
Abstract
Although rare, the presentation of the genetic disease spectrum associated with lysosomal acid lipase (LAL) deficiency, a disorder named cholesteryl ester storage disease (CESD) or the more severe form of Wolman's disease, is an important condition to recognise. LAL plays an essential role in lipid catabolism and a deficiency in this enzyme results in accumulation of cholesterol esters in multiple tissues. The first clinical manifestation is often hepatomegaly, despite the multi-system nature of the disorder. Other associated features include splenomegaly, adrenal calcification, malabsorption, hypercholesterolaemia or mixed hyperlipidaemia predisposing to premature atherosclerosis, as well as liver dysfunction, which can lead to cirrhosis and liver failure. Diagnosis can be made through genetic screening, LAL activity measurement or on liver biopsy. Recent advances in treatment of LAL deficiency have been made with development of a recombinant human LAL (sebelipase alpha). Adjunctive treatment with lipid-lowering therapy continues to be standard management.
Introduction
Cholesteryl ester storage disease (CESD) is a rare, autosomal recessive lysosomal storage disorder, resulting from a deficiency in lysosomal acid lipase (LAL) activity. LAL deficiency results in accumulation of cholesteryl esters and triglycerides in multiple body tissues, the clinical manifestations and complications of which can present from as early as the first year of life. Although a rare condition, the likelihood is that many patients go unrecognised or are misdiagnosed. In this report, we present a case of an adult female who initially presented, and was subsequently diagnosed, at the age of four years.
Case
At the time of presentation to the Division of Endocrinology & Metabolism at the Charlotte Maxeke Johannesburg Academic Hospital (CMJAH), the patient, a 31-year-old female, was soon to be married. She had been living (and receiving medical care) in Singapore but had recently relocated with her husband-to-be to Johannesburg and required ongoing medical care for her condition.
The patient is originally from Germany. At the age of four years, she presented with hepatosplenomegaly and, following a liver biopsy, was diagnosed with CESD. At that time, lipid-lowering therapy was initiated in the form of an HMG-CoA reductase inhibitor (simvastatin/atorvastatin), which she continued until the age of 19 years when she opted to stop treatment. Four years later, when her father died unexpectedly (at the age of 52 years) she restarted lipid-lowering therapy and was on atorvastatin 80 mg daily at presentation.
Further inquiry revealed that she had been a smoker, having stopped two years previously; had a modest alcohol intake and underwent surgical intervention for a left ovarian cyst five years earlier. She has no siblings and could provide little detail regarding her extended family's medical history. During her routine follow-up, she had been informed that the size of her liver and spleen had decreased on statin therapy. She had a normal effort tolerance and no history of angina pectoris.
Clinically, she displayed no arcus cornealis, xanthelasma or thickening of her Achilles tendons. She had a firm hepatomegaly palpable 3 cm below the costal margin but the spleen was not palpable. Significantly, she had a grade 3/6 ejection systolic murmur over the aortic area, radiating to the neck, suggestive of aortic stenosis.
Biochemical tests at the time of presentation revealed a total cholesterol (TC) level of 3.9 mmol/l; triglycerides (Trig) of 1.1 mmol/l; high density lipoprotein-cholesterol (HDLC) of 0.8 mmol/l and low-density lipoprotein cholesterol (LDLC) of 2.6 mmol/l. Furthermore, liver function tests showed mildly elevated transaminases, aspartate transaminase (AST) of 69 U/l (RR 5–40) and alanine transaminase (ALT) of 92 U/l (RR 5–40) with normal levels of alkaline phosphatase (ALP) of 48 U/l (RR 40–120) and gamma-glutamyl transferase (GGT) of 29 U/l (RR 0–35).
At 24 years of age, she had undergone an ultra-fast CT scan, which confirmed the presence of coronary calcification. At this point, the course of action decided upon was to aim to decrease the LDLC level further to < 2.6 mmol/l and to consider ezetimibe therapy in conjunction with high-intensity statin therapy. Furthermore, a cardiac assessment was requested, genetic testing discussed, and she was advised to discontinue statin therapy for the first trimester of a planned pregnancy.
The patient fell pregnant seven months later, having discontinued statin therapy in the same month. She was advised to remain off statin therapy for the duration of her pregnancy and to reinitiate lipid-lowering therapy post-partum. The pregnancy progressed well although symptomatic palpitations necessitated a cardiac assessment, which confirmed some calcification and thickening of the aortic valve and ascending aorta, causing minimal stenosis and an ejection fraction of 70%.
A healthy baby boy entered the patient's life. During pregnancy her cholesterol levels progressively increased and her lipogram at delivery, off any lipid-lowering therapy, revealed a TC of 16.53 mmol/l, Trig of 4.41 mmol/l, HDLC of 0.64 mmol/l and an LDLC of 13.85 mmol/l. Having been advised to restart the statin three months post-delivery, her repeat lipogram and liver function just prior to re-initiation of therapy showed markedly elevated cholesterol levels and increased transaminases (TC 14.38 mmol/l; Trig 2.84 mmol/l; HDLC 0.82 mmol/l; LDLC 12.27 mmol/l; ALT 103 IU/l; AST 74 IU/l; ALP 113 IU/l; GGT 34 IU/l). Atorvastatin 80mg daily was restarted.
Post-partum cardiac assessment revealed no significant difference in her status and a carotid duplex doppler study displayed minimal plaque bilaterally with no increase in intimal thickening. Within six months of restarting the statin, her LDLC dropped by almost fourfold, with an associated improvement in her triglyceride and liver function (TC 4.48 mmol/l; Trig 1.43 mmol/l; HDLC 0.75 mmol/l; LDLC 3.08 mmol/l; ALT 67 IU/l; AST 48 IU/l).
Just over a year after restarting statin treatment, the patient opted to interrupt therapy again in order to try for a second child. Unfortunately, the second pregnancy remained elusive and after having been off all lipid-lowering therapy for a full year and watching rising lipid levels, lipid-lowering therapy was recommenced.
Despite high-dose atorvastatin, her LDL remained persistently above 3 mmol/l. It was decided to change to rosuvastatin in an attempt to further lower the LDLC level. Disappointingly, the patient seemed to respond sub-optimally to high-dose rosuvastatin (40 mg), with her LDLC rising to 5.4 mmol/l, and she was switched back to atorvastatin.
After halting therapy for a time once more, in an attempt to achieve conception, the patient subsequently continued atorvastatin without further significant improvement in her LDLC levels (hovering around 3 mmol/l). Further cardiac review confirmed premature atherosclerosis with coronary calcification and calcific aortic stenosis, as well as plaque in both her carotid and femoral arteries. It was therefore considered essential that her hypercholesterolaemia be treated more aggressively. Ezetimibe 10 mg daily was thus added to her therapy. At this time her levels reflected the following: TC 4.8 mmol/l; Trig 1.4 mmol/l; HDLC 1 mmol/l; LDLC 3.3 mmol/l; ALT 63 IU/l; AST 55 IU/l.
Approximately a year later, a significant improvement in both lipid and liver function tests was apparent. Her lipid profile showed a TC of 3.3 mmol/l, Trig of 1 mmol/l, HDLC of 1mmol/l and a LDLC of 1.8 mmol/l, while her transaminases were only minimally elevated (ALT 44 IU/l; AST 39 IU/l).
For the past two years, the patient has been on a combination of atorvastatin and ezetimibe with her lipid levels at target. She continues to enjoy good cardiac health, with mild progression of the aortic valve disease but, importantly, no evidence of underlying ischaemia. She will require ongoing follow-up and may require aortic valve replacement at some point in future. As prior genotyping has not been performed for this patient, it may be a further consideration in view of her son's future risk ().
Table 1: Trend of case patient's biochemical parameters
Discussion
CESD is an autosomal recessive lysosomal storage disorder that arises from loss of function mutations in the lysosomal acid lipase gene (LIPA), which causes a deficiency in LAL activity. LAL plays an essential role in lipid catabolism. It is a lysosomal enzyme that hydrolyses cholesteryl ester and triglycerides delivered to the lysosome ().Citation1,Citation2
Figure 1: Cellular cholesterol homeostasis in healthy individuals and patients with LAL deficiency.
Abbreviations: ACAT = acyl-CoA: cholesterol acyltransferase; CE = cholesterol ester; FA = fatty acid; FC = free cholesterol; HMG-SoAr = 3-hydroxy-3-methyl-glutaryl.CoA reductase; LDL-C = low-density lipoprotein cholesterol; LDLR = low density lipoprotein remnant; SREBPs = sterol regulatory element binding proteins; TG = triglyceride; VLDL-C = very low-density lipoprotein cholesterol.Citation3
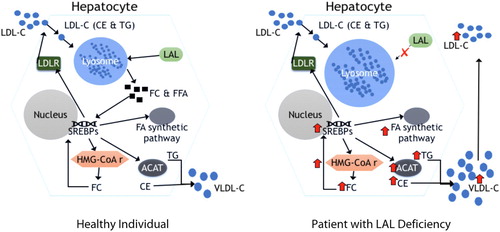
A deficiency in LAL results in accumulation of cholesteryl esters and, to a lesser extent, triglycerides in hepatocytes, spleen, adrenal glands, intestines and the macrophage-monocyte system of the body. The extent of involvement of the organs reflects their contribution in the receptor-mediated endocytosis and lysosomal degradation of lipoproteins.Citation1
CESD is a rare, under-diagnosed disease with estimates of prevalence ranging from 1/40 000–1/300 000.Citation4 The disorder can phenotypically manifest in two ways, namely the fulminant, infantile-onset Wolman's disease (WD) and the later-onset spectrum of CESD.
WD occurs when LAL activity is completely absent or less than 1% of normal activity. It presents within the first few months of life with extensive lysosomal accumulation of cholesteryl esters and triglycerides, resulting in hepatosplenomegaly, with associated liver disease, and malabsorption from intestinal villi involvement. Feeding difficulties, persistent emesis and malabsorptive diarrhoea results in malnutrition and growth retardation. Approximately half of patients have adrenal calcification resulting in adrenal insufficiency. The severity of the disorder is reflected by ultimate mortality within the first year of life unless successfully treated with haematopoietic stem cell transplantation.Citation1, Citation5
CESD, later in onset, can present as a spectrum of disease depending on the extent of LAL deficiency. It may present in early childhood with features similar to WD with failure to thrive and delayed milestones. More often, it remains unrecognised, and the patient may present with elevated serum lipids (LDLC and Trig) and/or hepatosplenomegaly that may be associated with elevated liver enzymes. This usually significantly predates the diagnosis. The morbidity and mortality of CESD arises from the premature atherosclerosis resulting in coronary artery disease or cerebrovascular events; associated liver dysfunction and ultimately liver failure; complications of secondary hypersplenism as well as malabsorption from lipid deposition in the intestinal tract.Citation1,Citation5
Due to the broad clinical spectrum of the disease as well as its rarity, CESD often goes unrecognised or diagnosis is delayed, hence it is thought to be an under-diagnosed condition. Patients typically present with hepatomegaly and liver dysfunction (elevated serum transaminases) or type IIB hyperlipoproteinemia (elevated serum LDLC and Trig, with normal to low HDLC). Continuous lipid deposition leads to hepatic fibrosis, micronodular cirrhosis and, eventually, liver failure.
The unexplained liver abnormalities may lead to a liver biopsy. Grossly, the liver appears bright yellow-orange in colour and, histologically, enlarged lipid-laden hepatocytes and macrophages with microvesicular steatosis are seen. A pathognomonic feature, which can be elucidated by ultrastructural examination, is the presence of the needle-shaped, lysosomal CE crystals. Through immunostaining for the membranous and luminal lysosomal proteins, LAMP2 and cathepsin D respectively, the histologic diagnosis of CESD can be improved upon.Citation1
It is, however, possible that the diagnosis may be missed, being commonly mistaken and misclassified as either non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), or even cryptogenic liver disease. NAFLD is usually associated with central obesity. It is, therefore, imperative that clinical aspects are taken into account—particularly waist circumference and body mass index, especially when considering such a diagnosis in a lean individual.Citation5
The clinical manifestations of CESD allow for a number of other differentials. In light of the hepatosplenomegaly, these may include other storage disorders, including other lysosomal storage disorder (such as mucolipidosis II, mucopolysaccharidoses, Gaucher's disease and Nieman–Pick disease types A and B) as well as glycogen storage diseases. In view of the lipid abnormalities, familial hypercholesterolaemia and autosomal recessive hypercholesterolaemia may be considered but hepatomegaly does not occur in these conditions.Citation5
Associated clinical and biochemical features can be used to distinguish the diagnosis. For example, although the lipid profile may be similar in Nieman–Pick, the interstitial lung disease and the ophthalmologic findings are not seen in LAL deficiency. In Gaucher's disease the lipid abnormalities, bone manifestations and adrenal calcifications are not apparent. And while the contractures, skeletal dysplasia and coarse facial features are typical of mucolipidosis II and mucopolysaccharidoses, they are not features of CESD. The same can be said of the hypoglycaemia, kidney dysfunction and cardiomyopathy associated with the glycogen storage diseases.Citation5
The diagnosis of CESD can be confirmed through genetic screening and identification of either biallelic pathogenic variants of LIPA or testing for deficiency in LAL enzyme activity. As over 40 loss-of-function mutations in the LIPA gene have been reported, one could consider a number of molecular testing methods. First, single gene testing, which consists of sequence analysis of the LIPA gene. Second, a multigene panel, which assesses the LIPA gene and other genes of interest. And third, comprehensive genomic testing can be considered in patients with features of LAL in whom the single/multi-gene testing were unsuccessful in identifying a mutation. If available, this includes genome sequencing as well as mitochondrial sequencing.Citation5, Citation6
In assessment of deficiency in LAL enzyme activity, the diagnostic test is the assay measuring LAL activity in peripheral blood leukocytes. However, enzyme activity can also be measured in hepatocytes, fibroblasts or dried blood spots. Residual LAL activity varies according to the severity of the disease with Wolman's disease usually estimated at less than 5% of normal activity and CESD ranging between 2% and 11%.Citation5–7
Unfortunately, neither the genetic nor the enzymatic testing is available in South Africa. Although cost is likely prohibitive, it is possible for the enzymatic test to be performed on a dried blood spot sample sent to an international laboratory. Once the diagnosis is made non-invasively, a liver biopsy may be done for staging and to monitor progression of the disease. It is, however, more likely, considering our resources in South Africa, that both diagnosis and staging will have to be confirmed by liver biopsy.
Treatment of patients with LAL deficiency, until recently, mainly focused on symptomatic management of the disorder. It was only in 2015 that the United States Food and Drug Administration (FDA) approved enzyme replacement therapy (ERT) with sebelipase alpha. Sebelipase alpha is a recombinant human LAL, with the same amino acid sequence as that of human LAL. The enzyme is attached to mannose, allowing delivery to the lysosome via mannose-6-phosphate receptors.Citation5, Citation8
In a randomised placebo-controlled, phase 3 study in children and adults with LAL deficiency, sebelipase alpha treatment was associated with significantly higher rates of normalisation in ALT and AST levels compared with placebo. Furthermore, a reduction in hepatic fat content and improvement in LDLC, HDLC and Trig levels was demonstrated. It was shown that ERT may be life-saving in WD and both improve and prolong life in CESD.Citation9 Recently, a study evaluating sebelipase alpha's impact on atherogenic biomarkers in children and adults with LAL deficiency appeared positive.Citation8 Treatment with sebelipase alpha (irrespective of lipid-lowering therapy use) reflected improved atherogenic biomarkers, particularly significant decreases in mean LDL particle number, LDLC levels and ApoB levels as well as significant increases in mean HDLC and ApoA1 levels compared with placebo. This reflects an improvement in the atherogenic profile of patients with LAL deficiency, and hence improved cardiovascular disease risk.Citation8 Sebelipase alpha is, unfortunately, currently not available in South Africa.
There was a paucity of treatment guidelines for LAL deficiency prior to the FDA approval of sebelipase alpha. Lipid-lowering therapy, in the form of statins, ezetimibe and/or cholestyramine, as well as a low-fat, low-cholesterol diet, was (and still are) often used to treat the dyslipidaemia associated with the disorder and alleviate the atherogenic risk. In advanced disease, liver and haemopoietic stem cell transplantation have been undertaken with significant associated morbidity and limited success. It must be noted that all these therapeutic interventions do not address the underlying mechanism or the multisystem nature of the disease and, hence, have limited efficacy.Citation5,Citation8
Furthermore, the importance of genetic counselling has to be stressed in order to equip both the patient and the family with adequate knowledge of the nature, inheritance and implications of the disease. This will also facilitate the screening of offspring of the affected patients.
When discussing rare diseases such as LAL deficiency, one has to consider the cost that diagnosis and management would entail, especially in a resource-constrained setting such as South Africa. Drugs that are developed for rare diseases are often extremely expensive on a cost-per-patient basis. Complications that arise from such a disease, such as liver failure or cardiovascular complications, also require significant resource outlay. Analysis of the pharmacoeconomics associated with the cost-effectiveness of a treatment such as ERT would thus be of interest.Citation10 A recent study in the United Kingdom evaluated the costs of managing children and adults with LAL deficiency in a tertiary centre, and found these to be staggering. In light of this, consideration needs to be given to rare diseases in policy and budgetary decision-making.Citation11
Conclusion
LAL deficiency is a rare genetic, multi-systemic disorder. It is an unusual cause of liver dysfunction, associated with dyslipidaemia and increased atherogenic risk, that eventually results in liver failure. Increased awareness of LAL deficiency by healthcare workers including hepatologists, pathologists, endocrinologists, cardiologists and neurologists will result in improved detection of patients, leading to earlier treatment and improved outcome. A high index of suspicion in lean patients who present with the constellation of signs including hypercholesterolaemia, hepatomegaly and abnormal liver enzymes may make early identification of this disease more likely. Establishing the diagnosis is imperative, now more than ever, due to the development of a new therapeutic approach in the form of recombinant human LAL, an enzyme replacement therapy.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Bernstein DL, Hülkova H, Bialer MG, et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58(6):1230–43. doi: 10.1016/j.jhep.2013.02.014
- Zhang H. Lysosomal acid lipase and lipid metabolism. Curr Opin Lipidol. 2018;29(3):218–23. doi: 10.1097/MOL.0000000000000507
- Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency – an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235(1):21–30. doi: 10.1016/j.atherosclerosis.2014.04.003
- Vinje T, Wierød L, Leren TP, et al. Prevalence of cholesteryl ester storage disease among hypercholesterolemic subjects and functional characterization of mutations in the lysosomal acid lipase gene. Mol Genet Metab. 2018;123(2):169–76. doi: 10.1016/j.ymgme.2017.11.008
- Hoffman EP, Barr ML, Giovanni MA, et al. Lysosomal Acid Lipase Deficiency. Gene Reviews. 2015 Jul (Updated 2016 Sep). Bookshelf URL: https://ncbi.nlm.nih.gov/books/
- Masi S, Chennamaneni N, Turecek F, et al. Specific substrate for the assay of lysosomal acid lipase. Clin Chem. 2018;64(4):690–6. doi: 10.1373/clinchem.2017.282251
- Lipiński P, Ługowska A, Zakharova EY, et al. Diagnostic algorithm for cholesteryl ester storage disease: clinical presentation in 19 polish patients. J Pediatr Gastroenterol Nutr. 2018;67(4):452–7. doi: 10.1097/MPG.0000000000002084
- Wilson DP, Friedman M, Marulkar S, et al. Sebelipase alfa improves atherogenic biomarkers in adults and children with lysosomal acid lipase deficiency. J Clin Lipidol. 2018;12(3):604–14. doi: 10.1016/j.jacl.2018.02.020
- Burton BK, Balwani M, Feillet F, et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med. 2015;373(11):1010–20. doi: 10.1056/NEJMoa1501365
- Frampton JE. Sebelipase alfa: a review in lysosomal acid lipase deficiency. Am J Cardiovasc Drugs. 2016;16(6):461–8. doi: 10.1007/s40256-016-0203-2
- Guest JF, Ingram A, Ayoub N, et al. Healthcare resource use and costs of managing children and adults with lysosomal acid lipase deficiency at a tertiary referral centre in the United Kingdom. PLoS One. 2018;13(2):e0191945. doi: 10.1371/journal.pone.0191945