
ABSTRACT
Background
Imatinib (IM) is the primary treatment for patients with chronic-phase CML (CML-CP). However, an increasing number of CML-CP patients have developed resistance to IM. Our study aims to explore the expression of miR-629-5p in extracellular vesicles (EVs) from both IM-sensitive (K562) and resistant (K562-Re) CML cell lines and to investigate the impact of regulating miR-629-5p expression on the biological characteristics of K562 and K562-Re cells.
Methods
Assess miR-629-5p expression levels in IM-sensitive and resistant CML cell lines. Separate EVs and verify it. EVs from K562-Re cells were co-cultured with K562 cells to detect the expression level of miR-629-5p. Target genes of miR-629-5p were determined and validated through luciferase experiments. Examined by manipulating miR-629-5p expression in cells using transfection techniques. The expression level of phosphorylated proteins in the PI3K/AKT/mTOR signaling pathway after IM was detected in CML cell lines. In K562-Re cells, the expression level of phosphorylated protein in the PI3K/AKT/mTOR signaling pathway was detected after single transfection of miR-629-5p inhibitor and cotransfection of miR-629-5p inhibitor and siSENP2.
Results
Increasing concentrations of EVs from K562-Re cells elevated miR-629-5p expression levels. The expression levels of miR-629-5p in CML cells varied with IM concentration and influenced the biological characteristics of cells. SENP2 was identified as a target gene of miR-629-5p. Furthermore, miR-629-5p was found to modulate the SENP2/PI3K/AKT/mTOR pathway, impacting IM resistance in CML cells.
Conclusion
EVs from IM-resistant CML cells alter the expression of miR-629-5p in sensitive cells, activating the SENP2/PI3K/AKT/mTOR pathway and leading to IM resistance.
1. Introduction
Chronic myeloid leukemia (CML) is a type of malignant proliferative disorder of the bone marrow that is characterized by the formation of the BCR::ABL1 fusion gene through translocation between chromosome 9 and chromosome 22. This fusion gene possesses tyrosine kinase activity, which activates downstream signaling pathways, promoting cell proliferation and inhibiting apoptosis. CML accounts for approximately 15% of adult leukemia cases and has an annual incidence rate of 1–2 per 100,000 individuals [Citation1]. The three phases of CML include chronic phase (CP), accelerated phase (AP), and blast phase (BP). Imatinib (IM), a tyrosine kinase inhibitor (TKI), is the first-line therapy for CML as it specifically targets the BCR::ABL1 fusion gene. However, approximately 10% of CML patients do not achieve optimal response during treatment, and drug resistance poses a significant challenge [Citation2,Citation3].
Exosomes are extracellular vesicles (EVs) secreted by living cells that are crucial for intercellular communication [Citation4]. These vesicles have a typical lipid bilayer structure and a diameter ranging from 40 to 160 nm [Citation5]. They contain abundant microRNAs (miRNAs), which can participate in the process of chemo-resistance or enhance therapeutic efficacy in tumors. Exosome-based therapies hold more promise than cell-based therapies [Citation6,Citation7]. Research has shown that miR-629-5p, which is found within exosomes derived from K562 cells, may promote CML development by affecting the interaction between CML cells and their microenvironment. Additionally, miR-629-5p has been found to be upregulated in exosomes derived from K562 IM-resistant cells (K562-Re) compared to those from K562 cells, suggesting its involvement in drug resistance [Citation8–10].
MiR-629-5p has been found as an oncogenic promoter in various cancers. Yan et al. [Citation11] found that enhanced miR-629 significantly promotes the oncogenicity and lung metastasis of pancreatic cancer cells, and negatively regulates the cancer gene FOXO3 by binding to the 3'UTR of FOXO3 gene in pancreatic cancer cells. Meanwhile, the expression of p-AKT, p-GSK3β, and β-catenin is positively correlated with the expression of miR-629 in pancreatic cancer clinical specimens. In renal clear cell carcinoma, miR-629 is significantly elevated and regulates the migration and invasion of tumor cell lines. It enhances the sensitivity of renal clear cell carcinoma to TGFβ by downregulating TRIM33, thereby modulating the TGFβ/Smad signaling pathway [Citation12]. In colorectal cancer, miR-629-5p has been found to regulate cell proliferation, migration, and apoptosis, and directly downregulate CXXC4 [Citation13]. Additionally, miR-629-5p has been found to promote cell proliferation, invasion, and migration in hepatocellular carcinoma by activating the β-catenin signaling pathway through binding to the 3'UTR of SFRP2 [Citation14]. MiR-629-5p also plays a critical role in lung cancer, gastric cancer, and breast cancer [Citation15,Citation16].
Small ubiquitin-related modifier (SUMO)ylation is a post-translational protein modification that plays a crucial role in eukaryotic cells [Citation17]. DeSUMOylation, which is catalyzed by SUMO-specific proteases, participates in the maturation process of SUMO precursor proteins [Citation18]. In higher eukaryotes, SENPs consist of SENP1-3 and SENP5-7, each with distinct biological functions. Among them, SENP2 acts as a tumor suppressor by maintaining protein stability and inhibiting the activation of tumor-related signaling pathways [Citation19]. The PI3K/AKT/mTOR signaling pathway is one of the crucial intracellular signaling pathways involved in the occurrence and development of various hematological malignancies [Citation20]. Study has shown that SENP2 can inhibit the AKT phosphorylation and suppress glycolysis in breast tumor [Citation21].
In this study, we investigated the mechanism by which CML IM-resistant cells induce IM resistance in sensitive cells. We found that miR-629-5p, transferred from IM-resistant (K562-Re) cell-derived exosomes to IM-sensitive (K562) cells, directly targets the 3'UTR of SENP2 mRNA, activating the PI3K-AKT-mTOR pathway and conferring IM resistance to K562 cells. This regulatory mechanism may play a significant role in IM resistance in CML.
2. Materials and methods
2.1. Cell culture and IM treatment
The K562 cell line and K562-Re cell line (human chronic myeloid leukemia cell line) were acquired from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI-1640 medium (Shanghai Donghuan Biotechnology) supplemented with double-antibody (penicillin–streptomycin) and exosome-depleted serum (SBI System Biosciences, USA), and maintained at 37°C in a 5% CO2 humidified incubator. IM mesylate (STI571) was dissolved in DMSO (Shanghai Donghuan Biotechnology) to prepare a 10 mM stock solution, which was further diluted with a culture medium to obtain a 100 μM working solution before use.
2.2. CML chronic phase and blast crisis patient samples
Eleven newly diagnosed chronic phase CML patients and six blast crisis patients who developed during IM treatment were recruited from the Department of Hematology, Guizhou Provincial People's Hospital, between January 2020 and December 2020. Clinical data of the patients are presented in . Informed consent was obtained from all participants, and the study was approved by the Ethics Committee of Guizhou Provincial People's Hospital (No.2022-444).
Table 1. Demographic and clinical characteristics of 17 patients with CML.
2.3. CCK8 assay for cell IC50 and cell viability
K562 and K562-Re cells were seeded in 96-well plates at a density of 1 × 105 cells/mL in 100 μL culture medium. Each group had 3 replicate wells, and the outer wells were filled with 100 μL PBS. Different concentrations of IM were added to the K562 and K562-Re cells. After 48 h of incubation, 10 μL of CCK-8 reagent was added to each well and incubated 2–4 h. The absorbance values at 450 nm were measured by a microplate reader.
2.4. Quantitative real-time PCR (qRT-PCR)
Cells and exosomes were collected, and total RNA was extracted using Trizol (Shanghai Donghuan Biotechnology). The OD value at A260 nm/A280 nm was measured, and 1000 ng of total RNA was used for cDNA synthesis. Reverse transcription from RNA to cDNA was performed using a reverse transcription kit (Takara, Japan), followed by RT-qPCR using 2×SYBR Green qPCR Master Mix (Shanghai Donghuan Biotechnology). The comparative threshold cycle (Ct) method was used to determine the analysis and fold change. The primer sets used for RT-qPCR are listed in .
Table 2. Primer sequence.
2.5. Isolation of extracellular vesicles (EVs)
Cell culture supernatant was collected and processed according to the instructions provided in the Umibio Exosome Isolation Kit (Shanghai Umibio). The isolated EVs were then purified using the Exosome Purification Filter (4°C, 3000 g, 10 min).
2.6. Electron microscopy observation of EVs
EVs from K562 and K562-Re cells were deposited on copper grids, followed by staining with phosphotungstic acid. After air-drying at room temperature, the samples were observed and imaged using an electron microscope (Hitachi) operated at 80 kV.
2.7. Particle size analysis of EVs
The performance of the nanoparticle tracking analyzer (Malvern) was tested using standard particles, and then 30 μL of EVs from K562 and K562-Re cells were analyzed to obtain information on particle size and concentration.
2.8. Uptake experiment of EVs
EVs were labeled with a red fluorescent dye (PKH26) according to the instructions provided by Umibio (Shanghai Umibio). The labeled EVs were then co-incubated with K562 cells and observed under a fluorescence microscope (OLYMPUS) for uptake experiments.
2.9. Extraction of total proteins
Cells or EVs were washed twice with PBS. Total proteins were extracted using lysis buffer (Shanghai Shenggong Biotechnology), and the protein concentration was analyzed with a BCA assay kit (Shanghai Donghuan Biotechnology). Equal amounts of protein were separated by SDS/PAGE and transferred to a PVDF membrane (Merck Millipore, Germany). The membrane was blocked with 5% skimmed milk, followed by overnight incubation with primary antibodies (CD63, Alix, HSP70, Tublin, β-Actin, SENP2, P-PI3K, PI3K, P-AKT, AKT, P-mTOR, mTOR, Abcam, USA) at 4°C. The membrane was incubated with secondary antibodies (Abcam), and the protein bands were visualized using a chemiluminescent substrate (Shanghai Donghuan Biotechnology) and detected using an imaging system (Tianeng).
2.10. Co-culture and determination of extracellular vesicle concentration
Different concentrations of K562-Re cell-derived extracellular vesicle solutions (2.5, 5.0 and 10.0 ng/mL) were added to the culture medium of K562 cells, with PBS as a blank control. After 48 h, the extracellular vesicle concentration was measured using a BCA assay kit (Shanghai Donghuan Biotechnology) before subsequent experiments.
2.11. EdU detection of cell proliferation in different groups
Cells were seeded in 24-well plates and incubated with EdU assay (Shanghai Donghuan Biotechnology) for 3 h. The cells were fixed, permeabilized, stained, and observed under a fluorescence microscope.
2.12. TUNEL assay
Cells were fixed, permeabilized, and subjected to TUNEL labeling according to the instructions provided by the TUNEL assay kit (Shanghai Donghuan Biotechnology), followed by counterstaining with Hoechst and observation under a fluorescence microscope.
2.13. Transfection of miR-629-5p mimics and inhibitor
Cells were transfected with miR-629-5p mimics/inhibitor using LipofectamineTM 2000 mediated transfection (Invitrogen) following the manufacturer's instructions, and subsequent experiments were conducted after 24–48 h.
2.14. Plasmid construction and synthesis
The construction of pcDNA3.1 and psiCheck2 vectors was designed by Shanghai Donghuan Biotechnology and the synthesis was completed by Shanghai Jima Pharmaceutical Technology. The vector plasmids were purchased from Shanghai Jima Pharmaceutical Technology.
2.15. Luciferase reporter assay
Targetscan, DIANA, and mirDIP databases were used to search for target genes that could bind to miR-629-5p, and the binding sites were predicted using Targetscan software. The wild-type and mutant plasmids of the wt-SENP2 gene were constructed and co-transfected with miR-629-5p mimics or mimics NC into HEK293 cells. The dual-luciferase reporter gene assay (Promega) was performed to detect luciferase activity and analyze the results.
2.16. Construction of cell lines for knocking out and overexpressing
Cells were transfected with miR-629-5p mimics/inhibitor, mimics/inhibitor NC, SENP2 overexpression plasmid, pcDNA3.1 NC, siSENP2, or siNC using LipofectamineTM 2000 (Invitrogen) mediated transfection, and subsequent RNA and protein extraction was performed after 24–48 h.
2.17. Statistical analysis
SPSS, ImageJ, and GraphPad Prism were used for data analysis. Data with a normal distribution were expressed as mean ± SD, and t-tests were used to compare intergroup differences. A P-value less than 0.05 was considered statistically significant.
3. Results
3.1. Differential expression of miR-629-5p in K562 and K562-Re cells
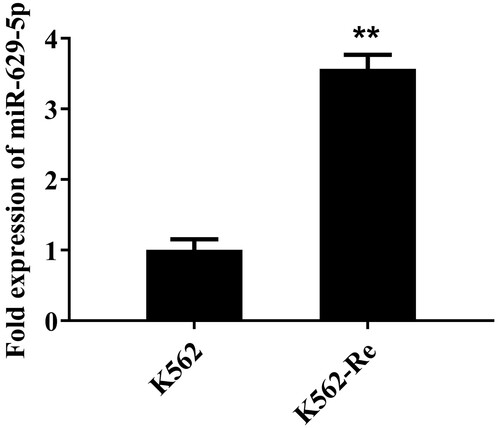
We conducted RT-qPCR to explore the variance in miR-629-5p expression between K562 and K562-Re cells. The findings indicated a substantial increase in miR-629-5p expression in K562-Re cells compared to K562 cells ().
Figure 1. The RT-qPCR results illustrating the expression of miR-629-5p in K562 cells and K562-Re cells (**P<0.01).
3.2. Identification of EVs from K562 and K562-Re cells
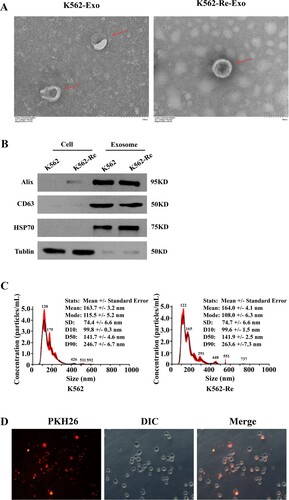
We identified EVs (K562-Exo and K562-Re-Exo) from both cell types using transmission electron microscopy, nanoparticle tracking analysis (NTA), and Western blot. Our observations revealed that both K562-Exo and K562-Re-Exo exhibited vesicular morphology with typical rounded or elliptical cup-shaped structures, and similar sizes ranging from 70 to 200 nm ((A and C)). Additionally, Western blot analysis confirmed the presence of Alix, CD63, and HSP70 proteins in both K562-Exo and K562-Re-Exo ((B)). Furthermore, we observed the uptake of resistant cell-derived EVs (labeled with PKH26) by sensitive cells after co-culture for 3 h ((D)).
Figure 2. (A) Transmission electron microscopy identify the morphology of exosomes; (B) Western blot identify the Expression of exosomal protein and gray values (**P<0.01); (C) NTA identify the diameter of exosomes; (D) PHK26 confirms that K562-Re-Exo is taken up by K562 cells (DIC is the cell, PKH26 red is the exosome).
3.3. Differential expression of miR-629-5p in cells and EVs
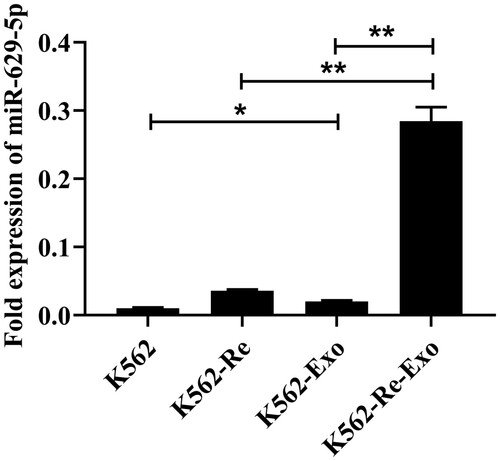
Our RT-qPCR analysis confirmed a significant increase in miR-629-5p expression in K562-Re-Exo compared to K562-Exo (P < 0.01). Additionally, miR-629-5p expression was significantly elevated in K562-Exo compared to K562 cells (P < 0.05), as well as in K562-Re-Exo compared to K562-Re cells (P < 0.01) ().
Figure 3. The RT-qPCR results illustrating the differential expression of miR-629-5p in K562, K562-Re, K562-Exo, K562-Re-Exo (*P<0.05, **P<0.01).
3.4. Promotion of IM and regulation of miR-629-5p expression by EVs secreted from K562-Re cells
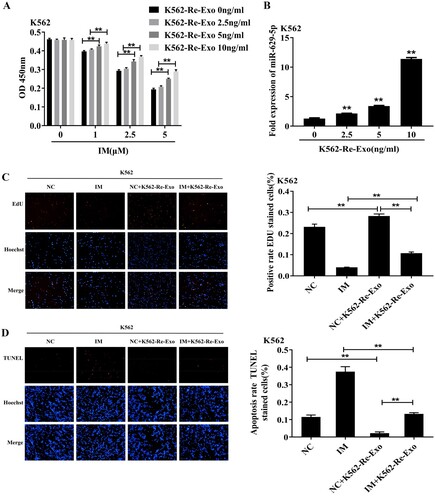
Co-culturing K562 cells with varying concentrations of K562-Re-Exo (0, 2.5, 5.0, 10 ng/mL) for 48 h resulted in increased cell viability (P < 0.01) and elevated miR-629-5p expression levels (P < 0.01) with increasing K562-Re-Exo concentration ((A and B)). Subsequent co-culture with 5.0 ng/ml K562-Re-Exo and 2.5μmol/L IM demonstrated enhanced proliferation (P < 0.01) and reduced apoptosis (P < 0.01) compared to the control group ((C and D)). However, co-culturing with IM led to decreased proliferation (P < 0.01) and increased apoptosis (P < 0.01) compared to the extracellular vesicle group, while co-culturing with IM and EVs resulted in increased proliferation (P < 0.01) and reduced apoptosis (P < 0.01) compared to IM alone ((C and D)).
Figure 4. (A) CCK8 detected the cell activity of K562 cells after ingesting different amounts of drug-resistant exosomes(**P < 0.01); (B) RT-qPCR detected the expression of miR-629-5p in K562 cells after ingesting different amounts of drug-resistant cell exosomes(**P < 0.01); (C) EdU detected the proliferation of K562 cells after ingesting drug-resistant exosomes(**P < 0.01); (D) TUNEL detected the apoptosis of K562 cells after ingesting drug-resistant.
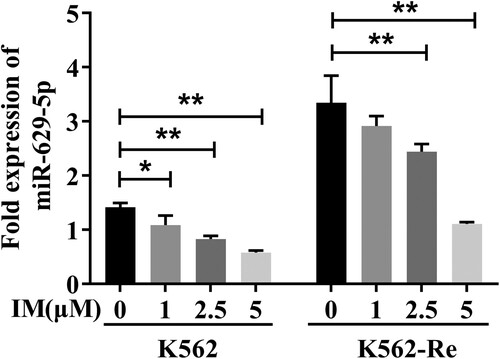
3.5. Differential expression of miR-629-5p in K562 and K562-Re cells stimulated with different IM concentrations
Stimulating K562 and K562-Re cells with varying IM concentrations (0μmol/L, 1μmol/L, 2.5μmol/L, 5μmol/L) for 48 h revealed a gradual decrease in miR-629-5p expression with increasing IM concentration in K562 cells (P < 0.01) and a significant reduction in miR-629-5p expression in K562-Re cells with 2.5μmol/L and 5μmol/L IM stimulation (P < 0.01) ().
Figure 5. Analysis of miR-629-5p expression in K562 cells and K562-Re cells treated by IM with 0μmol/L, 1μmol/L, 2.5μmol/L and 5μmol/L for 48 h (*P < 0.05, **P < 0.01).
3.6. Influence of miR-629-5p regulation on CML cell sensitivity to IM
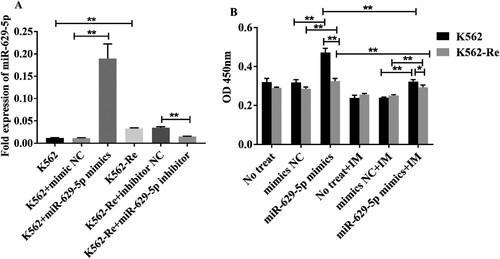
Transfecting miR-629-5p mimics and inhibitors in K562 and K562-Re cells for 48 h resulted in a significant increase in expression in the mimics group (P < 0.01) and a decreased expression in the inhibitor group (P < 0.01) ((A)). Subsequent CCK8 assays showed increased cell viability in both cell types transfected with miR-629-5p mimics (P < 0.01) and decreased sensitivity to IM ((B)).
Figure 6. (A) Analysis of miR-629-5p expression in K562 cells and K562-Re cells transfected with miR-629-5p mimics and inhibitor(**P < 0.01). (B) CCK8 detected cell activity of K562 cells and K562-Re cells transfected with miR-629-5p mimics(*P < 0.05, **P < 0.01).
3.7. Influence of miR-629-5p on CML cell activity
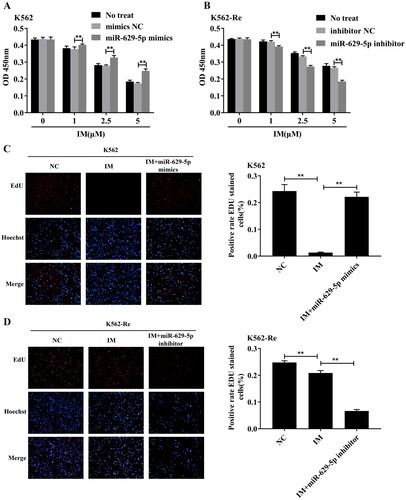
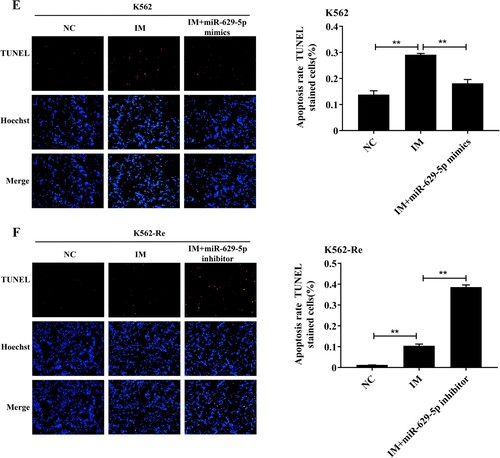
After treating K562 cells with different concentrations of IM (0μmol/L, 1μmol/L, 2.5μmol/L, 5μmol/L) for 48 h, their proliferation significantly increased (P < 0.01) in a concentration-dependent manner. Conversely, the downregulation of miR-629-5p in K562-Re cells resulted in decreased cell viability (P < 0.01) in a concentration-dependent manner. (A and B) illustrate these effects. Additionally, the EdU and TUNEL assays confirmed changes in proliferation and apoptosis, indicating the reversible effect of upregulated miR-629-5p on IM action in K562 cells and the enhanced effect of IM in K562-Re cells. These results are depicted in (C–F).
Figure 7. (A, B) CCK8 detected cell activity of K562 cells and K562-Re cells transfected with miR-629-5p mimics and inhibitors after treatment with different concentrations of IM(**P < 0.01); (C, D) EDU detected the proliferation of K562 cells and K562-Re cells(**P < 0.01); (E, F) TUNEL detected the apoptosis of K562 cells and K562-Re cells(**P < 0.01).
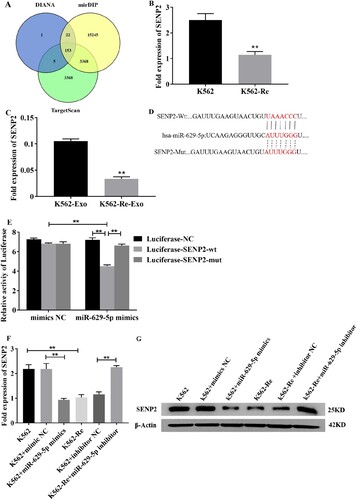
3.8. Prediction of miR-629-5p target genes
To investigate the role of miR-629-5p in regulating target genes associated with IM resistance in CML cells, we utilized bioinformatics tools, including TargetScan, DIANA, and mirDIP, to predict target genes. Through this analysis, we identified 153 common target genes among the three prediction platforms. High-scoring and tumor suppressor-expressing genes in CML cells were then selected. Among these, SENP2 was predicted as a target gene of miR-629-5p ((A)). RT-qPCR analysis confirmed significant downregulation of SENP2 expression in K562-Re cells and their extracellular vesicles (EVs) compared to K562 cells and their EVs (P < 0.01) ((B and C)). Luciferase reporter assays further validated the inhibitory effect of miR-629-5p on the 3'UTR of SENP2 mRNA ((D and E)). Functional studies demonstrated that miR-629-5p regulates SENP2 at both the mRNA and protein levels in K562 and K562-Re cells ((F and G)).
Figure 8. (A) Venn diagram showed the intersection of miR-629-5p target genes predicted by the three website. (B, C) Analysis of SENP2 gene expression in K562 cells, K562-Re cells, K562-Exo, K562-Re – Exo. (D) Prediction of miR-629-5p binding site at SENP2 3′-UTR. (E) Luciferase reporter assays were performed in K562 cells co-transfected cells with miR-629-5p mimic and wild-type (wt) or mutant (mut) SENP2 3′-UTR-luciferase reporter. (F, G) Transfect K562 cells and K562-Re cells with miR-629-5p inhibitor and mimics, and analyze the mRNA and protein expression of SENP2 using RT qPCR and Western blotting.
3.9 IM regulates the gene expression of SENP2 in CML cells
Treatment with different concentrations of IM significantly increased SENP2 mRNA and protein expression in both K562 and K562-Re cells, indicating the influence of IM on SENP2 expression ().
Figure 9. (A) Analysis of SENP2 mRNA expression in K562 cells and K562-Re cells treated with various concentrations of IM using RT-qPCR. (B) Analysis of SENP2 protein expression in K562 cells and K562-Re cells treated with various concentrations of IM using Western blotting.
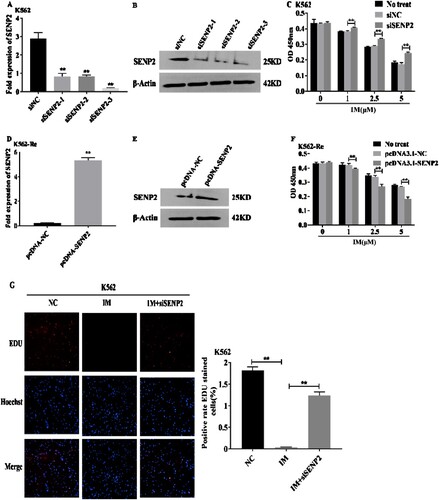
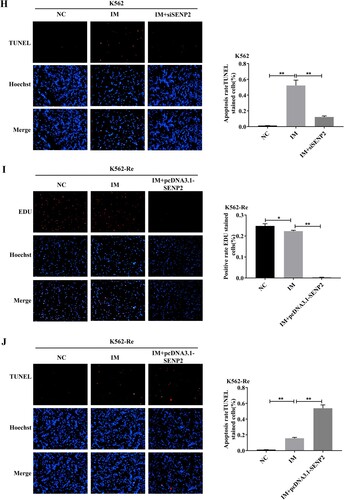
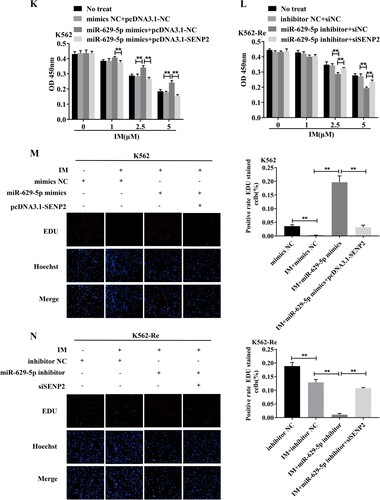
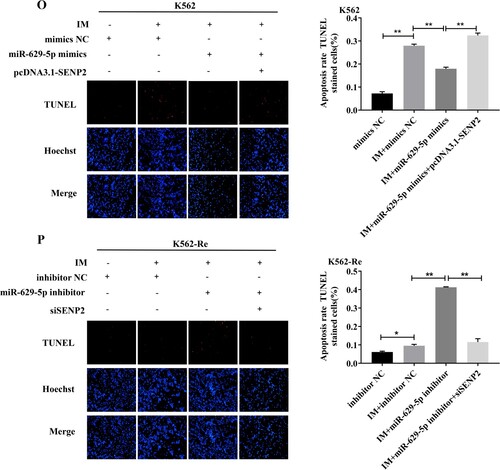
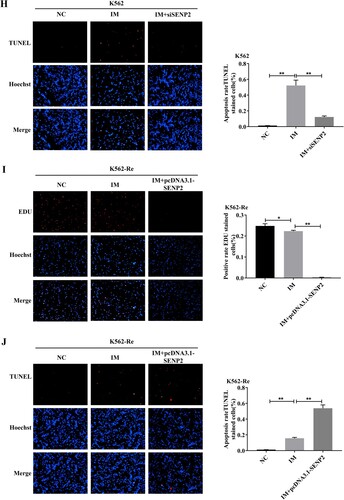
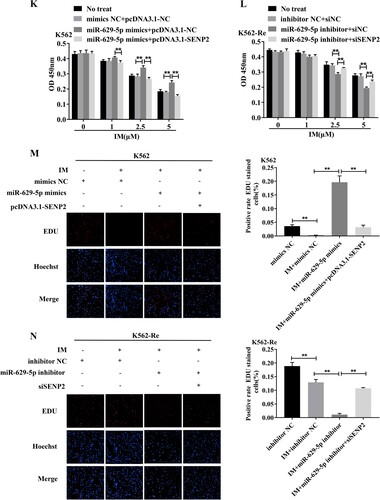
3.10 Involvement of SENP2 in CML cell resistance to IM
Knockdown of SENP2 expression in K562 cells resulted in increased cell viability and decreased sensitivity to IM. Conversely, overexpression of SENP2 in K562-Re cells led to decreased cell viability and increased sensitivity to IM. Co-transfection experiments further demonstrated the reversible effect of SENP2 on miR-629-5p-mediated IM resistance in CML cells ().
Figure 10. (A, B) Detection of SENP2 gene levels in transfected K562 cells using siNC, siSENP2-1, siSENP2-2, and siSENP2-3, followed by analysis using RT-qPCR and Western blotting. (C) Measurement of cell viability in transfected K562 cells using siNC and siSENP2 via the CCK8 assay. (D, E) Detection of SENP2 gene levels in transfected K562-Re cells using pcDNA3.1-NC and pcDNA3.1-SENP2, followed by analysis using RT-qPCR and Western blotting. (F) Measurement of cell viability in transfected K562-Re cells using pcDNA3.1-NC and pcDNA3.1-SENP2 via the CCK8 assay. (G, H) Evaluation of proliferation and apoptosis in K562 cells with reduced SENP2 expression by EdU and TUNEL assays. (I, J) Evaluation of proliferation and apoptosis in K562-Re cells with increased SENP2 expression by EdU and TUNEL assays. (K) Measurement of cell viability in co-transfected K562 cells with miR-629-5p mimic and pcDNA3.1-SENP2 via the CCK8 assay. (L) Measurement of cell viability in co-transfected K562-Re cells with miR-629-5p inhibitor and siSENP2 via the CCK8 assay. (M, O) Evaluation of proliferation and apoptosis in K562 cells co-transfected with miR-629-5p mimic and pcDNA3.1-SENP2 by EdU and TUNEL assays. (N, P) Evaluation of proliferation and apoptosis in K562-Re cells co-transfected with miR-629-5p inhibitor and siSENP2 by EdU and TUNEL assays.
3.11. MiR-629-5p regulates IM resistance in CML cells through the SENP2/PI3K/AKT/mTOR pathway
Downregulation of miR-629-5p inhibited phosphorylation of the PI3K/AKT/mTOR pathway, and this effect was further enhanced by the knockdown of SENP2 in K562-Re cells. These findings suggest that miR-629-5p is involved in influencing IM resistance through the regulation of the SENP2/PI3K/AKT/mTOR pathway ().
Figure 11. (A) Analysis of PI3K/AKT/mTOR signaling pathway protein expression in K562 cells and K562-Re cells treated with various concentrations of IM using Western blotting. (B) Analysis of PI3K/AKT/mTOR signaling pathway protein expression in K562-Re cells transfected with miR-629-5p inhibitor and co-transfected with miR-629-5p inhibitor and siSENP.
3.12. Differential expression of miR-629-5p in newly diagnosed CML patients and CML patients with disease progression
Analysis of peripheral blood mononuclear cells from newly diagnosed CML-CP patients and CML patients with disease progression revealed a significant increase in miR-629-5p expression in the latter group, indicating its potential role in disease progression and resistance to treatment ().
Figure 12. RT-qPCR analysis demonstrated the expression of miR-629-5p in newly diagnosed CML patients and drug-resistant CML patients, revealing a significant difference (**P < 0.01).
4. Discussion
Resistance to the drug Imatinib (IM) in Chronic Myeloid Leukemia (CML) can be classified into two categories: BCR::ABL1-dependent resistance and BCR::ABL1-independent resistance [Citation22]. Extracellular vesicles (EVs) are small vesicles secreted by cells that contain various genetic information, including RNA, proteins, and lipids. They play a crucial role in intercellular communication and have been implicated in tumor drug resistance in various cancers such as breast cancer, colon cancer, and glioblastoma [Citation23–25]. EVs can transmit information and induce signaling exchange between cells, affecting the recipient cells’ status [Citation6]. Therefore, our research group hypothesized that EVs are involved in CML drug resistance and conducted experiments to validate this hypothesis. We initially extracted EVs using a reagent kit and confirmed their identity as EVs through electron microscopy, particle size analysis, and protein marker analysis. We then co-cultured EVs derived from drug-resistant K562-Re cells with K562 cells, and Cell Counting Kit-8 (CCK8) assay results showed that higher levels of EVs led to increased activity of K562 cells, indicating their contribution to IM resistance. We proposed that EVs potentially transfer certain drug-resistant genes to recipient cells, enhancing their resistance to IM.
In the tumor microenvironment, EVs mediate factors that promote tumor occurrence, metastasis, and resistance [Citation26]. Among the contents of EVs, microRNAs (miRNAs) are the most abundant and play a significant role in cancer cell communication, influencing the tumor microenvironment [Citation27]. EV-derived miRNAs have been implicated in drug resistance in various tumors [Citation28], providing a new avenue for tumor diagnosis, treatment, and prognosis evaluation. EV-derived miRNA has been implicated in the drug resistance process of tumors such as neuroblastoma, colorectal cancer, and diffuse large B-cell lymphoma [Citation29–31]. Previous studies have found that EVs can influence CML cells to develop resistance to IM through the high expression of miR-629-5p [Citation14]. Based on these findings, our research group detected miR-629-5p expression in K562 cells, K562-Re cells, K562 cell-derived EVs, and K562-Re cell-derived EVs, revealing a significant increase in miR-629-5p expression in K562-Re cells compared to K562 cells, and a notable elevation in miR-629-5p expression in K562 and K562-Re cell-derived EVs compared to their cytoplasm. These findings align with previous microarray analysis results [Citation12,Citation13], leading to the speculation that EVs may mediate the involvement of miR-629-5p in the process of CML cell resistance to IM. To confirm this conjecture, our research group added different amounts of K562-Re cell-derived EVs to the co-culture with K562 cells and performed RT-qPCR to detect miR-629-5p expression, revealing an increase in miR-629-5p expression levels in K562 cells as the concentration of K562-Re cell-derived EVs increased. This experiment further validated that EVs can participate in CML resistance by mediating miR-629-5p.
MiR-629-5p has been shown to act as an oncogene in various solid tumors but limited research exists on its role in CML, especially in the context of drug resistance [Citation15,Citation16,Citation21]. To verify its biological function in CML, we measured miR-629-5p expression in response to IM treatment in K562 and K562-Re cells. IM reduced miR-629-5p expression in a dose-dependent manner. We further investigated the effects of different concentrations of IM on the viability of K562 cells overexpressing miR-629-5p and K562-Re cells with low miR-629-5p expression, which revealed that upregulation of miR-629-5p decreased the sensitivity of K562 cells to IM, while downregulation of miR-629-5p increased the sensitivity of K562-Re cells to IM. Moreover, overexpression of miR-629-5p resulted in increased proliferation and decreased apoptosis, while decreased expression of miR-629-5p led to reduced proliferation and increased apoptosis. These findings suggest that miR-629-5p participates in the development of IM resistance in CML.
SUMOylation modification plays a crucial role in regulating the activity, localization, and interaction of substrate proteins in many biological processes such as nuclear transport and transcription, DNA replication and repair, cell proliferation, and signal transduction [Citation32]. SENP2 is a critical protein involved in the SUMOylation modification process, which regulates the activity, localization, and interaction of substrate proteins in biological processes [Citation33]. SENP2 is linked to the regulation of multiple cellular signaling pathways and has been implicated in various tumors [Citation19]. In this study, prediction of target genes using bioinformatics websites TargetScan, DIANA, and mirDIP identified 153 common target genes, among which SENP2 scored high and was expressed as a tumor suppressor gene in CML cells. Furthermore, the expression of SENP2 was found to be decreased in K562-Re cells and K562-Re cell-derived EVs compared to K562 cells and K562 cell-derived EVs. To verify whether SENP2 is a target of miR-629-5p, we conducted luciferase reporter gene assays with mutated binding sites for miR-629-5p in the 3'UTR of SENP2 mRNA, confirming that miR-629-5p can target the 3'UTR of SENP2 mRNA in CML cells. To further confirm the regulatory role of miR-629-5p on SENP2 at the RNA and protein levels, our experimental group transfected K562 cells with miR-629-5p mimic and K562-Re cells with miR-629-5p inhibitor, resulting in decreased levels of SENP2 at the mRNA and protein levels in K562 cells and increased levels in K562-Re cells. These results suggest that in CML cell lines, miR-629-5p can target SENP2, and SENP2 is a downstream target of miR-629-5p. Subsequently, we examined whether IM could affect the mRNA and protein expression levels of SENP2 in CML cell lines by treating the cells with different concentrations of IM, revealing a significant increase in SENP2 mRNA and protein expression in K562 and K562-Re cells after IM treatment. These results indicate that IM can alter the expression of SENP2 in CML cell lines. To confirm the participation of SENP2 in the regulation of CML cell IM resistance by miR-629-5p, our experimental group co-transfected K562 cells with miR-629-5p mimic and pcDNA3.1-SENP2, and K562-Re cells with miR-629-5p inhibitor and siSENP2. The co-transfection experiments showed that in K562 cells, compared to the single transfection group, the co-transfected cells exhibited decreased viability, decreased proliferation, and increased apoptosis, while in K562-Re cells, the co-transfected cells showed increased viability, increased proliferation, and decreased apoptosis compared to the single transfection group. These results demonstrate that SENP2 can reverse CML cell resistance to IM mediated by miR-629-5p. Overall, it is concluded that targeted inhibition of miR-629-5p to suppress SENP2 expression regulates the sensitivity of CML cell lines to IM.
The PI3K/AKT/mTOR signaling pathway plays a significant role in the occurrence and development of CML [Citation34]. The PI3K/AKT/mTOR signaling pathway has become a therapeutic target for some malignant tumors, with PI3K inhibitors, AKT inhibitors, and rapamycin demonstrating good efficacy [Citation35]. Studies have shown that SENP2 can inhibit AKT phosphorylation in breast tumors [Citation33]. To confirm whether SENP2 and the PI3K/AKT/mTOR signaling pathway are involved in resistance to IM in CML, different concentrations of IM were used to treat CML cell lines, revealing that the levels of phosphorylated proteins in the PI3K/AKT/mTOR signaling pathway were reduced in K562 and K562-Re cells after IM treatment compared to the control group, indicating that IM can regulate the phosphorylation level of the PI3K/AKT/mTOR signaling pathway in CML cell lines. Furthermore, to verify whether miR-629-5p mediates the regulation of the SENP2/PI3K/AKT/mTOR pathway in CML cell IM resistance, we performed single transfection of miR-629-5p inhibitor and co-transfection of miR-629-5p inhibitor and siSENP2 in K562-Re cells, showing that reducing the expression of miR-629-5p could inhibit the phosphorylation of the PI3K/AKT/mTOR signaling pathway while knocking down SENP2 increased phosphorylation levels. Thus, it can be concluded that miR-629-5p can affect CML cell resistance to IM by regulating the SENP2/PI3K/AKT/mTOR pathway.
To validate our findings in patient samples, we assessed the expression levels of miR-629-5p in peripheral blood mononuclear cells of newly diagnosed CML-CP patients with good responses to IM treatment and patients who experienced blast crisis during IM treatment. The results indicated that the expression of miR-629-5p in patients resistant to IM was significantly higher than in patients responding well to IM treatment, consistent with the cell line research in this study. Thus, it is considered that miR-629-5p may participate in the formation of IM resistance in CML. However, limitations in EV extraction and low patient sample size are limitations of our study.
In conclusion, our study demonstrates that EVs and miR-629-5p play crucial roles in IM resistance in CML. We propose the miR-629-5p/SENP2/PI3K/AKT/mTOR pathway as a potential target for reversing IM resistance. These findings contribute to our understanding of the mechanisms underlying CML drug resistance and provide a basis for future research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Apperley JF. Chronic myeloid leukaemia. Lancet. 2015;385(9976):1447–1459. doi:10.1016/S0140-6736(13)62120-0
- Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of Imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376(10):917–927. doi:10.1056/NEJMoa1609324
- Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–984. doi:10.1038/s41375-020-0776-2
- Lin WC, Tsai CY, Huang JM, et al. Quantitative proteomic analysis and functional characterization of Acanthamoeba castellanii exosome-like vesicles. Parasit Vectors. 2019;12(1):467. doi:10.1186/s13071-019-3725-z
- Romanov V, McCullough J, Gale BK, et al. A tunable microfluidic device enables cargo encapsulation by cell- or organelle-sized lipid vesicles comprising asymmetric lipid bilayers. Adv Biosyst. 2019;3(7):1900010. doi:10.1002/adbi.201900010
- Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232. doi:10.1016/j.cell.2016.01.043
- Bach DH, Hong JY, Park HJ, et al. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int J Cancer. 2017;141(2):220–230. doi:10.1002/ijc.30669
- Bernardi S, Balbi C. Extracellular vesicles: from biomarkers to therapeutic tools. Biology (Basel). 2020;9(9):258. doi:10.3390/biology9090258
- Liu Y, Song BQ, Wei YM, et al. Exosomes from mesenchymal stromal cells enhance imatinib-induced apoptosis in human leukemia cells via activation of caspase signaling pathway. Cytotherapy. 2018;20(2):181–188. doi:10.1016/j.jcyt.2017.11.006
- Umezu T, Ohyashiki K, Kuroda M, et al. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene. 2013;32(22):2747–2755. doi:10.1038/onc.2012.295
- Taverna S, Giallombardo M, Pucci M, et al. Curcumin inhibits in vitro and in vivo chronic myelogenous leukemia cells growth: a possible role for exosomal disposal of miR-21. Oncotarget. 2015;6(26):21918–21933. doi:10.18632/oncotarget.4204
- Feng DQ, Huang B, Li J, et al. Selective miRNA expression profile in chronic myeloid leukemia K562 cell-derived exosomes. Asian Pac J Cancer Prev. 2013;14(12):7501–7508. doi:10.7314/APJCP.2013.14.12.7501
- Gao XT, Wan Z, Wei MY, et al. Chronic myelogenous leukemia cells remodel the bone marrow niche via exosome-mediated transfer of miR-320. Theranostics. 2019;9(19):5642–5656. doi:10.7150/thno.34813
- Min QH, Wang XZ, Zhang J, et al. Exosomes derived from imatinib-resistant chronic myeloid leukemia cells mediate a horizontal transfer of drug-resistant trait by delivering miR-365. Exp Cell Res. 2018;362(2):386–393. doi:10.1016/j.yexcr.2017.12.001
- Yan HJ, Li Q, Wu J, et al. MiR-629 promotes human pancreatic cancer progression by targeting FOXO3. Cell Death Dis. 2017;8(10):e3154. doi:10.1038/cddis.2017.525
- Jingushi K, Ueda Y, Kitae K, et al. miR-629 targets TRIM33 to promote TGFβ/Smad signaling and metastatic phenotypes in ccRCC. Mol Cancer Res. 2015;13(3):565–574. doi:10.1158/1541-7786.MCR-14-0300
- Sriramachandran AM, Dohmen RJ. SUMO-targeted ubiquitin ligases. Biochim Biophys Acta. 2014;1843(1):75–85. doi:10.1016/j.bbamcr.2013.08.022
- Zhou XF, Jiang YQ, Wang YW, et al. Endothelial FIS1 DeSUMOylation protects against hypoxic pulmonary hypertension. Circ Res. 2023;133(6):508–531. doi:10.1161/CIRCRESAHA.122.321200
- Tan MY, Zhang DG, Zhang EC, et al. Senp2 suppresses epithelial-mesenchymal transition of bladder cancer cells through deSUMOylation of TGF-βRI. Mol Carcinog. 2017;56(10):2332–2341. doi:10.1002/mc.22687
- Barrett D, Brown VI, Grupp SA, et al. Targeting the PI3K/AKT/mTOR signaling axis in children with hematologic malignancies. Paediatr Drugs. 2012;14(5):299–316. doi:10.2165/11594740-000000000-00000
- Lu JL, Lu SR, Li JZ, et al. MiR-629-5p promotes colorectal cancer progression through targetting CXXC finger protein 4. Biosci Rep. 2018;38(4):BSR20180613. doi:10.1042/BSR20180613
- Meenakshi Sundaram DN, Jiang XY, Brandwein JM, et al. Current outlook on drug resistance in chronic myeloid leukemia (CML) and potential therapeutic options. Drug Discov Today. 2019;24(7):1355–1369. doi:10.1016/j.drudis.2019.05.007
- Yu DD, Wu Y, Shen HY, et al. Exosomes in development, metastasis and drug resistance of breast cancer. Cancer Sci. 2015;106(8):959–964. doi:10.1111/cas.12715
- Liang GF, Zhu YL, Ali DJ, et al. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J Nanobiotechnology. 2020;18(1):10. doi:10.1186/s12951-019-0563-2
- Ding CY, Yi XH, Wu XY, et al. Exosome-mediated transfer of circRNA CircNFIX enhances temozolomide resistance in glioma [retracted in: Cancer Lett. 2022 May 1;533:215590. xxx]. Cancer Lett. 2020;479:1–12. doi:10.1016/j.canlet.2020.03.002
- Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32(3–4):623–642. doi:10.1007/s10555-013-9441-9
- Huang XY, Yuan TZ, Liang MH, et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur Urol. 2015;67(1):33–41. doi:10.1016/j.eururo.2014.07.035
- Tomasetti M, Lee W, Santarelli L, et al. Exosome-derived microRNAs in cancer metabolism: possible implications in cancer diagnostics and therapy. Exp Mol Med. 2017;49(1):e285. doi:10.1038/emm.2016.153
- Challagundla KB, Wise PM, Neviani P, et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst. 2015;107(7):djv135. Published 2015 May 13. doi:10.1093/jnci/djv135
- Liu T, Zhang X, Du LT, et al. Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol Cancer. 2019;18(1):43. doi:10.1186/s12943-019-0981-7
- Feng YH, Zhong MZ, Zeng S, et al. Exosome-derived miRNAs as predictive biomarkers for diffuse large B-cell lymphoma chemotherapy resistance. Epigenomics. 2019;11(1):35–51. doi:10.2217/epi-2018-0123
- Zhao XL. SUMO-mediated regulation of nuclear functions and signaling processes. Mol Cell. 2018;71(3):409–418. doi:10.1016/j.molcel.2018.07.027
- Tang S, Huang G, Tong XM, et al. Role of SUMO-specific protease 2 in reprogramming cellular glucose metabolism. PLoS One. 2013;8(5):e63965. doi:10.1371/journal.pone.0063965
- Nepstad I, Hatfield KJ, Grønningsæter IS, et al. The PI3K-Akt-mTOR signaling pathway in human Acute Myeloid Leukemia (AML) cells. Int J Mol Sci. 2020;21(8):2907. doi:10.3390/ijms21082907
- Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther. 2015;3:2. doi:10.1186/s40591-015-0040-8