinical significance of TP53 germline mutations and their impacts on disease progression and therapeutic response need further research and clarification. Moreover, B-ALL patients have a higher incidence of TP53 mutation than T-ALL and that the mutation often suggests a shorter median survival [Citation17]. In adult B-ALL patients, Hyper-CVAD regimen was able to counteract the poor prognostic impact of TP53 and improve survival [Citation18]. However, the specific pathogenic significance of TP53 germline mutation in leukemia has not well been clarified. TP53 germline mutations rarely occur in adults [Citation19]. Heterozygous germline mutation can lead to autosomal dominant cancer susceptibility syndrome, Li-Fraumeni syndrome (hereditary tumor syndrome) [Citation20], while TP53 germline mutation can be used as a basis for targeted therapy in Li-Fraumeni syndrome [Citation21]. Li-Fraumeni syndrome is a rare, autosomal dominant, inherited condition characterized by an increased risk for certain forms of cancer, particularly ALL [Citation22]. However, the potential relationship between TP53 germline mutations and the development and prognosis of leukemia, particularly in adult-onset cases, requires further investigation. Although reports have shown that TP53 germline mutations can contribute to the development of myeloid and lymphoid hematopoietic tumors, the involvement of the TP53 gene mutation locus described in this case has not yet been investigated. The mutation found in this patient is a novel point mutation that has not been previously reported in any leukemia cases to date. This case report has the value of expanding the genetic map and providing insight into disease diagnosis. Further studies are required to investigate the clinical implications of this mutation in the management of leukemia in other affected individuals.
Approximately 30,000 patients globally undergo HSCT each year with the goal of curing high-risk hematologic malignancies [Citation23]. Regarding the selection strategies of HSCT donors, matched sibling donor (MSD) is preferred, while haploidentical donor (HID) could be the better choice for high-risk leukemia according to its graft-versus-leukemia effect. Furthermore, unrelated donor (URD) grafts may be used when MSD or HID grafts are not feasible. During the COVID-19 pandemic, patient’s relative donors are easier get access than URDs [Citation24,Citation25]. For our patient, family pedigree screening revealed that both her father and the younger son (non-donor) carried TP53 c.848G>A (p.Arg283His) heterozygous germline mutation, and no other somatic mutations of definite pathogenic significance were identified, so it could be conjectured that the TP53 c.848G>A (p.Arg283His) germline mutation may be associated with the pathogenesis of ALL and may have the potential to serve as a poor prognostic factor. Given the possible correlation between this locus and pathogenicity and prognosis, the son who did not progenitor carry the mutation in this locus was selected as the HSCT donor in this case, and finally obtained a good outcome. This case may provide a reference for HSCT donor selection.
In fact, the TP53 c.848G>A (p.Arg283His) mutation has been reported in various disease types such as astrocytoma, glioblastoma, and breast cancer [Citation26,Citation27]. However, the current interpretation of its pathogenicity in the ClinVar database includes two contradictory results: ‘likely pathogenic (1/9)’ and ‘uncertain significance (8/9)’, leaving the clinical significance of this variant uncertain. In addition, biological functional validation studies have been performed using TP53 transcription analysis in yeast to determine the temporal occurrence and allelic distribution of TP53 mutations present in patients, and functional analysis and structural modeling to characterize the mutations, and found that the TP53 p.Arg283His germline mutation transactivates CDKN1A but not the BAX gene and retains the ability to induce human glioblastoma cells growth arrest. In contrast, the R267W somatic mutation combined with the R283H germline mutation failed to inhibit the growth of human astrocytomas, suggesting a complex function of TP53 c.848G>A (p.Arg283His) [Citation28]. Yet, no association between this rare TP53 point germline mutation and leukemia has been reported, so further validation of its biological functions and the exact impact on the prognosis of leukemia in patients is necessary.
Pathogenic mutations have emerged as promising biomarkers for prognostic or therapeutic monitoring and have begun to be used as clinical diagnostic aids. Therefore, it is necessary to screen patients for mutations at the time of initial diagnosis. It is also important to differentiate germline mutations from somatic mutations. DPCR has started to be performed in clinical practice with its advantages of high sensitivity, accuracy and absolute quantification in recent years [Citation29]. DPCR-based point mutation detection is not only a reliable tool for monitoring the efficacy of ALL during follow-up, but also an invaluable approach for treatment response and disease monitoring, which can be applied to evaluate MRD of patients with hematological disorders after each treatment cycle [Citation30]. Some genes are recommended for MRD surveillance in leukemia, such as FLT3-ITD, NPM1, IDH1, etc. While some genes are not recommended, for instance, DNMT3A, TET2, ASXL1, etc., because of clonal hematopoiesis. DPCR of mutated TP53 for MRD monitoring is still controversial, even including germline mutated TP53. Based on our data and analysis of the patient's treatment history at all stages, the frequency of TP53 point mutations decreased significantly following HSCT and subsequently remained at a low level during later stages with no evidence of leukemic relapse. However, the clinical relevance of TP53 germline point mutation monitoring after HSCT is still not clear, and additional research is necessary to clarify its significance.
To conclude, we described a Chinese female ALL case with a rare heterozygous germline mutation c.848G>A (p.Arg283His) in the TP53 gene, which may be associated with ALL pathogenesis. The results of this case highlight the importance of screening for germline mutations at the time of initial leukemia diagnosis in order to select the optimal related donor for HSCT. The identification of germline mutations may inform the selection of a donor who is free of the mutation and thereby improve the chances of a successful transplant. Allo-HSCT is an effectively curative therapy for ALL caused by the TP53 mutations. Point mutation monitoring based on dPCR is a promising tool for post-transplantation efficacy assessment. Nevertheless, the detection of TP53 germline mutations as MRD after transplantation might be of certain clinical significance, they might cause unnecessary treatment due to misinterpretation. Post-transplant monitoring of these patients with germline mutation needs to be further explored.
Consent
The patients/participants provided written informed consent for the publication of this case, including the publication of images.
Authors’ contributions
FH and ZZ participated in the experiments and wrote this article. YH and GH were responsible for the sample and information collection. FH, ZZ, and HY guided the entire essay. All authors have read and approved the final manuscript.
Statement of Ethics
This study protocol was reviewed and approved by the ethics committee of the General Hospital of Western Theater Command.
Acknowledgements
We would like to thank all of the authors for their excellent work. We also thank Huaxi Kindstar Medical Diagnostics Co., Ltd (Sichuan, China) for the sequencing technical support.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated or analyzed during this study are included in this article. Further enquiries should be directed to the corresponding author.
Additional information
Funding
References
- Wahlin A. Accumulating evidence for a role of p53 in multiple drug resistant acute myeloid leukemia. Leuk Lymphoma. 2008;49(3):383–84. doi:10.1080/10428190801950041
- Bug G. Reactivating hope for tp53-mutated acute myeloid leukaemia? Lancet Haematol. 2023;10(4):e239–40. doi:10.1016/S2352-3026(23)00028-5
- Stengel A, Kern W, Haferlach T, et al. The impact of tp53 mutations and tp53 deletions on survival varies between aml, all, mds and cll: an analysis of 3307 cases. Leukemia. 2017;31(3):705–11. doi:10.1038/leu.2016.263
- Rucker FG, Schlenk RF, Bullinger L, et al. Tp53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–21. doi:10.1182/blood-2011-08-375758
- Chen S, Wu JL, Liang Y, et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell. 2021;39(2):225–39. doi:10.1016/j.ccell.2020.11.013
- Lasham A, Knowlton N, Mehta SY, et al. Breast cancer patient prognosis is determined by the interplay between tp53 mutation and alternative transcript expression: insights from tp53 long amplicon digital pcr assays. Cancers (Basel). 2021;13(7):1531. doi:10.3390/cancers13071531
- Hosking FJ, Dobbins SE, Houlston RS. Genome-wide association studies for detecting cancer susceptibility. Br Med Bull. 2011;97:27–46. doi:10.1093/bmb/ldq038
- Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–83. doi:10.1038/nrc1299
- Zhang P, Kitchen-Smith I, Xiong L, et al. Germline and somatic genetic variants in the p53 pathway interact to affect cancer risk, progression, and drug response. Cancer Res. 2021;81(7):1667–80. doi:10.1158/0008-5472.CAN-20-0177
- Halldorsdottir AM, Lundin A, Murray F, et al. Impact of tp53 mutation and 17p deletion in mantle cell lymphoma. Leukemia. 2011;25(12):1904–08. doi:10.1038/leu.2011.162
- Young KH, Leroy K, Moller MB, et al. Structural profiles of tp53 gene mutations predict clinical outcome in diffuse large b-cell lymphoma: an international collaborative study. Blood. 2008;112(8):3088–98. doi:10.1182/blood-2008-01-129783
- Xu-Monette ZY, Wu L, Visco C, et al. Mutational profile and prognostic significance of tp53 in diffuse large b-cell lymphoma patients treated with r-chop: report from an international dlbcl rituximab-chop consortium program study. Blood. 2012;120(19):3986–96. doi:10.1182/blood-2012-05-433334
- Pommert L, Burns R, Furumo Q, et al. Novel germline traf3ip3 mutation in a dyad with familial acute b lymphoblastic leukemia. Cancer Rep (Hoboken). 2021;4(3):e1335. doi:10.1002/cnr2.1335
- Franca R, Zudeh G, Lucafo M, et al. Genome wide association studies for treatment-related adverse effects of pediatric acute lymphoblastic leukemia. Wires Mech Dis. 2021;13(3):e1509), doi:10.1002/wsbm.1509
- Gambale A, Russo R, Andolfo I, et al. Germline mutations and new copy number variants among 40 pediatric cancer patients suspected for genetic predisposition. Clin Genet. 2019;96(4):359–65. doi:10.1111/cge.13600
- Postel M, Roosen A, Laurent-Puig P, et al. Droplet-based digital pcr and next generation sequencing for monitoring circulating tumor dna: a cancer diagnostic perspective. Expert Rev Mol Diagn. 2018;18(1):7–17. doi:10.1080/14737159.2018.1400384
- Stengel A, Schnittger S, Weissmann S, et al. Tp53 mutations occur in 15.7% of all and are associated with myc-rearrangement, low hypodiploidy, and a poor prognosis. Blood. 2014;124(2):251–58. doi:10.1182/blood-2014-02-558833
- Kanagal-Shamanna R, Jain P, Takahashi K, et al. Tp53 mutation does not confer a poor outcome in adult patients with acute lymphoblastic leukemia who are treated with frontline hyper-cvad-based regimens. Cancer. 2017;123(19):3717–24. doi:10.1002/cncr.30810
- Muhlbacher V, Zenger M, Schnittger S, et al. Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high tp53 mutation frequency of 93%. Genes Chromosomes Cancer. 2014;53(6):524–36. doi:10.1002/gcc.22163
- Brown NJ, Bhatia K, Teague J, et al. Report of a bi-allelic truncating germline mutation in tp53. Fam Cancer. 2019;18(1):101–04. doi:10.1007/s10689-018-0087-1
- Sato H, Matsuo S, Ando Y, et al. Germline tp53 c.566c>t mutation incidentally diagnosed during treatment for acute myeloid leukemia: a case report. Clin Case Rep. 2021;9(12):e5221. doi:10.1002/ccr3.5221
- Winter G, Kirschner-Schwabe R, Groeneveld-Krentz S, et al. Clinical and genetic characteristics of children with acute lymphoblastic leukemia and li-fraumeni syndrome. Leukemia. 2021;35(5):1475–79. doi:10.1038/s41375-021-01163-y
- Hill GR, Betts BC, Tkachev V, et al. Current concepts and advances in graft-versus-host disease immunology. Annu Rev Immunol. 2021;39:19–49. doi:10.1146/annurev-immunol-102119-073227
- Zhang XH, Chen J, Han MZ, et al. The consensus from the Chinese society of hematology on indications, conditioning regimens and donor selection for allogeneic hematopoietic stem cell transplantation: 2021 update. J Hematol Oncol. 2021;14(1):145. doi:10.1186/s13045-021-01159-2
- Algwaiz G, Aljurf M, Koh M, et al. Real-world issues and potential solutions in hematopoietic cell transplantation during the covid-19 pandemic: perspectives from the worldwide network for blood and marrow transplantation and center for international blood and marrow transplant research health services and international studies committee. Biol Blood Marrow Transplant. 2020;26(12):2181–89. doi:10.1016/j.bbmt.2020.07.021
- Saito K, Yokogami K, Maekawa K, et al. High-resolution melting effectively pre-screens for tp53 mutations before direct sequencing in patients with diffuse glioma. Hum Cell. 2021;34(2):644–53. doi:10.1007/s13577-020-00471-2
- Marker DF, Agnihotri S, Amankulor N, et al. The dominant tp53 hotspot mutation in idh -mutant astrocytoma, r273c, has distinctive pathologic features and sex-specific prognostic implications. Neurooncol Adv. 2022;4(1):b182), doi:10.1093/noajnl/vdab182
- Fulci G, Ishii N, Maurici D, et al. Initiation of human astrocytoma by clonal evolution of cells with progressive loss of p53 functions in a patient with a 283 h tp53 germ-line mutation: evidence for a precursor lesion. Cancer Res. 2002;62(10):2897–905.
- Wang K, Li B, Guo Y, et al. An integrated digital pcr system with high universality and low cost for nucleic acid detection. Front Bioeng Biotechnol. 2022;10:947895. doi:10.3389/fbioe.2022.947895
- Frazzi R, Bizzarri V, Albertazzi L, et al. Droplet digital pcr is a sensitive tool for the detection of tp53 deletions and point mutations in chronic lymphocytic leukaemia. Br J Haematol. 2020;189(2):e49–52. doi:10.1111/bjh.16442