
ABSTRACT
The pharmacodynamic activity on the organic structure of benzoyl peroxide has been analysed by molecular spectroscopical tools (IR, Raman, nuclear magnetic resonance and UV–visible). Simultaneously, the results obtained in the experimental process are verified by performing Quantum Gaussian computational calculations with higher-order basis sets. The actual positions of internal compositions and purity of the compound are verified with the observations of fundamental and group frequency of the recorded pattern of the FT-IR and FT-Raman spectra. The chemical environment of different carbons existing in various entities for approving a drug property is keenly identified and distinguished. The energy level degeneracy among different Frontier molecular orbitals is viewed from the orbital overlapping interaction contour. The biological activity of the present compound is emphasized and correlated with the hyperpolarizability profile of the internal coordinate system of a molecular structure arrangement. The involvement of non-bonding molecular orbital for the inducement of drug reactivity is monitored from the observation of cluster electron transitions. The Gibbs energy for chemical reaction with augmented temperature is relatively discussed, and the continuum of chemical reaction is observed.
1. Introduction
Benzoyl peroxide [(C6H5CO)2O2] (BP) is an organic compound belonging to the peroxide family and it consists of two benzyl rings which are coupled by a peroxide group. It is a colourless crystalline solid and its melting point lies between 104°C and 106°C, and it will explode when its boiling point is at 107°C. It is easily soluble in alcohol, ether and water and its solubility value is 9.1 mg/lit at 25°C in water. BP is widely used as an initiator and catalyst in the food preservative industry [Citation1]. BP is a vital therapeutic agent for acne vulgaris due to its in vitro antimicrobial activity against Propionibacterium acnes [Citation2]. Owing to the use of topical and systemic antibiotics for acne vulgaris, the incidence of antibiotic-resistant Propionibacterium acnes is increasing worldwide.
The efficacy of BPO can be enhanced when used in combination with topical retinoids, antibiotics and tertiary amines. It contains combinations that induce effective bacterial resistance and that are important in bacterial infections’ treatment for moderate to heavy acne vulgaris. Of late, BPO has been commonly used as a topical drug for the treatment of acne vulgaris worldwide [Citation3–4]. In addition to that, BP also has antimicrobial activity against P. acnes and moreover possesses keratolytic/comedolytic and anti-inflammatory effects [Citation5]. BP data compiled from recent research [Citation6,7] indicate that it is a safe drug.
In spite of its important antibiotic and anti-inflammatory applications, BP has not been subjected to systematic investigation on the structure activity associated with its pharmaceutical potential. Therefore, the present investigation was conducted for the strong interpretation of the structure activity connected with the inducement of active drug property of the compound using spectroscopic data and computational results.
2. Experimental profile
Physical state
The prepared compound is in the solid phase and is found to be pure and of spectroscopic grade.
Recording profile
The FT-IR and FT-Raman spectra of the compound were recorded using a Bruker IFS 66V spectrometer and the instrument adopted with an FRA 106 Raman module equipped with aNd : doped yttrium aluminum garnet laser source operating at 1.064 µm line widths with 200 mW power [Citation8]. A standard optical system was used with KBr windows for data collection over a spectral range of 8300–350 cm−1 at the best resolution of 0.5 cm−1. Which is standard, high-performance, room-temperature LiTaO3 (lithium tantalate) MIR detector with an signal to noise ratio of 9300 : 1.
The high-resolution hydrogen nuclear magnetic resonance (1HNMR) and carbon nuclear magnetic resonance (13CNMR) spectra were recorded using the 300 MHz and 75 MHz FT-NMR spectrometer [Citation9]. The instrument was provided with Standard 5 mm nuclear magnetic resonance (NMR) tubes and less than 1.0 Hz (20 ppb) at full width at half maximum.
The UV–visible spectrum was recorded in the range of 200 to 800 nm, with the scanning interval of 0.2 nm, using the UV-1700 series instrument [Citation10].
3. Computational profile
In order to design the structure precisely, calculate geometrical parameters, display the Mulliken charge levels, study the vibrational spectral properties, observe the molecular orbital interactions, examine the frontier molecular transitions on the electronic structure, the entire quantum chemical computations were performed using the Gaussian 09 D. 01.version software program in a core i7 computer [Citation11]. The computational calculations were performed over all the geometrical parameters, vibrational frequencies, simulation of molecular structure and spectra using B3LYP and B3PW91 methods adopted with 6–31++G(d, p) and 6-311++G(d,p) basis sets.
Particularly, for aromatic polyatomic complex molecules, the density functional theory (DFT) methods lead the prediction for finding more accurate molecular structure and vibrational frequencies. In DFT methods, Becke’s three-parameter hybrids’ function combined with the Lee–Yang–Parr correlation function (B3LYP) precisely gives good molecular structures, vibrational frequencies and charge densities in strongly bounded systems. Becke’s three-parameter exact exchange function (B3) combined with the gradient-corrected correlational function of Lee, Yang and Parr (LYP) and Perdew and Wang (PW91) predicts the best results for molecular geometry and vibrational wave numbers for a moderately larger molecule.
The optimized molecular structure is presented in three different forms in . The energy absorbance by the present compound related with electronic spectra and the non bonding orbital (NBO) calculation are performed by using the NBO 6.1 program and HOMO–LUMO high occupied molecular orbitals (HOMO)–low unoccupied molecular orbitals (LUMO) depletion energies were calculated using the time-dependent self consistent field method with the best fit basis set. In the same way, the 1H and 13C NMR chemical shifts with respect to tri methyl saline (TMS) were calculated by the gauge independent atomic orbital method using the integral equation formalism variant polarizable continuum model in combination with B3LYP/6-311++G(2d,p). The Mullikan charge assignment on different parts of the compound was calculated and was purposely elucidated for the determination of the key factor for pharmaceutical activity of the compound. The dipole moment, linear polarizability and the first-order hyper polarizability in different coordinates of the compound were computed using the B3LYP method with the 6-311++G(d,p) basis set. The electronic circular dichroism and vibrational circular dichroism (VCD) spectra were simulated from available frequencies, the optical chirality was studied and the mechanism for masking the toxicity was interpreted.
Figure 1. The optimized molecular structure of benzoyl peroxide.
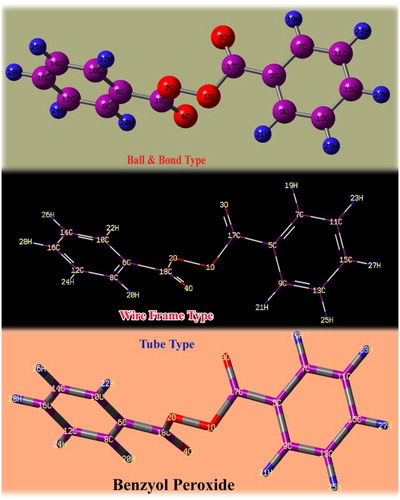
4. Results and discussion
4.1. Structural property and molecular geometry deformation analysis
The molecular properties of the present compound have been studied by the calculated specific absorption rate parameters, obtained using HyperChem 8.0.6 software which is depicted in . Topological polar surface area of the compound is a significant parameter which is used to predict the ability of drug transport properties. This factor is basically associated with the rate of human intestinal membrane penetration and the Caco-2 monolayer’s permeability. Here, the parameter value was found to be 52.60 A2, due to which it possessed enhanced intestinal absorption. If the molecular mass is less than 500 Da, it will be able to pass through the cell membrane. Here, the value was identified to be 242.05 g/mol, which is half of the limit and was permeable with the cell membrane. The present molecule has H-bond donor and acceptor count 0 and 4, respectively, and consequently the present drug conformed to Lipinski's rule and success to display drug likeness.
Table 1. Physical parameters of benzoyl peroxide.
The molecular deformation view is shown in and the present molecule possesses bi-symmetry by which it belongs to the C2V point group symmetry. The optimized geometrical parameters have been presented in . The two phenyl rings are connected with one another by peroxide groups. In order to equal the existing electrochemical moment, both the benzene rings are placed perpendicular to each other with respect to the ligand groups. Normally, the migration of electrons from the bonds makes the covalent to coordinate with the covalent bond. This effect induces some peculiar chemical property in the compound [Citation12]. Here, after attaining the optimized molecular structure, the covalent bond between O1 and O2 is observed to be coordinate covalent bond. Such a change in the compound is the root cause of inducement of effective bacterial resistance in the compound. At the centre, two phenyl rings are conjugated by the symmetrical ligand groups of C=O–O and thus two carbonyls strongly coupled with two benzene rings. In the left moiety, the bond lengths C6–C8 and C6–C10 are stretched by 0.009 Å than C8–C12 and C10–C14. Simultaneously, the bond lengths C12–C16 and C14–C16 are stretched by 0.005 Å than the others in the right moiety of the ring.
Figure 2. Molecular structure in the Cartesian coordinate system.
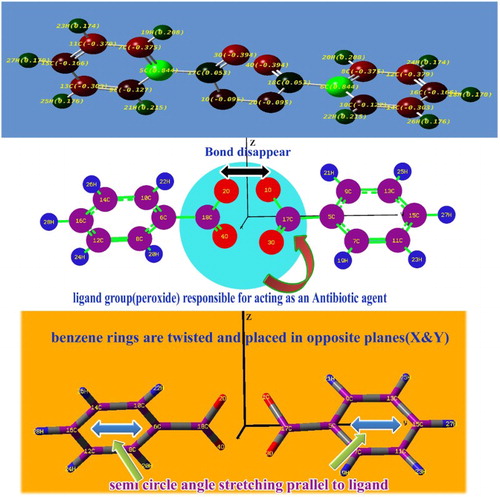
Table 2. Optimized geometrical parameters for benzoyl peroxide (BPO) computed at HF/DFT (B3LYP&B3PW91) with 6-31++G(d,p) and 6-311++G(d, p) basis sets.
Similarly, in the second ring, the bond lengths C5–C9 and C7–C5 are 0.009 Å greater than C11–C7 and C9–C13 of the left moiety and the bond lengths C13–C15 and C11–C15 are 0.004 Å greater than C7–C11 of the right moiety. Here, the substitutional groups are peroxide ligand, due to the insertion of the groups in both the benzene rings, the hexagonal frame is found to be little contravention. The reception of the peroxide groups is evidenced from the semicircular distortion in the rings. The bond length of C5–C17 and C6–C18 are 0.087 Å greater than carbon carbon (CC) of the ring. The bond lengths of the peroxide groups are consistently same since the ligand groups are highly symmetrical. Due to the injection of substitutions, the bond angles C9–C5–C7 and C8–C6–C10 are squeezed by 0.29° since both the rings pulled away from the substitutions. Thus, the bond length and bond angle changes in the compound, emphasizing that the exchange of electron clouds between the rings via peroxide groups causes the drug property in the compound.
4.2. Mulliken charge distribution
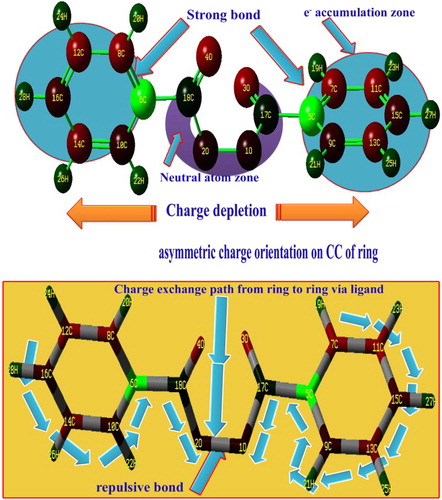
The charge distribution among the molecular orbitals described the energy transitions for generating the desired molecular properties such as physical as well as chemical properties. The consumption of energy during the vibrations showed the orientation of electron potential in different parts of the compound. Normally, the electron cloud is oriented on the side of the peroxide group since it contains four oxygens. Here, the electron clouds are directly concentrated over the carbons of the benzene rings and are directed from the peroxide groups via the peroxide-connected carbon of the top moiety of the ring to create the wonderful drug property for the title compound. Due to the orientation, the carbons of the peroxide groups become neutral, whereas the electron cloud remains concentrated on π-bonded 3O and 4O in order to maintain the chemical equilibrium. In addition to that, strong dipole bonds (C=O) are created symmetrically on the peroxide groups. The negative charge distribution appears on both sides of the molecule and the depletion zone is centred at the peroxide group due to which the strong drug property is consistently emphasized. The protonic content is richer in one of the carbons of both benzene rings than the others since the electronic charges oscillate back and forth with respect to the peroxide group. This form of asymmetric charge orientation causes the anti-inflammatory application. The asymmetric is charge a separation in the present compound is clearly shown in .
Figure 3. The asymmetric Mulliken charge separation of benzoyl peroxide.
4.3. Vibrational assignments
The fundamental group frequencies of organic complex: benzoyl peroxide has been assigned according to its characteristic vibrational regions with a high degree of accuracy. Since the entire molecule belongs to multiple planes due to the different breaking regions, the point group is supposed to change from C2V to CS. Due to this the molecule belongs to the CS point group symmetry which contains 28 atoms in different planes; so it has 78 normal vibrational modes. Out of that, the fundamental vibrations of the present molecule are distributed as 53 in-plane vibrations denoted by A′ species and 25 out-of-plane vibrations denoted by A″ species, i.e. Γvib = 53A′ + 25A″. The observed FT-IR and FT-Raman frequencies and calculated fundamentals have been presented in . Comparing the calculated frequencies with the experimental values showed the over-estimation of the calculated vibrational modes due to the violation of the state of the real system. Inclusion of electron correlation in the DFT makes the frequency values smaller in comparison with the Hartree Fock (HF) calculated data. The vibrational pattern generated by the FT-IR and FT-Raman spectrometer is presented in and , respectively ().
Figure 4. FT-IR vibrational spectral pattern of benzoyl peroxide.
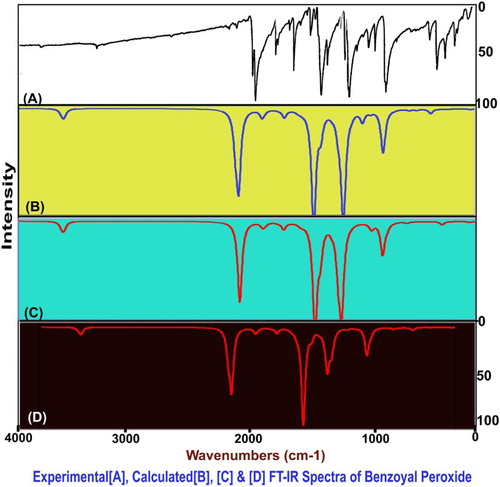
Figure 5. The FT-Raman spectra of benzoyl peroxide.
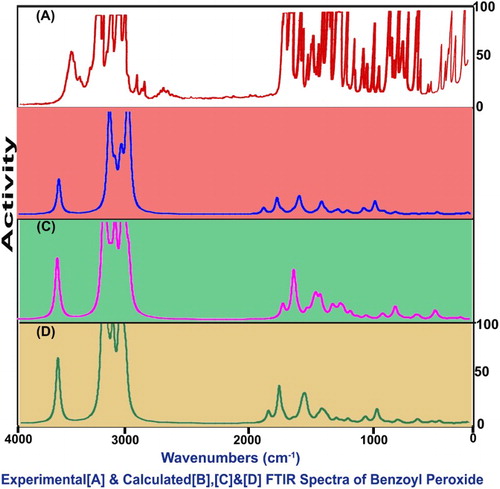
Table 3. Observed and HF and DFT (B3LYP & B3PW91) with 6-31++G(d,p) and 6-311++G (d,p) level calculated vibrational frequencies of benzoyl peroxide (BPO).
In order to compensate for deficient energy calculations on the tail end of an excited state and the loosely bonded electrons, computational scientists usually use diffuse functions. One needs to include diffuse functions (functions with small exponents, hence large radial extent) to predict properties of anions accurately. Diffuse functions are also necessary to describe certain non-bonding interactions. These basis sets utilize very small exponents to clarify the properties of the tail. Differences between diffuse basis sets are mostly due to the differences in their core and not in their diffuse component.
4.3.1. C–H vibrations
In all organic ring compounds, the purposive insertion of ligand groups for attaining desired drug properties was always pronounced in the fundamental modes of ring vibrations. Here, the peroxide ligand only the substitutional group which is acting as symmetrical bridge and link two benzene rings. With the symmetrical linking of benzene rings with peroxide groups, both the rings are twisted perpendicular to each other. The impact of injection of the substitutional group can be identified by the rate of suppression of vibrational modes of ring vibrations. Here, due to the mono substitution, 10 C–H bonds were identified and proportionate stretching vibrations were possible and consequently 20 bending vibrations will be observed continuously. In general, the C–H stretching vibrations are observed in the region 3000–3100 cm−1 for benzene derivatives [Citation13–15]. Accordingly, in this case, the C–H stretching peaks were available with very strong to very weak intensity at 3090, 3070, 3060, 3050, 3040, 3010, 2980, 2940 and 2900 cm−1. Except one, all the bands are found in Raman spectrum which contradicts previous works [Citation16]. Here, it is found that, except three peaks, all the vibrational bands are observed within the expected region which showed the lesser impact was pronounced on the ring vibrations due to the substitutions. Apart from that, three infected bands inferred that such bonds are placed beside the substitutional groups.
Here, the C–H in-plane bending modes are found at 1070, 1030, 1005, 1000, 950, 940, 895, 890, 885 and 840 cm−1. The out-of-plane bending is observed at 795, 705, 700, 695, 690, 660, 650, 620, 610 and 510 cm−1. Regularly, two different vibrational bending bands were identified in the region 1300–1000 cm−1 and 1000–750 cm−1, respectively [Citation17–19]. The part of in-plane bending modes is pulled well below the expected region whereas, except one, all the out-of-plane vibrations are moved down to the lower end of the expected region. Unlike stretching, the bending modes have much influence since the adoption of the ligand group. The entire ring C–H vibrations have suffered much. This view clearly described that the energy of ring modes are utilized by the ligand vibrations which leads to the inducement of a new property of the compound.
4.3.2. CC vibrations
Generally, the phenyl core CC (C=C and C–C) stretching vibrations are observed in the region 1600–1400 cm−1 [Citation20–22], in which the frequencies in the region 1600–1500 cm−1 are fundamentally assigned to C=C stretching and the rest to C–C stretching conservatively. In those bonds, since C=C and C–C bonds are unpredictable within the ring, three bonds of each of two rings are to be assigned and proportionately three stretchings of each bond are possible. Therefore, the C=C and C–C stretching bands are found at 1600, 1590, 1490, 1445, 1400 and 1380 cm−1 and 1370, 1320, 1280, 1230 and 1220 cm−1, respectively. Due to the strong bonding of the ligand with the core directly, the ring stretched diagonally. Except one band related to C=C, all stretchings are significantly found well below the expected region whereas the entire C–C stretching modes moved away from the characteristic region of the spectra. This appearance indicated that most of the potential energies are exchanged between rings via ligand for the enhancement of drug property of the compound. The ring carbon carbon carbon (CCC) in in-plane and out-of-plane breathing has been found at 470, 460, 450, 440, 400 and 360 cm−1 and 320, 300, 290, 250, 240 and 220 cm−1, respectively. Even a single ring breathing mode has not been identified within the limit of the expected region. From this condition, it is concluded that, due to the strong binding of the ligand group, the ring might not be breathing well.
4.3.3. Peroxide group vibrations
All acid peroxides have a weak absorption band in the region 900–800 cm−1 [Citation13,Citation23] due to the O–O stretching vibration. Acid peroxides also have two strong bands at 1820–1810 cm−1 and 1800–1780 cm−1 (saturated aliphatic) due to their C=O stretching vibration. Usually, the intensity of those bands is very high. For aryl and α, β-unsaturated acid peroxides, these bands occur at 1805–1780 cm−1 and 1785–1755 cm−1. The nature and position of the substituent(s) in the aromatic portion of acid peroxides may significantly influence the position of these bands. Here, the O–O stretching vibration is found with medium intensity at 800 cm−1. Its corresponding in-plane and out-of-plane bending bands are found at 200 and 100 cm−1, respectively. The band is located at the tail end of the expected region which is mainly due to uncertainty of the bond and high homo nuclear character. The C=O is the major part of the peroxide group and its stretching vibrations are observed at 1790 and 1770 cm−1 in the IR spectrum. These modes are −20 cm−1 dislocated from the expected region which demonstrates their involvement in transformation of electronic potential for generating the desired property. The C=O in-plane and out-of-plane bending modes appeared at 500 and 475 cm−1 and 190 and 170 cm−1, respectively. The C–O stretching vibrations are found in the region 1300–1050 cm−1 which are not very useful in the characterization of acid peroxides. The C–O stretching peaks appeared at 1180 and 1165 cm−1 which are positioned in the middle part of the spectrum, emphasizing its domination over the O–O bond in order to hold the ring proximity. The C–O in-plane and out-of-plane bending modes were identified at 365 and 345 cm−1 and 160 and 155 cm−1, respectively.
4.4. NMR assessment
The observed and simulated 13C and 1H NMR spectra of the BP compound are presented in and their results are depicted in . The 13C spectral is significantly vital in order to identify the chemical pattern of biochemically important molecules [Citation24]. In this compound, the carbon is situated in three different atmospheres in which the complete pattern is observed. Here, the carbons presented in base as well as substitutional groups and are differentiated with respect to their positions. The calculated chemical shift of C5 and C6 are 142 ppm whereas the observed shift is found to be 134 and 135 ppm. The calculated shift is more than observed values since their phases differ. The chemical shift of adjacent carbons C7 and C8 is 165 ppm and it is observed to be more than other carbons in the ring which is mainly due to asymmetrical breaking of the proton shield. It is also noted that C7 and C8 are more deshielded since the energy exchange is taking place between rings via the ligand through these carbons.
Figure 6. The observed and simulated 13C and 1H NMR spectra of benzoyl peroxide.
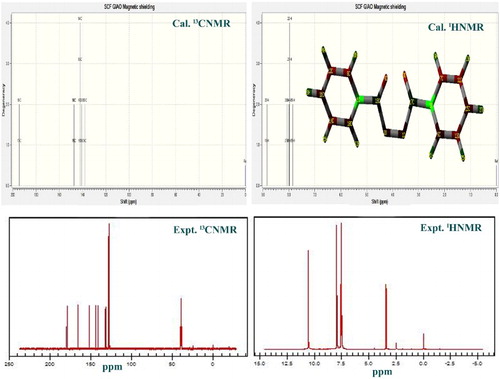
Table 4. Experimental and calculated 1H and 13C NMR chemical shifts (ppm) of benzoyl peroxide (BPO).
The calculated and observed chemical shifts of carbons C17 and C18 are found to be 193 ppm and 180 and 182 ppm, respectively. The chemical shifts of these carbons (C17 and C18) are greater than other carbons present in the molecule due to the asymmetrical breaking of the paramagnetic shield. This is obviously observed due to the holding the peroxide group and also bridge point of exchange of electron clouds between rings. The chemical shift of the remaining carbons in the ring ranges from 128 to 152 ppm, which fluctuate with a huge difference of values du to the arbitrary movement of electron clouds with large potential. This view emphasized the process of making compound be drug. Similar to the carbons in the ring, the chemical shift of H in the rings fluctuated from 7.9 to –8.8 ppm. The abrupt transformation of energy via the core carbons also affects the chemical shift of ring hydrogens in the ring.
4.5. Frontier molecular interaction examination
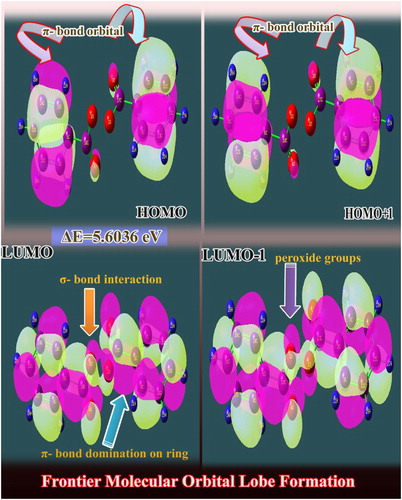
The formation of molecular orbitals from the atomic orbital is recognized by two sets of orbitals HOMO and LUMO in which the energy of the molecular orbitals was rearranged from a higher value to a lower energy value [Citation25]. Usually, the maximum energy value is found to be prearranged from the first HOMO to the last LUMO level. The chemical energy value of the compound is directly proportional to the energy value difference between HOMO and LUMO [Citation26], and the potential energy was exchanged to produce the desirable chemical behaviour.
The Frontier molecular orbital (FMO) interaction is shown in . Here, the Lewis base; (electron-pair donor) HOMO appears over semicircle of core CC bonds of benzene entities. Here, the orbital electrons are shared their orbital lobes and it is possible to donate the electron clouds to produce the chemical energy for the process of drug property. In the carbonyl group, the π-interaction flavour becomes visible. Simultaneously, the positive and negative iso surfaces are found to be reversed in the high molecular orbital (HMO-1) level which showed the reverse orbital interaction coupling. In Lewis acid (electron acceptor); LUMO, the π and σ-orbital interaction orbitals appeared in the ring and peroxide group, respectively. The σ-orbital interaction process takes place around the C of the ring and C of the peroxide group and O of the same groups. In LUMO+1, the intensity of σ-orbital interaction increases and the cascade process is extended with some more orbitals. Thus the two rings are connected with peroxide groups by transformation of electron cloud through σ and π interaction lobes and besides the chemical property is mixed and new drug property is produced.
Figure 7. The FMO interaction profile of benzoyl peroxide.
4.6. UV–visible absorption analysis
The electronic excitation parameters are presented in and the FMO levels of IR and UV–visible regions are depicted in . The CT complex absorption spectra of the compound are important to know the details of exchange of electronic energy between two significant entities (donor and acceptor) for occurring chemical reaction to generate desired chemical properties. The CT complex transfer spectra also directly describe that part of the compound that cause the formation of new physical as well as chemical properties [Citation27,Citation28]. Usually, the charge transfer progression takes place between base ligand groups in the chemical compound since the ligand groups are the major source of formation of the needed chemical property.
Table 5. Theoretical electronic absorption spectra of benzoyl peroxide (BPO) (absorption wavelength λ (nm), excitation energies E (eV) and oscillator strengths (f)) using the TD-DFT/CAM B3LYP/6-311++G(d,p) method.
Table 6. Frontier molecular orbitals of benzoyl peroxide (BPO) with energy levels.
Here, the benzene rings act as base compound and peroxide groups act as ligand groups. Necessarily, the CT transformation takes place between them and is directly evidenced from Mulliken charge levels. Here, the transition is represented by n → π* and observed at 271 nm on the energy gap of 4.565 eV with oscillator strength of 0.478. This absorption band is designated by 92% of H-L level and such a band (R-band-German, radikalartig) is assigned to the UV region of the spectrum. In the solvent phase (DMSO and CCl4), the same band was found at 271 and 269 nm on the energy gap of 4.56 and 4.60 eV with oscillator strength of 0.52 and 0.55, respectively. According to , there are several molecular orbitals overlapping that appeared between the ligand and base rings and the observed transition is found to be very strong and emphasized the energy exchange between peroxide groups to benzene rings from side to side. Usually, if the CT band is identified in Quartz-UV, the compound will be drug active [Citation29]. Here, a similar drug property is observed in this compound; in addition to that, the charge transfer is restricted within the closed path between rings via the peroxide group for deducing consistent drug action.
4.7. Molecular electrostatic potential (MEP) maps
Usually, after the formation of the chemical compound by fusing the base with ligand groups, the chemical charges are enforced to organize in order to equalize the induced dipoles. Thereby, two opposite extremely charged regions (protonic and electronic) are alienated with respect to the rate of chemical-potential energy which is utilized for displacing electron clouds. The depleted MEP appearance on various parts of the molecule is shown in .
Figure 8. The MEP diagram of benzoyl peroxide.
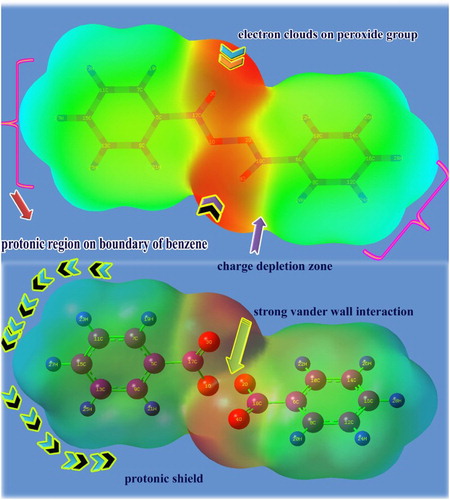
The source of rich protonic region is represented by a rich green shade whereas the origin of the enriched electronic region is designated by the red region of the colour spectrum. In this case, the protonic region is symbolized over hydrocarbons around both benzene rings by a high degree of green shadow and is shielded by the positive charges rigidly. The electronic clouds are concentrated in the mid region of the molecule (red region) which divides the positive boundary symmetrically. The charge depletion zone is found with a yellow colour region which lies between the red and green regions. Two dipoles are observed between the green and red regions of the molecule altogether which seems to indicate that the protons are bundled by an electron rope. From this view, it can be concluded that the strong electrophilic-nucleophilic dipole is established between rings and peroxide groups. This colour structure of the molecule is able to maintain the drug consistency and this system of the molecule acted as a good receptor ligand for the concerned protein absorption.
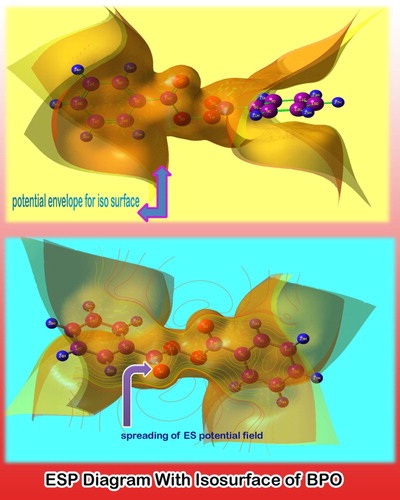
The potential envelope grid points are internally connected and the corresponding iso surface clearly appears as in . In the figure, the potential field is found to be spread out from the peroxide group and core of benzene rings aggressively. Normally, the potential field starts from the main group of the molecule which causes the generation of a peculiar drug property. Accordingly, in this case, the peroxide group linked with symmetrical benzene rings is the root cause of biological activeness.
Figure 9. ESP view of benzoyl peroxide.
4.8. Physico-chemical properties
The chemical properties related to molecular structure and Frontier molecular energy levels of the present compound are computed and the entire parameters are presented in .
Table 7. Calculated energies, chemical hardness, electro negativity, chemical potential, electrophilicity index of benzoyl peroxide (BPO) in the UV–visible region.
The zero point vibrational energy of the compound is found to be −840.25 K cal/mol and is same in both the IR and UV–visible regions. This circumstance shows the wide range of chemical consistency. The resultant dipole moment of the molecule as a whole is the gauge of asymmetric charge displacement and the same is found to be 1.19 and 4.94 dyne in the IR and UV–visible region, respectively. The actual dipole moment is greater in the UV–visible region than the IR region which shows the rich biological activity in UV–visible boundary. In this case, the base compound is a couple of benzenes; its dipole moment is almost zero whereas when peroxide groups are substituted between two rings, the total computed dipole moment is found to be very high and it is stressed that the asymmetric charge orientation reflected the desired pharmaceutical property.
Chemical hardness is one of the highly useful concepts which enable chemists to understand reactivities of a chemical compound. The chemical stability of the aromatic chemical species is normally determined from generated molecular orbitals’ (HOMO and LUMO) energy gap. In this case, it is found to be 2.83 and 2.49 eV in the IR and UV–visible regions, respectively. If the measured value of the molecule is greater than unity, the compound will be chemically strong and it will be having good reactivity. Here, the observed values showed reasonable chemical stability and it is also much reactive in the Quartz UV region.
The electron affinity of the molecule is very vital for finding whether the reaction is exothermic or endothermic [Citation30,Citation31] and measured reaction ability is found to be 4.7 and 4.9 in the IR and UV–visible region. The observed value showed the reaction is exothermic and a prominent amount of energy is released during the reaction.
The ionization potential energy is one of the primary energy considerations used in quantifying chemical bonds and characterizes the electron’s bond strength [Citation32]. The direction and cause of polarity of the molecule can be identified strongly. The ionization potential is observed to be 1.93 and 2.49 which is moderate and it is adequate to maintain the chemical bond stability. Electronegativity is a measure of the ability of an atom in a molecule to draw bonding electrons to itself. An important application of electronegativity is in the prediction of the polarity of a chemical bond [Citation33]. Here, both parameters were found to be 4.76 and 4.99 in the IR and UV region, respectively. The observed values are so high and demonstrate the fine reactive nature and its structure form leads the dual bonds between two benzene rings and peroxide group and is strongly evidenced in the MEP diagram.
The electrophilicity index is a gauge of rate of potential energy flow through generated interactive molecular orbitals. Here, the electrophilicity index is 4.00 and 4.99 eV in the IR and UV–visible region correspondingly. But, it is found to be 2.09 eV for the benzene ring. The resultant energy is considerably very high which shows a huge amount of energy flow through overlapped orbitals which is due to the symmetrical existence of the benzene rings about the peroxide group. The energy transformation is clearly evidenced from the electrophilicity charge transfer of the compound which is found to be +2.21. From this observation, it is inferred that such large amount of energy is needed to induce the antibiotics activity.
4.9. Polarization and hyperpolarization analysis
The chemical polarization index of the organic compound represents the molecular orbital arrangement in different internal coordinate systems. The hyper polarizability indicates the occurrence of a hyper interaction of complex molecular orbitals for inducing drug activity. The higher-order methods of computed polarizability and first-order hyperpolarizability indices are depicted in . The calculated dipole moment showed that the axis of molecular groups of the compound belongs to the z coordinate which is ensured by the high value of dipole moment on the same coordinate system. The calculated average polarizability and anisotropy of the polarizability is 270 × 10−30 and 353 × 10−30 esu, respectively. This is comparatively so high which is mainly due to strong coupling of benzenes with the peroxide group. The hyperpolarizability β is one of the imperative key systems which designate hyper interaction of molecular orbitals. Normally, the hyper interaction is very intensive in a drug complex. Here, the calculated first hyperpolarizability value (β) is 353.99 × 10−33 esu. From this observation, it is clear that the hyper asymmetrical polarization appears irrespective of base and substitutional groups which strappingly authorize the particular pharmaceutical property.
Table 8. The dipole moments µ (D), the polarizability α (a.u.), the average polarizability αo (esu), the anisotropy of the polarizability Δα (esu) and the first hyperpolarizability β(esu) of benzoyl peroxide (BPO).
4.10. Thermodynamical functions analysis
Normally, the thermodynamic analysis on an aromatic compound is very important since it provides the necessary information regarding the chemical reactivity [Citation34]. The thermodynamic functional parameters are depicted in . The variation of thermodynamic functional parameters with temperature is shown in . The calculated entropy, specific heat capacity and enthalpy were found to be varied with a positive temperature coefficient. When the temperature increased from 100 K to absolute temperature 298.15, the functional parameters were varied unhurriedly whereas from 350 to 1000 K, the thermodynamic functions seemed to swing in a linear pattern and were rather constant at maximum temperature. This view of variation showed the consistent chemical reactivity and considerable chemical hardness of the present compound. The Gibbs free energy is always a negative temperature coefficient and here since it was found to be true, the present compound has strong and unique chemical property and endless chemical reaction.
Table 9. Specific heat capacity, entropy and enthalpy at different temperatures for benzoyl peroxide (BPO).
The entropy is a measure of disorder of the chemical system with respect to temperature. It reflects the unavailable energy to do the chemical reaction. In this case, it is observed to be increased according to the temperature randomly which illustrates the consistency of the chemical reaction for the inducement of drug property. The enthalpy is the measurement of the energy transformation of the system externally or internally [Citation35]. The ΔH is a positive change in endothermic reactions, and negative in heat-releasing exothermic processes. In this case, the reaction magnitude is found to be positive and the chemical reaction is endothermic which describes the considerable energy is utilized for fusing peroxide group with phenyl rings. The specific heat capacity of the present compound also increased with respect to temperature. The kinetic energy of the atoms or molecules is increased across the chemical system boundary. From this condition, it is clear that the confined drug system is formed and the kinetics of internal parts of molecule is active. Gibbs free energy is a thermodynamic potential of thermodynamic chemical system that can be used to calculate the maximum of reversible work [Citation36]. In this case, the observed values for various temperatures are negative which means that the chemical reactivity is irreversible and permanent.
4.11. NBO transition analysis
The hybridization of molecular orbitals is classified into bonding and anti-bonding orbitals which describes the transformation of internal energy by electron transitions. Usually, the electron density is delocalized from occupied Lewis type (bond or lone pair (LP)) orbitals and unoccupied (anti-bonding and Rydberg) non-Lewis orbital in order to stabilize donor-acceptor interaction [Citation37–41]. The donor and acceptors of electronic orbitals are identified and their energy transitions are presented in .
Table 10. The calculated NBO of benzoyl peroxide by second-order perturbation theory.
In the peroxide group, the transition occurs from O1–O2 to C17 and C18 and it is assigned to σ–σ* by which 0.53 kcal/mol energy is transferred in order to connect the C and O, whereas the transition occurs from O3–C17 and O4–C18 to C5 and C6 and it is assigned π–π* interaction for which 1.28 kcal/mol energy is observed. The important transition is observed from peroxide O to C15, C16, C18, C5 and C6 with the energy gap of 21.81 a.u.; such amount of energy is absorbed from external energy for creating the desired chemical property. This transferred energy is further conducted through the ring for inducing drug property by the transitions (σ–σ* and π–π*) from C5 and C6 to C5–C7, C7–H18, C12, C6–C8, C6–C10, C9–C13 with 1.01 kcal/mol with energy gap of 12.50 a.u. The same transition (LP – σ* and π*) occurs from C12 and C13 to C8, C16, C12–C16, C14–C16, C9, C15 by intake of energy 2.37 kcal/mol. by crossing the energy gap of 11.98 a.u. The important transitions of σ–σ* (SP-hybridization) and π – π* (SP2-hybridization) are identified from C5–C17 and C6–C18 to O1 and O2 and also C5–C9, C5–C7, C6–C10, C6–C8, C8–C12 and so on, by back and forth oscillations of chemical kinetics in order to stimulate successful antibiotic activity. Though the lower order energies are observed for producing transitions, the electron clouds are crossed considerable energy gap for attaining enriched internal drug energy.
4.12. VCD verification
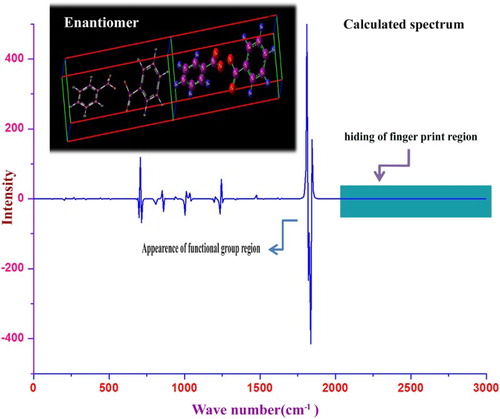
The VCD verification of the compound is mainly used for stereochemical examination; it is sensitive to the absolute configuration as well as to conformational features, which are often completely obscured in the ordinary absorption spectrum. It is greatly mapped to analyse some critical aspects regarding the interpretation of electronic CD spectra of organic compounds. The vibrational circular dichroism is the difference between the absorption of left and right circularly polarized lights through which the identity can be checked abruptly. The identity is used to elucidate the characteristics of chromophores causing the rate of drug purity and toxicity. The VCD spectrum of the present molecule is depicted in in which strong transmission and absorption peaks are obtained in various regions. Some peaks with same intensity are identified in the chromophores region (1700–1550 cm−1) which clearly indicate the greatest purity and less toxicity of the present compound. The hindering of fingerprint peaks in the higher wavenumber region showed the unavailable involvement of ring vibrations in the toxic effect of the molecule.
Figure 10. VCD spectrum of benzoyl peroxide.
5. Conclusion
The compound BP is the primary derivative of benzene. In order to predict the unknown chemical properties, different analyses have been made on the chemical structure. The molecular deformation analysis gave the complete information regarding the amendment of benzene structure for fixing the peroxide group to generate physical fitness for drug activity. The chemical charge equilibrium between bonded entities exposed the asymmetric movement of the charges which is favourable for inducement of a peculiar drug property. The vibrational pattern assignments of the molecular complex explicit the fundamental IR and Raman frequencies which are uniquely stressed the correct compositional bonds which self-possessed the compound. The chemical reaction path arrangement of different carbons is ensured from the discrete chemical shift and the fundamental cause is drawn. The orbital interaction lobe formation favoured the chemical process to produce a desirable drug property was predicted from the cascade arrangement of molecular orbital boundary. The chromophores reactivity on the base compound causing the electronic shift in UV–visible spectra is argued in detail. The CT band of electronic transitions between ligand and base compound is evaluated and the presence of the band in the appropriate region of the spectrum causing the drug property is determined.
Acknowledgements
The authors thank the computational consultancy, Department of Physics, A.V.C. College (Autonomous), Mayiladuthurai, Tamilnadu, India, for providing computational Lab facility to carry out the computational calculations.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Ponhong K, Supharoek S, Siriangkhawut W, et al. A rapid and sensitive spectrophotometric method for the determination of benzoyl peroxide in wheat flour samples. J Food Drug Anal. 2015;23:652–659. doi: 10.1016/j.jfda.2015.03.006
- Okamoto K, Ikeda F, Kanayama S, et al. In vitro antimicrobial activity of benzoyl peroxide against Propionibacterium acnes assessed by a novel susceptibility testing method. J Infect Chemother. 2016;22:426–429. doi: 10.1016/j.jiac.2015.12.010
- Strauss JS, Krowchuk DP, Leyden JJ, et al. Guidelines of care for acne vulgaris management. J Am Acad Dermatol. 2007;56:651–663. doi: 10.1016/j.jaad.2006.08.048
- Nast A, Dreno B, Bettoli V, et al. J Eur Acad Dermatol Venereol. 2012;26:1–29. doi: 10.1111/j.1468-3083.2011.04374.x
- Sagransky M, Yentzer BA, Feldman SR. Benzoyl peroxide: a review of its current use in the treatment of acne vulgaris. Expert Opin Pharmacother. 2009;10:2555–2562. doi: 10.1517/14656560903277228
- Bevington JC. Anomalous behaviour of benzoyl peroxide as an initiator of polymerization. CanJDyball Polymer. 1975;16:938–939. doi: 10.1016/0032-3861(75)90220-7
- Okieimen EF. Non-ideal kinetics in vinyl polymerization primary radical termination and chain transfer to solvent in benzoyl peroxide initiated polymerization of vinyl acetate in benzene. Polymer (Guildf). 1981;22:1737–1739. doi: 10.1016/0032-3861(81)90396-7
- Moorthy N, JobePrabakar PC, Ramalingam S, Pandian GV, Anbusrinivasan P. Vibrational, NMR and UV–visible spectroscopic investigation and NLO studies on benzaldehyde thiosemicarbazone using computational calculations. J Phys Chem Solids. 2016;91:55–68. doi: 10.1016/j.jpcs.2015.11.021
- Xavier S, Periandy S. Spectroscopic (FT-IR, FT-Raman, UV and NMR) investigation on 1-phenyl-2-nitropropene by quantum computational calculations. Spectrochim Acta, Part A. 2015;149:216–230. doi: 10.1016/j.saa.2015.04.055
- Moorthy N, JobePrabakar PC, Ramalingam S, et al. Spectroscopic analysis, AIM, NLO and VCD investigations of acetaldehyde thiosemicarbazone using quantum mechanical simulations. J Phys Chem Solids. 2016;95:74–88. doi: 10.1016/j.jpcs.2016.04.002
- Hiremath CS, Yenagi J, Tonannavar J. FT-Raman and infrared spectra and vibrational assignments for 3-chloro-4-methoxybenzaldehyde, as supported by ab initio, hybrid density functional theory and normal coordinate calculations. Spectrochim Acta A. 2007;68:710–717. doi: 10.1016/j.saa.2006.12.050
- Anand S, Sundararajan RS, Ramachandraraja C, et al. Molecular vibrational investigation [FT-IR, FT-Raman, UV–visible and NMR] on Bis(thiourea) nickel chloride using HF and DFT calculations. Spectrochimica Acta Part A. 2015;138:203–215. doi: 10.1016/j.saa.2014.11.032
- Socrates G. Infrared and Raman characteristic group frequencies (tables and charts). Chichester: Wiley; 2001.
- Varsanyi G. Assignments of vibrational spectra of 700 benzene derivatives. New York: Wiley;1974.
- Ebenezar JD, Ramalingam S, Raja RCR, et al. J Theoret Comput Sci. 2013;1:2.
- Lu Y-X, Zou J-W, Wang Y-H, Yu Q-S. Theoretical investigations of the C–X/π interactions between benzene and some model halocarbons. Chem Phys. 2007;334:1–7. doi: 10.1016/j.chemphys.2006.12.011
- Becke D. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. doi: 10.1063/1.464913
- Altun A, Golcuk K, Kumru M. Structure and vibrational spectra of p-methylaniline: Hartree-Fock, MP2 and density functional theory studies. J Mol Struct [Theochem]. 2003;637:155–169. doi: 10.1016/S0166-1280(03)00531-1
- Muthu S, Ramachandran G, Uma Maheswari J. Vibrational spectroscopic investigation on the structure of 2-ethylpyridine-4-carbothioamide. Spectrochimica Acta A. 2012;93:214–222. doi: 10.1016/j.saa.2012.02.107
- Dani VR. Organic spectroscopy. New Delhi: Tata McGraw Hill; 1995.
- Manohar S, Nagalkshmi R, Krishnakumar V. Crystal growth and characterization of N-hydroxyphthalimide (C8H5NO3) crystal. Spectrochimica Acta A. 2008;71:110–115. doi: 10.1016/j.saa.2007.11.023
- Fereyduni E, Rofouei MK, Kamaee M, Ramalingam S, Sharifkhani SM. Single crystal structure, spectroscopic (FT-IR, FT-Raman, 1H NMR, 13C NMR) studies, physico-chemical properties and theoretical calculations of 1-(4-chlorophenyl)-3-(4-nitrophenyl)triazene. Spectrochimica Acta A. 2012;90:193–201. doi: 10.1016/j.saa.2012.01.032
- Bellamy LJ, et al. Z Elektrochim. 1960;64:563–560.
- Fleming I, Williams DH. Spectroscopic methods in organic chemistry, IV ed. London (UK): McGraw-Hill; 1987.
- Yvesand J, Francois V. An introduction to molecular orbitals. Oxford: Oxford University Press; 2005.
- Catherine H, Alan S. Inorganic chemistry. 3rd ed. Upper Saddle River, NJ: Prentice Hall; 2007.
- Mulliken RS. Molecular compounds and their spectra. II. J Am Chem Soc. 1952;74:811. doi: 10.1021/ja01123a067
- Al-Hashimi NA, Hussein YHA. Ab initio study on the formation of triiodide CT complex from the reaction of iodine with 2,3-diaminopyridine. Spectrochim Acta A. 2010;75:198–202. doi: 10.1016/j.saa.2009.10.012
- Manzoor AM, George G, Ramalingam S et al., J Mol Struct. 2015;1099:463–481. doi: 10.1016/j.molstruc.2015.05.066
- Harwood P, Madura H. General chemistry principles & modern applications. Upper Saddle River, NJ: Prentice Hall; 2007.
- Thomas MR. The periodicity of electron affinity. J. Chem. Educ. 1990;67:307. doi: 10.1021/ed067p307
- Moore CE. Ionization potentials and ionization limits derived from the analysis of optical spectra. NSRDS-NBS 34. Washington, DC: Government Printing Office; 1970.
- Nivaldo TJ. Chemistry: A molecular approach. 2nd ed. Upper Saddle River, NJ: Pearson Prentice Hall; 2011.
- Zemansky MW. Chapter 11. In Heat and thermodynamics. 5th ed. New York, NY: McGraw-Hill; 1968. p. 275.
- Pierre P. Thermodynamics. Oxford: Oxford University Press; 1998.
- Na LJ, Rong CZ, Feng YS. J Zhejiang Univers Sci. 2005;68:584–592.
- Nakanishi K, Berova N, Woody RW. 1 circular dichroism: principles and applications. 2nd ed. New York: Wiley-VCH; 2000.
- Paul BK, Guchhait N. TD–DFT investigation of the potential energy surface for excited-state intramolecular proton transfer (ESIPT) reaction of 10-hydroxybenzo[h]quinoline: topological (AIM) and population (NBO) analysis of the intramolecular hydrogen bonding interaction. J Lumin. 2011;131:1918–1926. doi: 10.1016/j.jlumin.2011.04.046
- Paul BK, Guchhait N. A computational investigation on the intramolecular hydrogen bonding interaction and excited state intramolecular proton transfer process in 2-quinolin-2-yl-phenol. Comput Theoret Chem. 2011;978:67–76. doi: 10.1016/j.comptc.2011.09.040
- Paul BK, Mahanta S, Singh RB, et al. A DFT-based theoretical study on the photophysics of 4-hydroxyacridine: single-water-mediated excited state proton transfer. J Phys Chem A. 2010;114(7):2618–2627. doi: 10.1021/jp909029c
- Paul BK, Guchhait N. Phase transitions in [Co(NH3)6](ClO4)3 investigated by neutron scattering methods. Chem Phys. 2013;412:1–6. doi: 10.1016/j.chemphys.2012.12.006