
ABSTRACT
In this work, the molecular spectroscopic technique was used in a nonconventional way to predict the drug mechanism action of the drug compound, Ethionamide. The formatted molecular charge levels, due to the injection of substitutions in suitable positions, were measured. The molecular geometry distortion by the adoption of ligands in the ring system was noted at the nodal points of the core and the change of force constant of the heterogeneous intra-molecular assembly. The activity of the compositional parts of the molecule was purposively monitored from which the entire core and allied parts of the molecule were evaluated by the vibrational characteristics. The molecular reaction mechanism for saturating drug potential was observed from the chemical shift of core and substituted carbons. The fabricated charge transfer complex was identified and recognized from the base and ligand-binding structure. The major part of the exchange of electro-chemical quantized energy in non-bonding molecular orbitals for arranging biological activity in the compound was measured. The rule of five and Lipinski parameters to recognize the drug quality were checked and estimated.
1. Introduction
Ethionamide is called 2-Ethyl-4-thiopyridylamide or 2-ethylthioisonicotinamide, which is the combination of pyridine ring along with ethyl–methyl chain and thioamide [Citation1]. Isonicotinamide is an isomer of nicotinamide which has anti-inflammatory actions and primarily used for the treatment of niacin deficiency [Citation2]. When it is substituted with ethyl and thioamide groups, it forms Ethionamide and simultaneously its pharmaceutical characteristics are changed as antibiotics which are used for the treatment of tuberculosis [Citation3]. The drug Ethinamate is bacteriostatic against M. tuberculosis and resists the further growth of Mycobacterium tuberculosis organisms and terminates it from the lungs. It is a second-order anti-tubercular representative, which restrains mycolic acid amalgamation. It is also used for the treatment of leprosy [Citation4].
It is an antibacterial agent that treats the bacterial infections in the interior and exterior parts of the human body [Citation5]. It is an azacycle compound substituted with organo-sulphur and hydrocarbon groups. The antibiotic prototype special species, Ethionamide is an anti-tubercular prodrug used for the second-order therapy to cure the MDR-TB which is being bio-activated by the bio-sensitive mycobacterial mono-oxygenase [Citation6]. It is a novel pharmaceutical derivative, which is a potentially functional drug to fight tuberculosis also. The drug activity is enabled in the structural geometry by the application of ligand injection of sulphur group along with amine group [Citation7].
The computational tools in theoretical pharmacology play a vital role in recognizing the drug properties and enabling the interpretation on the drug applications, which is mainly used for the fabrication of a better new drug with multifunctional quality. The vibrational analysis, along with the structural activity, is successfully employed to identify the root cause of drug mechanism to act as useful antibiotic species. The benzene derivatives are normally the successful drugs and their drug potential is improved up to the level of antibiotic by the thio-bridge linked with amide bonds [Citation8].
In spite of its drug popularity, even making thorough investigation on literatures, there is no research work carried out to analyse the structural activity on the antibiotic and biological properties of Ethionamide. So, in this work, detailed vibrational, NMR, QSAR, hyperactive frontier molecular and NBMO studies were performed to interpret the entire drug properties.
2. Experimental profile
Physical state of compound:
The compound, Ethionamide was prepared in solid phase and it is found to be of spectroscopic grade.
Recording summary:
The FT technique used IR Bruker IFS 66V spectrometer to record spectra and FT-Raman module FRA 106 Raman was used to record FT-Raman spectra.
The high-resolution 1HNMR and 13CNMR spectra were recorded in different sequences for filtration using 300 and 75 MHz FT-NMR spectrometers.
The UV-Vis absorption spectrum was recorded with 100% transmission coefficient in the range of 100 nm to 700 nm, with the scanning interval of 0.5 nm, by using the UV-1700 sequence instrument.
3. Computational details
Computational software was used for carrying out the geometry calculations: Gaussian 09 D. 01.version software in core i7 computer.
Calculation Methods: B3LYP and B3PW91
Basis sets: 6–31++G(d,p) and 6-311G(d,p)
Spectral simulations: FT-IR, FT-Raman, UV-Visible, ECD, VCD and 1H and 13C NMR.
4. Results and discussion
4.1. Molecular geometry deformation analysis
The present molecular base was pyridine in which the ethyl–methyl chain and thiocarbonamide groups were injected at the first and third positions of the ring, as shown in Figure . According to the uncertainty of chemical bonds, the imine group was formed itself of the ring and relatively, two C=C bonds were formed. Usually, since the presence of nitrogen in the ring, pyridine is more reactive and enables to react with nucleophiles, causing medicinal effect naturally. Two important ligand groups were identified at ortho- and parapositions which normally produced more reactive antibiotic property in the compound [Citation9]. The present molecular structure is in (111) crystal view and is shown in Figure in which the planes of the molecule were arranged perpendicularly to the plane of the crystal. The optimized geometry of the molecular structure is presented in Table .
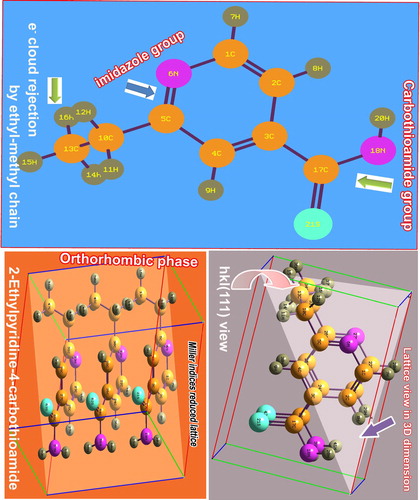
Table 1. Optimized geometrical parameters of Ethionamide.
The structural deformation usually intimates the impact of ligand groups in different positions of the ring base and the rate of change of elemental distortion. The substitutional impact is justified by the rate of change of bond length, bond angle and force constant of the specific inter- and intra-bonds in the molecule. In this alternation of geometrical parameters, the elemental instantaneous strain is observed varying with respect to the substituted atoms or molecules. Such electrostatic chemical strain usually initiates the chemical property and it can be measured from the force constant of the bonds [Citation10].
In the present case, the base was the pyridine ring in which the bond lengths of C1–N6 and C5=N6 were 1.332Å and 1.343Å, respectively. Since the first one was σ bond, its length should be greater than that of the second π-bond. But here, the second one was 0.011Å greater than that of the first bond which was due to the loading of ethyl and methyl chain at C5. In this case, the chemical strain was −4.31 mDyne/A which was more in C(5)–C(10) bond than other existed C–C bonds. In this point, the observed value showed the chemical strain, generated in bridge C–C bond between the chain and ring. Such induced strain releases the static chemical energy from the chain to the ring as a result of which the chemical property was blended with pyridine property.
The bond lengths of C2–C3 and C3–C4 were 1.399Å and 1.396Å, respectively, in which small difference was determined between them due to the σ and π bond flavour. These bonds also have been infected by the chemical strain which is enforced by the loading of thioamide. Here, the bond strain was 27.235 nDyne/A which is so high and it should be due to the exchange of large chemical energy between pyridine ring and thioamide group. Actually, the observed value was negative and the exchange energy was directed only from the ligand group to pyridine ring. The chemical strain generated by ethyl–methyl chain was lesser than that of the thioamide group, which showed the maximum part of change of molecular property by the thioamide group. In this exchange of chemical energy, the negative potential was observed from the ethyl–methyl chain, whereas the positive potential was recognized from the thioamide group. From this, it was well known that, the supplied chemical energy from thioamide to ring was reduced or suppressed by ethyl–methyl chain. In this energy scheme, it was inferred that the basic antiseptic property of pyridine ring was changed to the antibiotic property by controlling the chemical reactivity using ligand groups.
4.2. Mulliken- charged surface analysis
The Mulliken-charged distribution usually detailed the charge depletion taking place among the inter-molecular bonds that assisted to study the chemical reaction stream which facilitated to induce the drug chemical mechanism of action [Citation11]. It is a molecular charge displacement due to the chemical restoring energy or chemical equilibrium enforcement of molecules. It represents prepared chemical energy to equip the optimized chemical property which shapes up the drug potential in the compound.
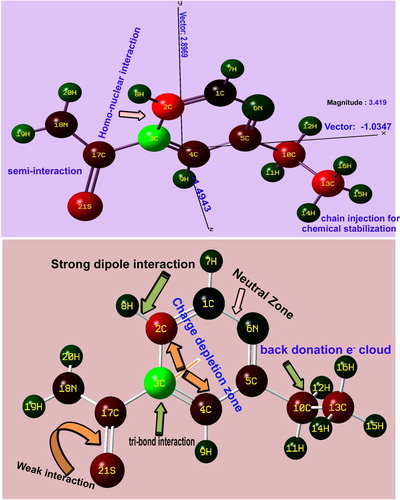
As the couple of ligand groups is injected into the major core compound, the arrangement of chemical potential in terms of asymmetric charge distribution takes place among the atoms or molecules. As in Figure , in the molecular coordinate system, the direction vector is found to be parallel to the plane of the pyridine ring in which the centre of symmetry was on the left moiety of the pyridine ring and it was 3.419 Newton/coulomb. It shows the asymmetrical charge displacement due to the non-primitive molecular charge gradients. The displacement gradient was 2.896 N/C in z-centre. Here, the C3 of core ring was shown to be positive due to the suction of molecular charge domain by the thioamide group from pyridine ring, whereas the C5 was shown to be negative due to the donation of charge realm from the chain to the pyridine ring. This diverted charge volume accumulated over the main core ring whose asymmetric charge reservoir is to produce the drug behaviour particularly, the stabilized antibiotic property. In the molecular topography, the magnitude of strong bonds were purposively organized which was greater than weak bonds in order to make consistency of drug potential. According to the charge gradient on the inter-molecular bonds, the prepared chemical property was found to be pharmaceutically rigid.
4.3. QSAR and biological properties analysis
Usually, the pharmacologically active molecular structure is optimized by physico-chemical properties that are to be sustained, as expressed by Lipinski's rule. The RO5 expected that the privileged absorption or permeation is more probable, while there are less than 5 Hydrogen bond donors, 10 Hydrogen bond acceptors, the monoisotropic mass is less than 500 and the computed hydrophilicity or MLogP is less than 4.15 [Citation12,Citation13]. For the present case, the above parameters have been calculated to be Hbd-1, Hba-2, the isotropic mass-166.05 g/mol. and MlogP-1.46, respectively, as shown in Table . The above-said parameters were found to be the expected limit of RO5 and the present compound was biologically and pharmaceutically active with the substance's toxicity.
Table 2. Structural and biological parameters of Ethionamide.
The biological target is used to describe the resident protein in the human body and its activity is modified by a drug, such as G protein-coupled receptors (GPCR), Enzyme inhibitor, Kinase inhibitor, Protease inhibitor, Ion channel modulator and nuclear receptor ligand.
The GPCR is a big protein family of receptors, which sense the drug molecules outside the cell and activate inside signal transduction pathways. In this case, the same was 0.97 which was moderately active and sufficient signal will be produced when the ligand binds. For the present case, the Enzyme inhibitor value was 0.53, which was observed to be sensitive and the present compound can inhibit an enzyme to control the activity and correct a metabolic imbalance.
The Ion channel modulator usually plays an important role for regulating many physiological and patho-physiological processes through which the disease is treated [Citation14]. Here, the same was calculated for this case to be 1.11. It was considerably high and it acted as a good therapeutic agent to cure the illness. The Kinase inhibitor of the Ethionamide was 1.63 and it was a sufficient parameter to control the kinase activities like cell growth, movement and death in the human body. The Protease inhibitor is the molecule and acted as drug that was found to be 1.68 for this case. It was prohibiting inflammatory diseases by making phosphorylation. The Nuclear receptor ligand is normally working with supplementary proteins to normalize the development, homeostasis, and metabolism of the organism. It was determined to be 1.55 which was greater than unity as in other parameters. It was observed to be a very rational value which can control the expression of genomic DNA.
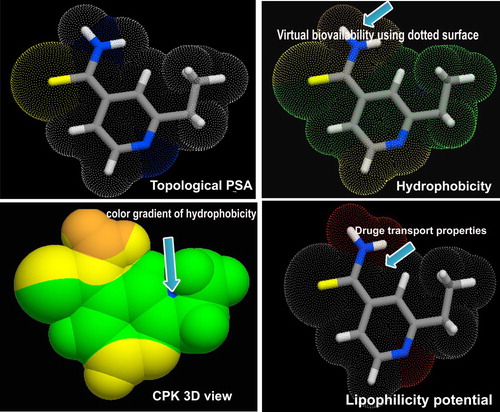
The topological surface area, hydrophobicity, lipophilicity potential and CPK view of the present drug are displayed in Figure . From the Figure, it was found that, the bio-availability was oriented in the amide group which was ensured in the biological parameters and it is also responsible for the major part of drug role. The lipophilicity potential of the compound was shown in and around the nitrogen and it was observed that the amine and imine group in the compound were active and the background of the antibiotic property. Other than such two groups, the potential was observed to be restrained which was shown by a different colour. The CPK view also called electrostatic chemical potential which showed the prepared chemical energy to produce drug activity. Here, it was represented by three different colour gradients which were clearly seen over ethyl–methyl chain, thioamide and ring, respectively.
4.4. Molecular vibrational analysis
4.4.1. Vibrational assignments
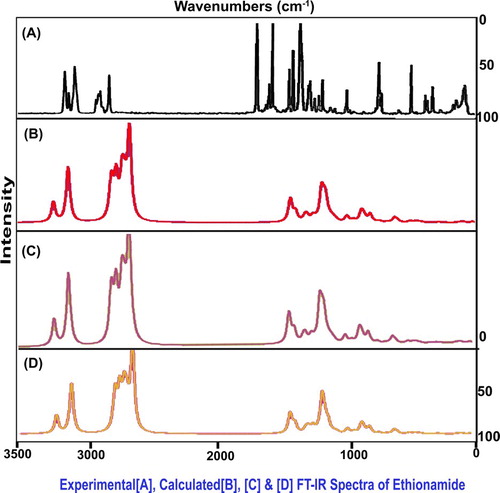
The present molecular structure was designed with 21 atoms which undergo 57 vibrations and all the vibrations were classified at CS point group of symmetry into A′ and A″ irreducible representations. According to the assignment rules, 21 stretching, 18 in plane bending and 18 out-of-plane bending modes of wavenumbers were observed. The experimental and calculated scaled frequencies are obtained in Table and their FT-IR and FT-Raman spectral patterns are displayed in Figures and , respectively.
Table 3. Observed and calculated (HF and DFT) vibrational frequencies of Ethionamide.
4.4.2. C–H vibrations
Even though the present compound is a heterocyclic ring, the C–H stretching multiple peaks are usually arranged in the spectral region 3100–3010 cm−1[Citation15,Citation16] as in the benzene ring. The difference of the expected wavenumber is usually about ± 10 cm−1 between pyridine and benzene rings. Here, the same wavenumbers were observed at 2970, 2950 and 2940 cm−1 which were found to be shifted to well below the expected level. Since two separate ligand groups were substituted ortho- and parapositions, this type of hold back in observed frequency was expected. The related in-plane and out-of-plane bending peaks were observed at 1060, 1010 and 1005 cm−1 and 865, 810 and 805 cm−1, respectively. But, the C–H in-plane and out-of-plane bending vibrations for pyridine ring were usually observed in the region 1100–1000 cm−1 and 850–690 cm−1, respectively [Citation17]. As per the expected region, both the bending modes have been experimentally restricted within the expected region which is dissimilar to the stretching modes. Usually, the impact of substitutions has been observed in the bending modes of core vibrations whereas in this case, the ligand impact was realized in stretching modes which was by the heteronuclear bridge effect.
4.4.3. CC and CN vibrations
Due to the nitrogen bonding, the heterocyclic compound, pyridine, has C=N and C=C stretching vibrations with a strong intensity about 100 cm−1 apart. These two absorption bands are normally found in the spectral region 1615–1575 cm−1 and 1520–1465 cm−1, respectively [Citation18,Citation19] and also the higher-frequency band often having another medium-intensity band on its low-frequency side which is found at 1590–1555 cm−1. Here, the C=N and C=C stretching modes were shown to be at 1660 cm−1 and 1655 and1610 cm−1, respectively. In this case, certain setback would have been expected in the core stretching bands since the impact of injected ligands on base ring. But, instead of that, the elevation in stretching bands was observed which showed the active core part and thereby it participated in the molecular property preparation.
The corresponding stretching of C–N and C–C stretching modes was taking place at 1400–1240 cm−1 and 1300–1100 cm−1, respectively [Citation20]. But, in this case, the same vibrations were taking place at 1600 cm−1 and 1550 and 1490 cm−1, respectively. These vibrational bands were also elevated to well above the expected region and this state in vibrational pattern illustrated that the chemical potential was partially transferred to the ring simultaneously which was supported by the vibrational domain.
4.4.4. Thioamide vibrations
When thio-carbonyl group is directly attached to the nitrogen atom; N–C=S, the stretching modes of the C=S portion are strongly coupled to that of C–N part of it is the direct consequence of which several bands, may, at least partly, be associated with the C=S stretching vibrations. Hence, the thioamide has C=S stretching mode in the region 1570–1395 cm−1[Citation21,Citation22]. For this case, the same stretching wavenumber was identified at 1480 cm−1 and it in plane bending mode was shown at 570 cm−1. Due to its strong position, its stretching band was observed at the exact location of expected limit. This emphasized the utmost involvement in the part of drug property. For thioamide case, the N–C=S stretching can be observed with a strong intensity in the region 1140–950 cm−1[Citation23] whereas for the present case, the same stretching mode was observed with a weak intensity at 1260 cm−1. This band was purposively moved up to the higher expected region and it was mainly due to sulphur support.
The secondary amines are having absorption bands in the region 3450–3300 cm−1 for N–H stretching wavenumber [Citation24]. In this molecule, the N–H stretching bands were observed with a strong intensity at 3140 and 3135 cm−1 in IR as well as Raman spectra. According to the literature, these bands were located well below the expected range of wavenumbers, which was due to the chemical energy absorption of C=S.
In the case of thioamide, the N–H scissoring and N–H out-of-plane vibrations (Twisting modes) ranged 1650–1590 cm−1 and 1305–1085 cm−1, respectively [Citation25]. According to the vibrational assignments of N–H group of the present case, the N-H in-plane and out-of-plane bending modes were found at 1420 and 1280 cm−1 and 875 and 870 cm−1, respectively. Both the vibrational bands were well dislocated from the expected region as in the case of NH stretching. The C=S suppressions on these bending vibrations were expected and the same trend was taking place.
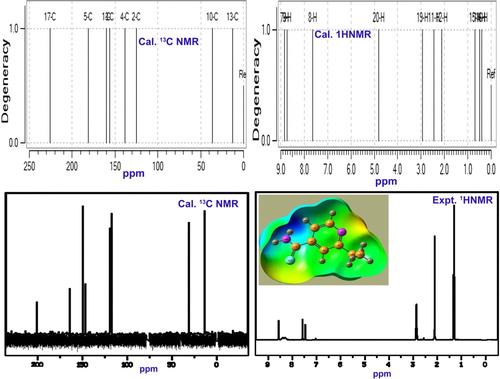
4.5. NMR chemical examination
Usually, the chemical reaction is taking place for the arrangement of chemical properties in the molecular complex by the addition of substitutional groups on the specific positions or active parts of the base molecule [Citation26]. The organized chemical property can be identified and measured from the obtained chemical shift of core as well as ligand carbons in the customized molecular architecture. In this case, the pyridine base was substituted by ethyl and thioamide groups by which the entire chemical environment was rearranged in terms of chemical shift.
The chemical shift of core carbons of pyridine rings; C1, C2, C3, C4 and C5 were 162, 124, 161, 125 and 199 ppm, respectively, as shown in Table . Here, C3 and C5 were more shifted than adjacent core parts which were mainly by the eventual breaking of paramagnetic shield due to ethyl and thioamide injection. Among these shifts, C5 has been much affected than C3 due to heavy atom coupling. Apart from that, thio injection is purposefully drawing possible electron cloud, whereas the ethyl chain is supplying electron content. Such donor and acceptor of electron cloud alternate the chemical environment which leads the pushing and pulling chemical effect. Thus, the resultant chemical property was induced. Already, the base has a chemical mechanism for generating anti-fungal activity in which by the addition of such ligand groups, it was diverted or transformed into antibiotic activity. The entire chemical mechanism was constructed and confined by the exchange of chemical potential through C3 and C5. Additionally, it was also observed that, the core frame carbons; C3 and C5 acted as the chemical exchange path on which the chemical kinetics were found to be activated.
Table 4. Experimental and calculated 1H and 13C NMR chemical shifts of Ethionamide.
Since no electron cloud was transferred from chain to ring, the lowest chemical shift was observed at C13 [Expt. (35 ppm) Cal. (36 ppm)] and C17 [Expt. (13 ppm) Cal.(12 ppm)]. But, the chemical shift of C17 was 205 and 225 ppm for the experimented and calculated, respectively. Here, the large chemical shift was observed at C17 due to the asymmetrical breaking of the paramagnetic shield. Chemical shifts of H, H7, H8 and H9 were found to be more shifted than that of H of ligand groups. This is because of the variable chemical strain produced in the ligand H. (Figures ).
4.6. Frontier molecular interaction
The σ, π and δ orbital overlapping appeared among bonding and antibonding orbital localization which is arranged in the compound by the physical mechanism of Frontier molecular polar orbital interactions. Figure show various characteristic interactions among different bonding and antibonding orbitals. In the figure, several in-phase and out-of-phase interactions were taking place with respect to degenerate and non-degenerate orbitals among different entities of the structure. From the transitions among interactive high occupied molecular orbitals (HOMO) and low unoccupied molecular orbitals (LUMO), the molecular reactivity mechanism can be measured which causes drug mechanical property.
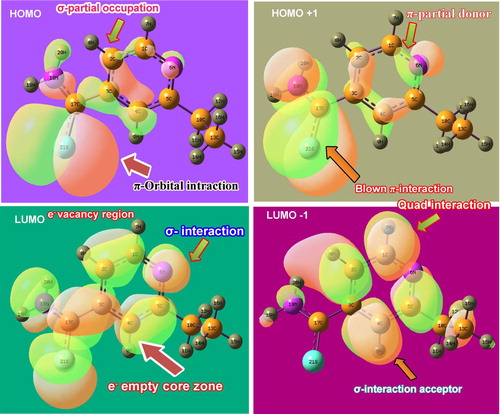
In the case of LUMO (bonding), the σ-orbital overlapping was dominated which demonstrated that the empty zones were identified as σ-bonding over which the excited electron cloud was transferred. Here, the methyl–ethyl chain was exempted, whereas thioamide and pyridine ring fully participated. In the second-order LUMO called LUMO+1, the σ-bonding was shown in which the pyridine ring fully participated, whereas the ethyl and thioamide partially participated. From this view, it was clear that, the σ-degenerate levels acted as a chemical energy domain in which transitions almost take place. In the case of HOMO (antibonding), π-orbital interactions take place and the electron cloud transited from the thioamide group. Additionally, some of the electron cloud from core C was ready to make transitions. In the next level of HOMO known as HOMO-1, apart from ethyl–methyl chain, the pyridine and thioamide group participated well in transferring the chemical potential for generating antibiotic activity.
In HOMO levels, the customized chemical potential in the form of excited electron cloud was transferred to LUMO levels. Here, the pyridine and thioamide flavour electron transitions were found to have appeared. So it was concluded that the dominated drug property of this compound was purely prepared by the pyridine ring and thioamide group and the prepared drug potential was stabilized by ethyl–methyl group.
4.7. CT complex analysis in UV-Visible spectra
The present aromatic drug compound, Ethionamide, was taken to the UV-Visible recording and its results were validated by the theoretical calculations performed by the Gaussian programme. The theoretical parameters are presented in Table and the experimental electronic-excitation analytical graph along with simulated peaks is presented in Figure . In every molecular composite, the CT complex was generated in the compound, causing molecular mechanism to enable the drug property in the compound under study.
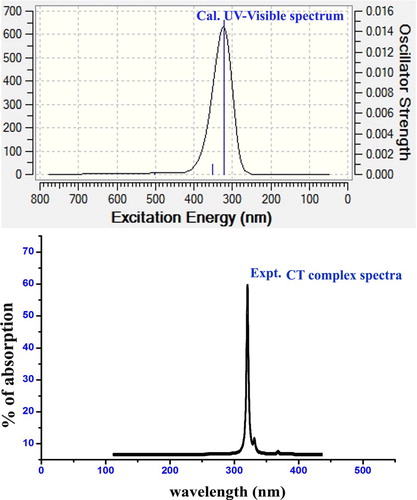
Table 5. Theoretical electronic absorption spectral parameters of Ethionamide.
In UV-Visible spectral pattern, three sets of excited transitions were observed for gas and solvent phase. Among three excited absorption peaks, the transition was in the region 328.12 nm with the energy gap of 3.850 eV at an oscillator strength of 0.0151. This absorption peak was assigned to n→π* transition in which its region was found to be Quartz UV and the chromospheres were designated as R-Bands (German, radikalartig). Similarly, in the solvent phase, the transition was found to be in the range of 322.28 nm with the energy gap of 3.847 eV at the oscillator strength of 0.016. This absorption was assigned to n→π* transition level and its regions were found to be Quartz UV and chromospheres were designated as R-Band (German, radikalartig). In the case of the chloroform solvent phase, the same trend was taking place. The singlet peak was observed with elementary transitions at 320 nm and 350 nm with minimum intensity. From the observed UV-Visible band, it was clear that the assigned absorption band was obtained from the π-bonding interaction system. Such π-bond molecular system was designated by the thioamide group which was found to be CT complex of the present aromatic molecular system. Here, the experimented peak was rather similar to the calculated peak by which the CT complex was validated.
In this case, if the thioamide group is not present in the present case, the imine group will be the CT complex. But, here, since the accumulation of negative chemical potential over S=C–NH2 group, the charge transfer resonance source was determined to be thioamide group. So, the antibiotic property was produced in the present molecule due to this existence and it was ensured from the obtained value of electrophilicity index.
4.8. Physico-chemical properties
The zero-point vibrational energy of the present molecular system was 818 Hartree by which it was clear that the customized drug structure was found to be optimized and the associated parameters were truly calculated. The electron affinity and ionization potential were 6.1 and 2.3 eV, respectively. Both were very unique and the band energy of electric and optic components was located in two extremes by which the present compound acted as a good drug and its reliability was so high on both regions. The dipole moment of the present case at both regions was 3.39 and 2.47 dyne, respectively. These values represented electromagnetic polarization gradient on two regions. The dipole moment in IR region was more than UV-visible region since the important ligand groups belong to the heteronuclear dipole region. The energy gap was 3.86 eV in IR which was more than the optical band gap of 3.71 eV.
The chemical hardness of the chemical compound was 1.931 which was moderate and its ability to react with other ligands was rather difficult. The chemical softness is the ability of the chemical species to react with the optimized structure with fast rate which was found to be 7.72. The obtained value was so high and the present compound can react easily with the injected ligand groups and produce a new tailored property. The electro negativity is an important factor, which estimates the required negative potential to make the chemical mechanism for generating optimized drug property. Here, it was 4.24 and 4.28, respectively and these values were more enough than required values to generate antibiotic potential. The physico-chemical parameters for the present case are calculated and depicted in Table .
Table 6. Physico-chemical parameters of Ethionamide.
The Electrophilicity index (ω) is a indicator which rightly estimated the charge transfer characteristics of aromatic system and here it was calculated to be 4.65 and 6.14 in IR and UV-Visible region, respectively. From the observed values, it was clear that the charge flow gradient of the present system was found to be high and it was from the main source of the thioamide group. The electrophilicity charge transfer was calculated to be 1.813 which was greater than unity which showed the charge transformation from the thio group to the pyridine group.
4.9. MEP investigation
The electrostatic potential map of the Ethionamide is displayed in Figure in which the electrophilic and nucleophilic influence is over the different compositional parts of the molecule [Citation27]. The colour gradients, such as the red region, showed the electrophilic attraction domain, whereas the blue colour showed the nucleophilic region and the intermediate region was represented by the green colour called depletion range [Citation28,Citation29]. These kinds of colour regions illustrated the balance orientation of electronic region against the protonic region. Here, the negative charge gradient continuum appeared on thio group and imine group of pyridine ring, while the intensive protonic region was found on NH2 group. In such a circumstance, the electrophilic region should be over the amide group. But in this case, the NH2 group was shown to be in the nucleophilic region which was due to the shifting of electronic configuration to C=S group for generating enriched electronic domain to produce purposive drug property; antibiotic.
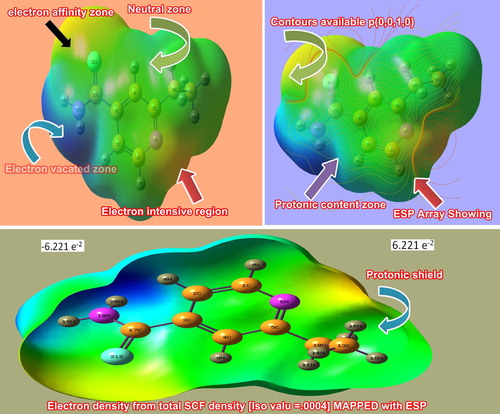
The contour field map showed the distribution of static field of electro-polarized component. Here, the origin of spreading of field was found at the imine group and the thio group. The incredible point of dispersion of field emphasized that two separate diagonal electrophilic regions were targeted by the nucleophilic region in tetragonal form. The triangle influence of electrostatic field chemical-potential in the compound induced the consistent antibiotic compound.
4.10. Polarization analysis
The average polarizability and anisotropy of the polarizability of the organic molecule facilitate to measure the rate of change of biological properties with respect to ligand groups. Similarly, and first-order hyperpolarizability is also a factor which measures the hyper action of electro-chemical process generated by electro-active mechanism. The ligand-induced electro-chemical potential is added with base chemical reactivity and produces hyperpolarizability gradient in the compound. The polarizability parameters are depicted in Table . Accordingly, the average polarizability and anisotropy polarizability are the factors which indicate biological flavour which was found to be 170 and 233.14 × 10−33 esu, respectively. From the observed values, it was confirmed that the compound under study was able to produce the strong fundamental biological mechanism for generating antibiotic nature. Similarly, the hyperpolarizability parameter that is used to measure the biological weightage value was calculated to be 264.97 × 10−33 esu which was able to make electrophilic aroma for inducing antibiotic properties.
Table 7. The polarizability and the first hyperpolarizability of Ethionamide.
4.11. NBMO analysis
The occurrence of a pair of electronic transitions in different donor and acceptor (Lewis acid and Lewis base) of non-bonding molecular orbitals is usually giving the information regarding the assignment of electronic chemical potential by the assembly of molecular interactive orbitals [Citation30,Citation31]. The interaction is taking place between degenerate orbitals equipped by occupancy orbitals of different entities of the chemical species of the complex compound. The calculated transitional energy by second-order Perturbation theory is arranged in Table . In the transitional process, the exchange of a large amount of chemical energy in important non-bonding orbitals of various entities in the present molecule is presented in Table .
Table 8. The calculated NBO of Ethionamide by second-order Perturbation theory.
Here, the transitions were identified between C1–C2 and C3–C4 and C5–N6 at which the energies of 11.99 and 8.94 kcal./mol were found to be transferred within the occupied energy of 1.987 kcal./mol. such that transitions were represented by π- π* interaction. Similarly, in the ring itself, the transitions were identified between C3–C4 and C1–C2 and C5–N6 with the energy absorption of 10.2 and 13.7 kcal./mol, respectively. These transitions were represented by π- π* interaction. The same transitions take place from C5–N6 to C1–C2 and C3–C4 with the transitional energy of 13.2 and 7.4 kcal./mol which were represented by π- π* interaction system.
The energy of 5.5 and 3.8 kcal./mol for the non-bonding orbitals of bonding segment from C5–C10 to C1–N6 and C5–N6, respectively. These transitions were assigned to π- π* system and this energy was found to be transferred from ethyl chain to ring. The transition takes place from lone pair to lone pair; S21 to C17 and C5 with the absorption energy of 6.1 and 6.4 kcal./mol, respectively. Similarly, the transitions take place from N6 to C4–C5 and C5–C10 with the energy band of 13.5 and 2.7 kcal./mol, respectively. These heterogeneous transitions represented were by lone pair to n*. In the ligand group, the transitions take place from N18 to C3–C17 and C17–S21 by absorbing the electronic energy of 7.9 and 6.1 kcal./mol.; another transition was observed from S21 to C3–C17 and C17–N by in taking energy of 18 11.7 and 18.4 kcal./mol, respectively. In the thio group, the electronic transition from C17–S21 to C3–C4 by consuming the energy of 11.4 kcal./mol and it was the important transition in which the thio group chemical potential was completely transferred to ring.
From those transitions, it was clear that the chemical potential of the ring was exchanged among the core carbons and the enriched chemical potential of the ligand groups was observed to be transferred to the ring unidirectional mode. The transferred potential was exchanged between the core nodes which generates hyperchemical mechanism for antibiotic property.
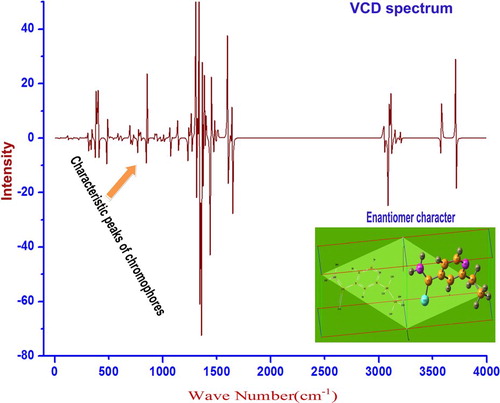
4.12. VCD spectral sign
The stoichiometric design of the chemical species was represented by vibrational circular dichroism which are the right- and left-hand sides of circular polarizations. Both the polarizations are circularly dispersed by electro-magnetic components of resultant multipole moments of the customized molecular structure [Citation32]. The VCD is nothing but vibrational absorption–emission spectrum. The VCD spectral pattern is the vibrational characteristic of chromophores present in the compound. It is also known that the lower symmetry property of ligand groups usually affects the entire symmetrical property of the complex compound. If the asymmetrical substitutions are present in the compound, they will affect the chirality of the compound and also influence the enantiomer form [Citation33]. The rate of change chirality character of the compound is directly proportional to the symmetrical form of VCD and thereby the toxicity can be measured [Citation34].
In this case, the vibrational spectrum is shown in Figure and the enantiomer view of the compound under study is also presented. Actually, the molecular structure of the compound was found to be not complicated even after the ligand groups were injected. Since the symmetry was not affected much, the enantiomer construction was appeared to be uncomplicated and hence the VCD spectral pattern was quite simple. From this view, it was clear that the compound was found to be pure and free from toxicity.
5. Conclusion
The antibiotic drug, Ethionamide, was characterized by FT-IR, FT-Raman, UV-Visible and NMR spectral tools and their results were validated by performing the theoretical calculations. The structural and biological studies were carried out to interpret the fundamental properties and constructed Physico-chemical properties. The forceful alternation of molecular charge levels of pyridine ring on par with the ligand groups was keenly observed to interpret the inducement of additional drug property. The origin of chemical potential to give out the customized property was found out and the movement of the chemical potential was found through nodal core segments. The chemical reactivity path via the core carbons was identified from the associated chemical shift. The frontier molecular interaction system was keenly observed from which how the cascading degenerate orbital overlapping helped to accumulate the chemical reactivity for inducing antibiotic activity.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
S. Ramalingam http://orcid.org/0000-0002-1758-3930
References
- Morlock GP, Metchock B, Sikes D, et al. Etha, inhA, and katG Loci of Ethionamide-resistant clinical mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2003;47(12):3799–3805. doi: 10.1128/AAC.47.12.3799-3805.2003
- Stuart MC, Kouimtzi M. WHO Model Formulary, World Health Organization, (2009) 496–500.
- Auclair B, Nix DE, Adam RD, et al. Pharmacokinetics of Ethionamide administered under fasting conditions or with orange juice, food, or antacids. Antimicrob Agents Chemother. 2001;45(3):810–814. doi: 10.1128/AAC.45.3.810-814.2001
- Smith and Reynard, The text book if pharmacology, Harcourt Publishers Ltd, (1992). 868
- Banerjee A, Dubnau E, Quemard A, et al. J Sci. 1994;14, 263(5144):227–230.
- Vale N, Correia A, Figueiredo P, et al. J Mol Struct. 2015;1105(7):286–292.
- Abdelghany S, Alkhawaldeh M, AlKhatib HS. Carrageenan-stabilized chitosan alginate nanoparticles loaded with Ethionamide for the treatment of tuberculosis. J Drug Deliv Sci Technol. 2017;39:442–449. doi: 10.1016/j.jddst.2017.04.034
- Diniz LF, Carvalho PS, de Melo CC, et al. Development of a salt drug with improved solubility: Ethionamide nitrate. J Mol Struct. 2017;1137(5):119–125. doi: 10.1016/j.molstruc.2017.02.036
- Ramalingam S, Periandy S, Mohan S. Vibrational spectroscopy (FTIR and FTRaman) investigation using ab initio (HF) and DFT (B3LYP and B3PW91) analysis on the structure of 2-amino pyridine. Spectrochim Acta, Part A. 2010;77:73–81. doi: 10.1016/j.saa.2010.04.027
- Madanagopal A, Periandy S, Gayathri P, et al. Spectroscopic and computational investigation of the structure and pharmacological activity of 1-benzylimidazole. J Taibah Univ Sci. 2017;11:975–996. doi: 10.1016/j.jtusci.2017.02.006
- Aarthi R, Ramalingam S, Periandy S, et al. Molecular structure-associated pharmacodynamic investigation on benzoyl peroxide using spectroscopic and quantum computational tools. J Taibah Univ Sci. 2018;12(1):104–122. doi: 10.1080/16583655.2018.1451116
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings 1PII of original article: S0169-409X(96)00423-1. The article was originally published in Advanced drug Delivery Reviews. Adv Drug Deliv Rev. 1997;23:3–25. 46 3-26: (2001), 3-26 doi: 10.1016/S0169-409X(96)00423-1
- Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discovery. 2007;6(11):881–890. doi: 10.1038/nrd2445
- Leo A, Hansch C, Elkins D. Partition coefficients and their uses. Chem Rev. 1971;71(6):525–616. doi: 10.1021/cr60274a001
- Ardyukova AF, et al. Atlas of spectra of aromatic and heterocyclic compounds. Novosibirsk: Nauka Sib. Otd.; 1973.
- Green JHS, Harrison DJ. Vibrational spectra of the trimethylpyridmes (collidines). Spectrochim Acta. 1973;29A:293–299. doi: 10.1016/0584-8539(73)80073-X
- Tripathi R, Indian 1. Pure Appl Phys. 1973;11:277.
- Wilmshurst K, Bernstein HJ. The vibrational spectra of pyridine, pyridine-4-d, pyridine-2,6-d>2>, and pyridine-3,5-d2. Can. l. Chem. 1957;35:1183. doi: 10.1139/v57-159
- Ramalingam S, Periandy S, Mohan S. Vibrational spectroscopy (FTIR and FTRaman) investigation using ab initio (HF) and DFT (B3LYP and B3PW91) analysis on the structure of 2-amino pyridine. Spectrochim Acta A. 2010;77:73–81. doi: 10.1016/j.saa.2010.04.027
- Sundaraganesan N, Meganathan C, Kurt M. Molecular structure and vibrational spectra of 2-amino-5-methyl pyridine and 2-amino-6-methyl pyridine by density functional methods. J Mol Struct. 2008;891:284–291. doi: 10.1016/j.molstruc.2008.03.051
- Bellamy LJ, Rogash PE. J. Chon. Soc. 1960: 2218. doi: 10.1039/JR9600002218
- Jensen KA, Nielsen PM. Infrared spectra of thioamides and selenoamides. Acta Chern. Scand. 1966;20:597. doi: 10.3891/acta.chem.scand.20-0597
- Socrates G. Infrared and Raman characteristic group frequencies. Third ed. New York: Wiley; 2001.
- Brown TL. The carbonyl intensities of some simple amides. J Phys Chem. 1959;63:1324. doi: 10.1021/j150578a033
- Stewart JE. Vibrational spectra of primary and secondary aliphatic amines. J Chem Phys 1959;30:1259. doi: 10.1063/1.1730168
- Silverstein RM, Webster FX, Kiemle DJ. Spectrometric identification of organic compounds. 7th ed. Wiley; 2005.
- Munoz-Caro C, Nino A, Sement ML, et al. Modeling of protonation processes in acetohydroxamic acid. J Org Chem. 2000;65:405. doi: 10.1021/jo991251x
- Scrocco E, Tomasi J. Topics in current chemistry, 42, Springer Verlag, Berlin, 1973, 95-101.
- Bader RFW. A quantum theory of molecular structure and its applications. Chem. Rev. 1990;91:893. doi: 10.1021/cr00005a013
- Woodford JN. Density functional theory and atoms-in-molecules investigation of intramolecular hydrogen bonding in derivatives of malonaldehyde and implications for resonance-assisted hydrogen bonding. J Phys Chem A 2007;111:8519. doi: 10.1021/jp073098d
- Palusiak M, Simon S, Sola M. Interplay between intramolecular resonance-assisted hydrogen bonding and aromaticity ino-hydroxyaryl aldehydes. J Org Chem 2006;71:5241. doi: 10.1021/jo060591x
- Berova N, Di Bari L, Pescitelli G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem Soc Rev 2007;36:914–931. doi: 10.1039/b515476f
- Mason SF. Molecular optical activity and the chiral discrimination. Cambridge: Cambridge University Press; 1982.
- Circular dichroism and the conformational analysis of biomolecules. New York: Plenum Press; ed. G. D. Fasman, 1996.