
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
This paper has investigated the synthesis and characterization of CuO-coated goethite and its application in the removal of mercury. The presence of CuO coating significantly modified the adsorption pattern of mercury on goethite. The synthesis was carried out in a trimetric manner and characterization was conducted using standard laboratory procedures. Batch mode technique under reducing conditions at ambient temperature was used to determine mercury adsorption on CuO-coated goethite. Reaction mechanism indicated a less than one proton coefficient (0.68), an intraparticle diffusion that was controlled by boundary layer, an intercept C equals to 444.45, ≠0 and mass transfer rates (8.752–6 cm h–1 and 7.216–7 cm h–1) higher than those of previous studies.
KEYWORDS:
1. Introduction
Mercury, especially in the forms of elemental and methyl mercury, is known to be a cumulative toxicant that affects several body organs. Minamata disease is a neurological disorder caused by serous mercury poisoning. Humans are exposed to mercury via the food chain. Exposures to metallic mercury are associated with mercury spillage and breakage of products containing mercury [Citation1]. There is an increased observation of mercury ions above-recommended levels (10 µg/L) in water bodies [Citation5]. Chronic health disorders arising from elemental mercury bioaccumulation in the environment have been reported [Citation6]. Brain damage arising from methyl mercury induced by microbial biomethylation of Hg (II) ions has been reported [Citation7].
Therefore, research that could lead to smarter and cost-effective processes of mercury (Hg2+) removal from the aquatic environment has gained considerable concern. The modern method for the release of mercury into the aquatic environment is by a hydrometallurgical process involving leaching of cinnabar ore. This process involves ore attack using hydrobromic acid. The solution was treated in order to recover the mercury and to regenerate hydrobromic acid [Citation2]. The hydrometallurgical recovery of mercury is done at pH less than 5. This process leads to the release of leached materials containing clay minerals and oxides of copper as composites. The toxic nature of these effluents of the ecosystem has attracted scientific interest [Citation3].
The Eh–pH of the hydrometallurgical wastes controls the leaching of elements. Therefore, before discharge into the aquatic environment, hydrometallurgical waste containing Hg2+ ions and other metals is contained in sealed plastic tanks under reducing conditions. This process results in the lowering of the metal load in the slurries and leaching mud. Under reducing conditions, leached out metals and compounds inclusive copper oxide (CuO)-goethite interface will be negligible [Citation4]. Therefore, there is need to test the removal of Hg2+ ions from hydrometallurgical contained-slurries using standard laboratory procedures.
In the treatment of mercury-contaminated aquatic environment, adsorbent and chemistry of solution are key controlling factors. A combination of several other factors regulates its removal from the aquatic environment. Contact time and pH are regulating factors in the hydrolysis of mercury ions and species solubility. Huang and Lin have reported an increase in adsorption of mercury by biomass as pH was increased [Citation3,Citation8,Citation9]. Particle concentration, particle sizes and chemistry of the adsorbent control the diminution of mercury removal from the aquatic environment [Citation10]. Goethite coated with aluminium on its surfaces is known to reduce adsorption [Citation11]. Outer-sphere complexation is linked to an increase in mercury removal as adsorbent particle concentration was increased.
However, increase in particle size and particle concentration does not necessarily lead to increase in mercury uptake all the time [Citation12]. Mercury uptake is controlled by the specific surface area and surface-active sites [Citation13]. Residence time or ageing enhances the reorganization of mineral surfaces in aqueous solution [Citation14]. Based on the cost and simplicity of design, adsorption is considered a simple technique for mercury-contaminated water [Citation15,Citation16]. A quantitative decrease in adsorption and vice versa is dictated by an increase in metal concentration [Citation10,Citation6]. The fast process of intraparticle diffusion and the slow process of outer-sphere complexation are components of reaction mechanism involved in mercury uptake in aqueous solution [Citation17,Citation18].
In addition, mercury removal from the aquatic environment is controlled by solution dilution and exchange stoichiometry [Citation5,Citation6]. The mechanism controlling mercury removal and the effect of pH, residence time, metal initial concentration and ageing on the removal of mercury(II) ions, using copper oxide (CuO)-coated, goethite have received limited studies. The use of untreated goethite in mercury removal had been the focus of previous studies [Citation5,Citation19]. Santana et al. [Citation20] reported that biomass materials are potential adsorbents for mercury(II) ion removal in aqueous solution, with acidified P. Aquatics Abel is the most efficient adsorbent. Untreated goethite possesses the capacity to adsorb heavy metals using cation exchange and inner-sphere complexes formation via the aluminol and silanol sites [Citation21,Citation10].
Chemical precipitation and electrochemical recovery are other conservative techniques used in the removal of mercury from aqueous solution [Citation22]. Four successive steps have been identified as a pathway for a solid-solution system containing a solute, undergoing adsorption. These include external mass transfer, intraparticle diffusion, protonation and adsorption of molecules of adsorbate [Citation23]. Therefore, models of reaction mechanism are necessary to determine the reactions involved.
This study was aimed at determining the role of synthetic and characterized CuO-coated goethite in the treatment of mercury-contaminated water in relation to pH, residence time, metal initial concentration, the dosage of adsorbent and ageing of the affective solution. Experiments were conducted using batch-mode-related techniques in relation to the variable parameters. Synthesis of the adsorbent, characterization of the system and the testing of CuO-coated goethite to remove mercury ions have been discussed. CuO coating supported on goethite for Hg2+ ion removal is a new novel adsorbent. This has been applied in the treatment of Hg2+ ions in aqueous solution. This is useful in the treatment of hydrometallurgical waste water. This CuO–goethite composite significantly enhanced adsorption of Hg2+ ions.
2. Materials and experimental methods
All reagents used were of analytical grade. Ochre (goethite) – also called PMM goethite – was collected from settling ponds at Parys Mountain mine in Anglesey, United Kingdom. The yellow amorphous ferric hydrous oxide precipitate was sieved to 100 µm. Mercuric chloride dissolved in double distilled water was used in the preparation of various concentrations of Hg(II) ions.
2.1. Adsorbent characterization
PMM goethite contained 0.30% SiO2, 17.00% Al2O3, 62.62% FeO, 0.23% CaO, 0.03% MgO, 0.00% Na2O, 0.02% K2O and 0.06% MnO. pH of PMM goethite suspensions and reacting solutions were evaluated using the Model 3340 Jenway ion metre. The cation exchange capacity (CEC) was evaluated by Na saturation method. The specific surface area of the PMM goethite was determined using the standard volumetric Brunauer, Emmett and Teller (BET) method [Citation24]. Here, the determination was by measuring the adsorption of N2 gas on the mineral solid phase at the boiling point of liquid nitrogen [Citation25]. Spectral analysis was done using a JEOL JSM 5900 LV scanning electron microscopy (SEM) with Oxford INCA energy dispersive spectroscopy (EDS) [Citation26,Citation27]. The samples were viewed, and secondary electron images were acquired at low-vacuum control pressure. The point of zero salt effect (PZSE) of the PMM goethite was carried out using standard laboratory procedures [Citation28,Citation29]. Potentiometric titration was conducted after equilibration of 1% (by mass) of PMM goethite suspensions. 1:1 electrolyte solutions initially adjusted to pH ranges near the PZSE were used as references.
2.2. Synthesis of CuO-coated PMM goethite
The procedure was as reported by Eren [Citation30]. The Setting of CuO-coated goethite was by precipitating the copper ions on the PMM goethite surface. Sodium hydroxide addition tracked by thermal treatment was conducted. PMM CuO-coated goethite was set by precipitating the copper ions on its surface. This was tracked by thermal treatment after treatment with sodium hydroxide [Citation31,Citation30]. 0.20 g of PMM goethite was mixed with 100 mL 1 M Cu(NO3)2 solution and 180 mL of 2 M NaOH solution. These were freshly prepared, and the reaction mixture maintained at 90°C for 48 h. The PMM goethite activated with the NaOH solution was dispersed into 150 mL of 0.10 M Cu(NO3)2 solution. 0.10 M NaOH aqueous solution of 300 μL was titrated at the rate of 1 mL/h. Carbonate salt formation was minimized, by titrating under nitrogen flow condition [Citation32,Citation33]. To free the content from NO3– ions, precipitate centrifugation and washing were carried out. Heating the solid for 4 h in air at 700 K led to the formation of CuO goethite. PMM goethite was verified from the X-ray diffraction (XRD) patterns of the product.
2.3. Batch mode adsorption experiments
Hg(II) solutions (10–40 mg L–1) were reacted with 1% each of PMM CuO-coated goethite suspension. To determine the effect of metal initial concentration, the suspension was made into 50 mL and subsequently equilibrated for 24 h at pH = 4–8. A range of solid concentrations of CuO-coated goethite (2 g L–1, –10 g L–1) made into 50 mL were reacted with solutions containing Hg (II) ions (10 mg L–1, –40 mg L–1) and equilibrated for 24 h at pH = 4–8. This was used to investigate the particle concentration effect (Cp). Mercury concentrations (10–40 mg L–1) was reacted with 1% PMM CuO-coated goethite at pH = 4–8. The suspensions were made into 50 mL and aged from 24 to 720 h. This was used to investigate the effect of ageing. All experiments in triplicates were conducted under reducing conditions at ambient temperature. Kinetic experiments carried out to deduce the reaction mechanisms involved in mercury removal involved several kinetic models. The proton coefficient otherwise known as the proton exchange isotherm was derived from the change of pH versus Log Kd plot. This was based on Freundlich isotherm. Freundlich isotherm was used to describe adsorption of Hg2+. This is due to the fact this isotherm is suitable for heterogeneous surfaces over a wide range of solute concentrations [Citation5,Citation34,Citation6] as given by
(1)
(1)
(2)
(2)
Here, SOH equals into the mineral surface-reactive site, SO– equals into the surface-bound Pb2+, log Kp equals into the apparent equilibrium-binding constant and α equals into the coefficient of protonation. This represents the number of protons displaced when one mole of metal binds to the mineral surface [Citation5,Citation35]. Proton coefficients were obtained by graphical analysis using logKd versus pH plots. This plot gave α the proton coefficient as its slope. To determine this coefficient, 1% PMM CuO-coated goethite suspension was regulated to required pH, made into 50 mL, and reacted with a mercury ion solution of 10 mg L–1. Secondly, the mass transfer rate and intraparticle diffusion were derived from Equations (3,4,5):
(3)
(3)
Here, C0 equals the initial metal concentration (mg L–1) at time t = 0; Ct equals the concentration (mg L–1) at time t; V equals the total CuO-coated goethite suspension volume and m is the weight of the adsorbent (g) [Citation5,Citation23]. The kinetics of adsorbate to the mineral surface binding sites was controlled by the mass transfer constant constant Kf. Here, Ct/C0 vs. time provided the slopes of the curves derived from Equation (4) [Citation36,Citation5]:
(4)
(4)
Here, Ct and C0 denote the initial concentrations of Hg(II) at time t, Ss equals the exposed specific surface area of the adsorbent, and Kf denotes the coefficient of mass transfer [Citation37]. These models as reviewed by Qiu et al. [Citation38] and derived from the Freundlich isotherm were adopted to describe adsorption of Hg(II) ions. These models were considered appropriate for heterogeneous surfaces over a wide range of solute concentrations [Citation23]. To investigate the action of intraparticle diffusion on Hg(II) adsorption, the Weber–Morris model was used [Citation39,Citation23] as given in Equation (5):
(5)
(5)
Here, Ki equals the intraparticle diffusion constant (mg g–1 min) and the intercept (C) represents the effect of the layer boundary. Ki value is derived from the slope (Ki) of the plots of qt vs. t0.5. A linear plot of qt versus t0.5 indicates diffusion of intraparticle was involved in the process of adsorption. The rate-controlling step was regulated by intraparticle diffusion if these lines pass through the origin. Otherwise, this is indicative of some degree of boundary layer control. For these reaction mechanisms, 1% CuO-coated goethite was reacted with 10 mg L–1 Hg2+ ions solution. The content was made into 50 mL and regulated to required ph. Amounts of Hg(II) ions remaining in solution was determined after 2nd, 4th, 6th, 8th, 12th, 18th, and 24th hour. These studies were done in triplicates at ambient temperature. A 0.2 µm pore size cellulose acetate filter was used in the supernatant and content analysed for Hg(II) ions, using a Hitachi Atomic Absorption Spectrophotometer (HG-AAS).
3. Results and discussion
In this study, PMM goethite contained 0.14 ppm Cu, 0.18 ppm Zn, 0.01–100.00 µm particle size range 1.83% colloid, 3.00 pH, 97.00 specific surface area(m2/g), 56.00 and 7.13 point of zero salt effect. The systems involved in this study have been characterized and summarized (Table and Figures ). The X-ray diffraction spectrum indicated goethite as the key constituent. The EDS spectrum and SEM morphology indicated the presence of hydrous Iron Oxide and sulphur as a trace element. This agrees with the chemical constituents of the PMM goethite and micron range values of the particle sizes. The point zero charge pHpzc is also known as the point of zero salt effect and the surface area of adsorbents are important characterization concerning adsorption. Here, the adsorbent surface negative charge equals into the positive charges. Therefore, it presents the divide of the positive and negative charge on the external layer of the adsorbent. The adsorbent external layer governed the surface of mineral accessible for reaction.
Figure 1. X-ray diffraction for PMM goethite.
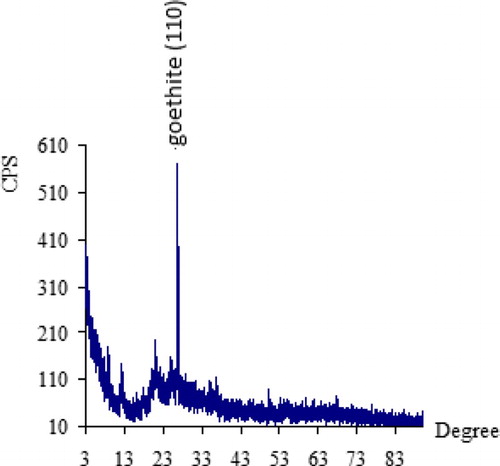
Figure 2. (a)EDS-SEM for PMM goethite showing element peaks and particle sizes. (b): X-ray diffraction of synthetic CuO–goethite showing peaks.
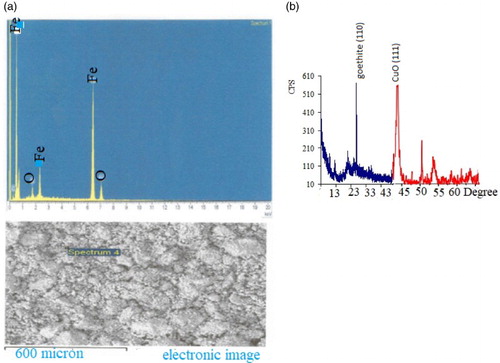
Figure 3. Particle size distribution of PMM goethite at suspension pH.
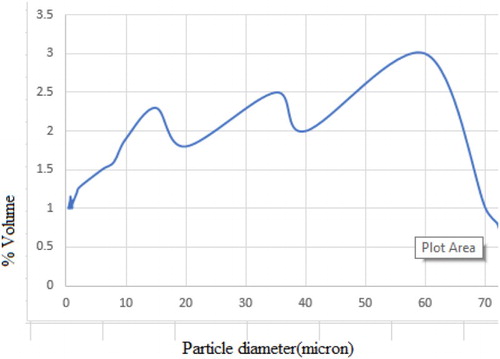
Figure 4. Plot of net proton charge versus suspension pH for PZSE.
Table 1. Characteristics of PMM goethite.
Proton coefficient ((α)) was based on a theoretical framework given by Equations (1, 2), predicted and derived from the plot (Figure ), (Table ). The value was 0.68, <1. Intraparticle diffusion was based on a theoretical framework given by Equation (5), predicted and derived from the plot (Figure andTable ). The intraparticle diffusion constant derived from the slope was 13.36 (mg g–1) min0.5 and the intercept C was 444.45, ≠ 0. This plot consists of two linear parts with the first part representing the external mass transfer. The S-curve adsorption pattern plateaued after the 720th minute. The second part represents the intraparticle diffusion.
Figure 5. Plot of Log Kd (adsorption capacity) versus final pH for proton coefficient.
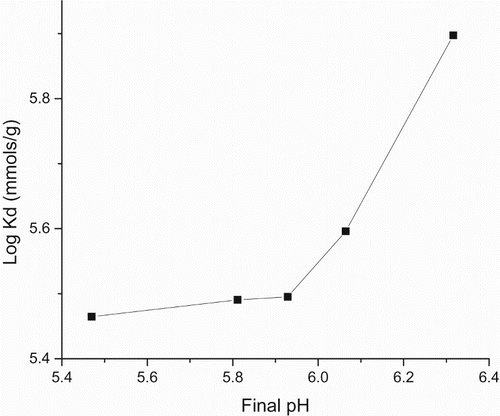
Figure 6. Plot of adsorption capacity versus time for intraparticle diffusion.
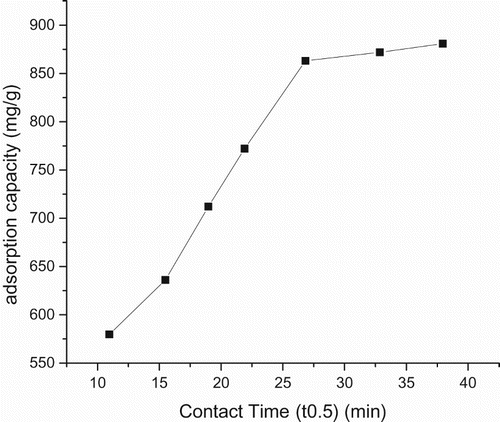
Table 2. Statistical presentation of proton coefficient derived from Figure .
Table 3. Statistical presentation of intraparticle diffusion data derived from linear fit of Figure .
The mass transfer rate predicted from Equations (3, 4) and derived from Figure is given (Tables –). Also this consists of two linear parts with the first part lower than the second. The second linear part started after the 15th hour. Figure consists of three linear parts. There was an increase in capacity of adsorption as metal initial concentration was increased. Adsorption capacity increased from 185 to 300 mg g–1over the range of metal concentration investigated. The maximum adsorption capacity occurred at 40 mg L–1. Figure consists of four linear parts occurring in a sinusoidal pattern. There was a decrease in capacity of adsorption as particle concentration was increased into 4 g L–1, thereafter increasing as solid concentration was increased into 6 g L–1, decreasing thereafter onto 8 g L–1 and increasing over the rest of the solid concentration. The highest adsorption capacity was at 10 g L–1. Residence time, i.e. ageing, increased with an increase in adsorption capacity. This plot consists of two linear parts. The S-curve adsorption pattern plateaued after the 576th hour. (Figure ). The rate of removal of Hg(II) ions increased from 570 to 870 mg g–1over the range of residence time investigated. Adsorption capacity generally increased with an increase in pH. In acidic medium, the adsorption capacity was low, but increased with an increase in pH. The rate of removal of Hg(II) ions increased from 720 to 790 mg g–1over the range of pH investigated (Figure ).
Figure 7. Plot of Ct/C0 versus contact time for mass transfer rates.
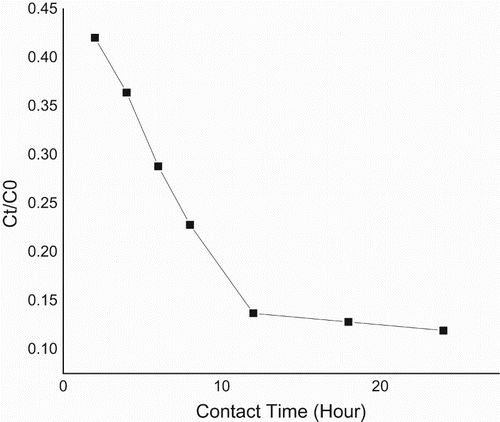
Figure 8. Plot of adsorption capacity versus initial metal concentration at pH = 4 and solid concentration = 10 g/L.
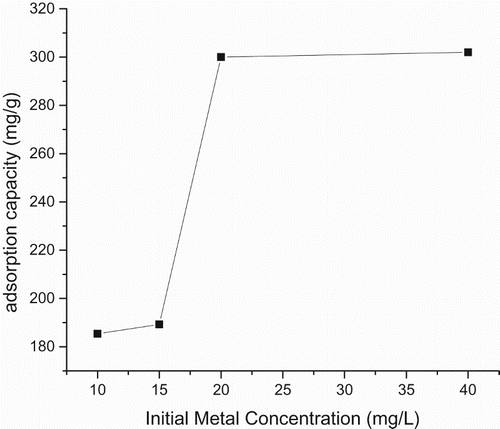
Figure 9. Plot of adsorption capacity versus Cp at pH = 4.
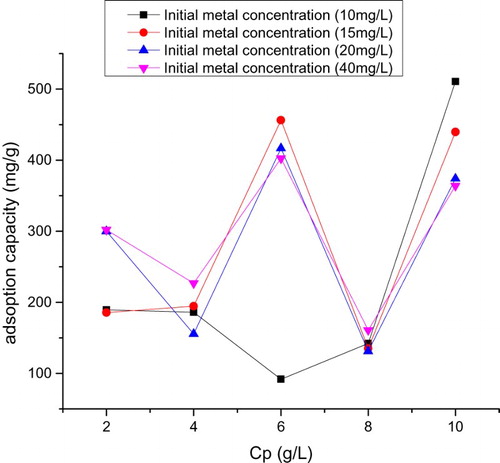
Figure 10. Plot of adsorption capacity versus residence time–ageing for mineral systems at pH = 4 and 10 mg L–1 metal concentration.
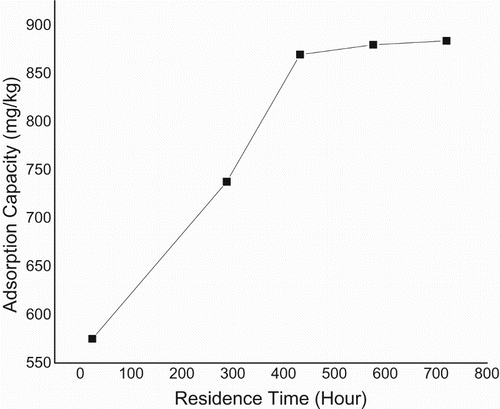
Figure 11. Plot of adsorption capacity versus initial pH at 10 mg L–1 metal concentration.
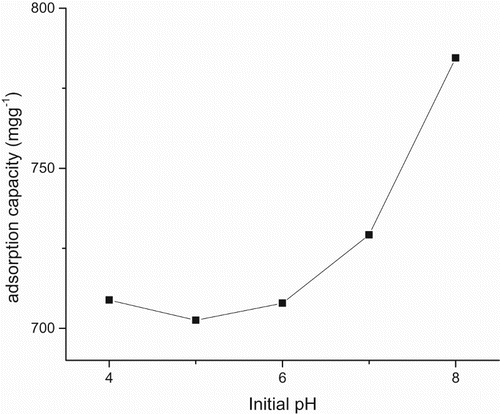
Table 4. Mass transfer rates derived from Figure .
Table 5. Gauss fit for Figure .
In comparison with cited literature data, CuO–goethite composite stands out as a novel adsorbent, useful in the treatment of mercury-contaminated aqueous solution. The proton coefficient, intraparticle diffusion and mass transfer rates were used to discuss the reaction mechanism. In previous studies deprived of coated CuO on goethite, proton coefficient was greater than one [Citation5,Citation6]. Here, α for CuO-coated goethite was less than one and slightly greater than the value recorded by Egirani et al. [Citation6] for goethite (Table ). This suggested that protonation was controlled by strongly acidic sites in the CuO goethite. There was an indication that protonation was lowered in the presence of CuO coating. A coated medium could mask the sites of acidity on the edges and planar surfaces of goethite. Intraparticle diffusion was involved in the adsorption process (Figure and Table ). However, this was not a rate-limiting reaction. Also, there was an indication of boundary layer control. When compared with previous studies [Citation19,Citation6], the slope and intercept for the uncoated goethite were higher than the coated goethite, thus suggesting that the presence of CuO coating enhanced intraparticle diffusion. When compared with previous studies, the mass transfer rates for the uncoated goethite were lower than results for the coated goethite [Citation6], thus suggesting the enhancement of mass transfer of adsorbate to the external layer of the adsorbent (Figure ). Based on the plot of adsorption capacity versus time, it was obvious that mercury adsorption was dependent on contact time (Figure ). The dependence on time attribute of Hg(II) removal was assessed from 2 to 24 h at ambient temperature. This was at an initial mercury concentration of 10 mg L–1 at pH 4. This reaction pattern plateaued after the 12th-hour at 860 mg g–1.
Based on Figure , the capacity of adsorption increased with an increase in contact time and the optimum adsorption (i.e. 875 mg g–1) was at the 24th hour. The adsorption rate was initially fast and the capacity of adsorption increased over time. The initial speedy adsorption of Hg(II) ions in the first step may be ascribed to larger numbers of active adsorption sites and were related to outer-sphere complexation [Citation5,Citation40]. The investigation of different metal concentrations was relevant in this report because most contaminated aquatic systems offer different concentrations of metal ions. The increase in adsorption capacity as metal concentration was increased indicated that the active and reactive sites of the PMM CuO-coated goethite were not yet saturated (Figure ). Egirani et al. [Citation6] reported a decrease in mercury adsorption as initial metal concentration was increased for CuO-coated goethite. It could be suggested that the mass transfer rate of metal ions in the solid-solution phase was controlled by a concentration gradient. Here, the number of active sites is fixed and Akpomie et al. [Citation10] reported a decrease in adsorption capacity for some heavy metals adsorbed on montmorillonite as the metal initial concentration was raised from 100 to 300 mg L–1. In this report, the change in the linear plot after the 20 mg L–1 point indicated a gradual plateauing of the capacity of adsorption due to gradual saturation of the active and reactive sites.
The complex characteristics of adsorption over the range of Cp investigated were different from the report of Egirani et al. [Citation19] for uncoated goethite (Figure ). In addition, this was different from the report of Egirani et al. [Citation6] for CuO-coated goethite, where an increase in the concentration of particle led to a decrease in adsorption. The Decrease in adsorption capacity, in this case, suggested an increase in particle size and aggregation of the mineral system as the reaction proceeded. In addition, there was a decrease in the surface area and the reactive sites on the surfaces of PMM CuO- goethite. Increase in adsorption capacity may be assigned to effect of concentration gradient effect. This means that for every reaction pressure exacted on the system due to adsorption, there was a corresponding concentration gradient effect on the system. 10 mg L–1 metal concentration provided the highest adsorption capacity of 500 mg g–1 and 20 mg L–1 offered the lowest adsorption capacity of 150 mg L–1, thus indicating that this metal concentration was the minimum for a minimal adsorption capacity of mercury ions in this regard.
Egirani et al. [Citation19] reported that adsorption capacity increased as ageing increased (). This was for over the same range of residence time investigated. The increase was from 972 mg g–1to 982 mg g–1. In this report, adsorption capacity was higher over the same range of residence time investigated. This higher magnitude was essentially controlled by hydrolysis and reactive support of CuO coating inclusive increased reorganization of active sites. Given that mercury removal was pH dependent, its investigation was necessary in this report. The protonation and hydroxylation of a mineral surface controlled the adsorption process as pH was increased (Figure ). This was similar to the reports from Egirani et al. [Citation5–Citation7]. Also, Akpomie et al. [Citation10] reported a similar trend for adsorption of heavy metals on uncoated montmorillonite. As pH was increased, there was a decrease in protonation and enhancement in hydroxylation. This favoured mercury adsorption. Increase in the adsorption of mercury by biomass, maximizing at pH = 8 and plateauing thereafter has been reported [Citation9]. This paper reports a similar pattern of adsorption. A huge amount of quantities of protons competed with the mercury ions for reactive and active sites at low or acidic pH. However, there was a decrease in the number of available protonation sites as pH was increased. This gave rise to increased hydroxylation on the CuO-coated goethite surfaces.
4. Conclusions
The magnitude of adsorption pattern as reaction time, pH and mercury concentration was increased indicated that these were controlled by reactive support of CuO coating. pH provided the highest adsorption capacity of 784.5 mg g–1 at pH = 8 followed by ageing with an adsorption capacity of 575 mg g–1. The effect of solid concentration provided the least adsorption capacity. The synthesis of PMM CuO on goethite was done and characterization conducted using standard laboratory procedures. Batch mode systems were used to test the adsorption of Hg(II) ions by PMM CuO-coated goethite. Reaction mechanism of adsorption tested included proton coefficient that was less than one, an intraparticle diffusion that was controlled by boundary layer and mass transfer rates that were lower than those of previous studies. There was an increase in adsorption capacity as metal concentration was increased, thus indicating that the active and reactive sites of the PMM CuO-coated goethite were not yet saturated. The sinusoidal characteristics of adsorption over the range of Cp investigated indicated that for every reaction pressure exacted on the system due to adsorption, there was a corresponding concentration gradient effect on the system. Decrease in adsorption capacity in this scenario suggested an increase in particle size and aggregation of the mineral system as the reaction proceeded. Ageing increased adsorption of mercury ions. The rate of removal of Hg(II) ions increased from 570 to 870 mg g–1 over the range of residence time investigated. The magnitude of adsorption, when compared with that of the previous studies, indicated that it was essentially controlled by hydrolysis, reactive support of CuO coating and increased reorganization of active sites. In acidic medium, the adsorption capacity was low, but increased with increases in pH. Here, adsorption increased from 720 to 790 mg g–1 over the range of pH investigated. As pH was increased, there was a decrease in protonation and enhancement in hydroxylation. This led to an increase in adsorption capacity of mercury ions.
Acknowledgements
The authors are grateful to the Niger Delta University for the usual research allowances provided in completing this research.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Nanfe. R. Poyi http://orcid.org/0000-0001-9220-2176
Napoleon Wessey http://orcid.org/0000-0001-8168-1116
Shukla Acharjee http://orcid.org/0000-0001-5426-1087
References
- Goel J, Kadirvelu K, Rajagopal C, et al. Removal of mercury(II) from aqueous solution by adsorption on carbon aerogel: response surface methodological approach. Carbon N Y. 2005;43:97–200. doi: 10.1016/j.carbon.2004.08.002
- Ballester A, Otero E, González F. Mercury extraction from cinnabar ores using hydrobromic acid: previous studies. Hydrometallurgy. 1988;21:127–143. doi: 10.1016/0304-386X(88)90001-1
- van der Sloot HA. Systematic leaching behavior of trace elements from construction materials and waste materials. Stud Environ Sci. 1991;48:19–36. doi: 10.1016/S0166-1116(08)70388-1
- Husein DZ. Adsorption and removal of mercury ions from aqueous solution using raw and chemically modified Egyptian mandarin peel. Desalin Water Treat. 2013;51:6761–6769. doi: 10.1080/19443994.2013.801793
- François F, Lombard C, Guigner J-M, et al. Isolation and characterization of environmental bacteria capable of extracellular biosorption of mercury. Appl Environ Microbiol.; 78(2011):1097–1106.
- Tunsu C, Ekberg C, Retegan T. Characterization and leaching of real fluorescent lamp waste for the recovery of rare earth metals and mercury. Hydrometallurgy. 2014;144:91–98. doi: 10.1016/j.hydromet.2014.01.019
- Egirani DE, Andrews JE, Baker AR. Mercury removal from aqueous solution by mixed mineral systems I. Reactivity and removal kinetics. IOSR-JESTFT. 2013a;4(2):73–81. doi: 10.9790/2402-0427381
- Egirani DE, Andrews JE, Baker AR. Mercury removal from aqueous solution using mixed mineral systems injected with iron sulfide under sulfidic- anoxic conditions 1: reactivity and removal kinetics. J Chem Biol Phys Sci. 2014;4(2):991–1005.
- Egirani DE, Wessey N, Aderogba A. Effect of mineral systems on mercury removal from aqueous solution part ii. J Appl Chem Sci Int. 2016;5:188–194.
- Raza MH, Sadiq A, Farooq U, et al. Phragmites karka as a biosorbent for the removal of mercury metal ions from aqueous solution: effect of modification. J Chem. 2015. doi: 10.1155/2015/293054
- Huang S, Lin G. Biosorption of Hg(II) and Cu(II) by biomass of dried Sargassum fusiforme in aquatic solution. J Environ Health Sci Eng. 2015;13:21. doi: 10.1186/s40201-015-0180-4
- Akpomie KG, Dawodu FA, Adebowale KO. Mechanism on the sorption of heavy metals from binary-solution by a low-cost montmorillonite and its desorption potential. Alexandria Eng J. 2015;54:757–767. doi: 10.1016/j.aej.2015.03.025
- Keren R. Reduction of the cation-exchange capacity of goethite by take-up of hydroxy-a1 polymers. Clays Clay Miner. 1986;34:534–538. doi: 10.1346/CCMN.1986.0340506
- Eze SO, Igwe JC, Dipo D. Effect of particle size on adsorption of heavy metals using chemically modified and unmodified fluted pumpkin and broad-leafed pumpkin pods. Int J Biol Chem Sci. 2013;7:852–860.
- Mishra R, Padil VVT, Militky J. Removal of mercury from aqueous environment by jute nanofiber. J Fiber Bioeng Inform. 2013;6(2):175–184. doi: 10.3993/jfbi06201306
- Bonnissel-Gissinger P, Alnot M, Lickes JP, et al. Modeling the adsorption of mercury (II) on (Hydr)oxides II: alpha-FeOOH (Goethite) and amorphous silica. J. Colloid Interf Sci. 1999;215:313–322. doi: 10.1006/jcis.1999.6263
- Arshadi M, Khalafi-Nezhad A, Firouzabadi H, et al. Adsorption of mercury ions from wastewater by a hyperbranched and multifunctionalized dendrimer modified mixed-oxides nanoparticles. J. Colloid Interf Sci. 2017;505:93–306. doi: 10.1016/j.jcis.2017.05.052
- Hua M, Zhang S, Pan B, et al. Heavy metal removal from water/wastewater by nanosized metal oxides: a review. J Hazard Mater.; 211–212(2012):317–331.
- Yadav SK, Singh DK, Sinha S. Adsorptive removal of Hg(II) from synthetic and real aqueous solutions using modified papaya seed. J Dispers Sci Technol.; 37(2014):1613–1622.
- Dos SMB, Lea KZ, Oliveira FJ, et al. Efficient removal of mercury from aqueous solutions and industrial effluent. J. Environ Sci Health. 2015;50(12):1230–1240. doi: 10.1080/10934529.2015.1055147
- Egirani DE, Baker AR, Andrews JE. Mercury removal from aqueous solution by mixed mineral systems II. The role of solution composition and ageing. IOSR-JESTFT. 2013b;4:49–55. doi: 10.9790/2402-0424955
- Santana AJ, dos Santos WN, Silva LO, et al. Removal of mercury(II) ions in aqueous solution using the peel biomass of Pachira aquatica Aubl: kinetics and adsorption equilibrium studies. Environ Monit Assess. 2016;188(5):293. doi: 10.1007/s10661-016-5266-7
- Saleh TA, Seri A, Tuzen M. Optimization of parameters with experimental design for the adsorption of mercury using polyethyleneimines modified-activated carbon. J Environ Chem Eng. 2017;5:1079–1088. doi: 10.1016/j.jece.2017.01.032
- Uzum Ç, Shahwan T, Eroğlu AE, et al. Synthesis and characterization of goethite-supported zero-valent iron nanoparticles and their application for the removal of aqueous Cu2+ and Co2+ ions. Appl Clay Sci. 2009;43:172–181. doi: 10.1016/j.clay.2008.07.030
- Anagnostopoulos VA, Panagiotis ID, Karapanagioti HK, et al. Removal of mercury from aqueous solutions by malt spent rootlets. Chem Eng J. 2012;213:135–141. doi: 10.1016/j.cej.2012.09.074
- Brunauer S, Emmett PH, Teller E. Adsorption of gases in multimolecular layers. J Am Chem Soc. 1938;60:309–319. doi: 10.1021/ja01269a023
- Lowell S, Shields JE. Powder surface area and porosity. Springer; 1991.
- Karickhoff SW, Bailey GW. Optical absorption spectra of clay minerals. Clays Clay Miner. 1973;21:59–70. doi: 10.1346/CCMN.1973.0210109
- Janaki RSG, Sreenivas K, Sivasamy R. Hyperspectral analysis of clay minerals. Int Archives photogr Remote SensSpatial Inform Sci. 2014;40:443–446. doi: 10.5194/isprsarchives-XL-8-443-2014
- Alves ME, Lavorenti A. Point of zero salt effect: relationship with clay mineralogy of representative soils of Sao Paulo State, Brazil. Pedosphere. 2005;15:545–553.
- Bolan NS, Syers JK, Tillman RW. Ionic strength effects on surface charge and adsorption of phosphate and sulphate by soils. Eur J Soil Sci.; 37(1986):379–388.
- Eren E. Removal of heavy metal ions by Unye (Turkey) bentonite in iron and magnesium oxide-coated forms. J. Hazard. Mater. 2009;165:63–70. doi: 10.1016/j.jhazmat.2008.09.066
- Phiwdanga K, Suphankija S, Mekprasarta W, et al. Synthesis of CuO nanoparticles by precipitation method using different precursors. Energy Procedia. 2013;34:740–745. doi: 10.1016/j.egypro.2013.06.808
- Maruthupandy M, Zuo Y, Chen J-S, et al. Synthesis of metal oxide nanoparticles (CuO and ZnO NPs) via biological template and their optical sensor applications: the key laboratory of environment friendly. Applied Surf. Sci. 2017;397:167–174. doi: 10.1016/j.apsusc.2016.11.118
- Wang D, Lin Z, Wang T, et al. Where does the toxicity of metal oxide nanoparticles come from: the nanoparticles, the ions, or a combination of both? J. Hazard. Materials. 2016;308:328–334. doi: 10.1016/j.jhazmat.2016.01.066
- Campos K, Guibal E, Peirano F, et al. Mercury sorption on chitosan. Adv Mater Res. 2007;20-21:635–638. doi: 10.4028/www.scientific.net/AMR.20-21.635
- Contescu C, Jagiello J, Schwarz JA. Heterogeneity of proton binding sites at the oxide/solution interface. Langmuir. 1993;9(7):1754–1765. doi: 10.1021/la00031a024
- Egirani DE, Baker AR, Andrews JE. Copper and zinc removal from aqueous solution by mixed mineral systems I: reactivity and removal kinetics. J. Colloid Interf Sci. 2005;291:319–325. doi: 10.1016/j.jcis.2005.05.007
- Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24:1504–1534. doi: 10.1002/adma.201104763
- Qiu H, Lu LV, Bing-cai PAN, et al. Critical review in adsorption kinetic models. J. Zhejiang Univ Sci A. 2009;10:716–724. doi: 10.1631/jzus.A0820524
- Feng Q, Lin Q, Gong F, et al. Adsorption of lead and mercury by rice husk ash. J Colloid Interf Sci. 2004;278:1–8. doi: 10.1016/j.jcis.2004.05.030
- Tuazon BG, Yalçin E, Başaran G, et al. Equilibrium and kinetic studies on biosorption of Hg(II), Cd(II) and Pb(II) ions onto microalgae Chlamydomonas reinhardtii. J Environ Mgt. 2005;77:85–92. doi: 10.1016/j.jenvman.2005.01.028