
Abstract
S. gregaria is able to swarm and disseminate bacterial load over vast areas. Adult locusts were collected from Kingdom of Saudi Arabia (KSA), individual locust was divided into head, thorax and abdomen. External and internal cultivable bacteria were cultured, cell and colony characteristics were investigated. Pure colonies were sequenced using 16S rRNA. Sequences and phylogeny were analyzed. Using morphological and molecular results, 9 bacterial genera belonging to 5 families, 3 orders, 2 classes, and 2 phyla were recorded. Ribosomal Database Project (RDP) Classifier succeeded to confirm 8 out of 9 genera. The ninth isolate was designated as unclassified. To specify a genus to its closest species, 16S rRNA genes of the type species have been downloaded. Phylogenetic tree was generated using all available sequences. Similarity measures and phylogenetic trees were used to place each genus to its closest species. Six genera created similarities less than 98% to related species. Generally, analyses of 16S rRNA gene succeeded to identify 3 isolates to species level, 5 isolates to genus level and designated one isolate “unclassified”. Investigations are recommended to place all genera to their operational taxonomic units (OTU). Significance of our findings relies on the impact of locust-microbiota on heath of people who consumed insects as food. Bacterial species which were internally, isolated from abdomen were described beneficial commensals. Bacterial species which were internally, isolated from thorax and head were described non-pathogenic beneficial endosymbionts, except Bacillus sp. Salmonella sp. was isolated as external species and was described external contaminant of the locust.
Introduction
A tenth of the world’s population was endangered in their sustenance by locust plagues. Regarding locust swarms in the Middle East between 1860 and 2005, Saudi Arabian peninsula was the most influenced region, ever. Successive rainfalls at the winter of 1996–97, created optimal breeding conditions (lasting for 6 months) for S. gregaria along the Red Sea coasts of KSA [Citation1,Citation2]. Following rainy seasons, locust reproduction is an annual consequence. Cyclones in 2018, warm climate at the end of 2019 and abnormal rate of torrential downpours shaped favorable conditions of the present outbreak of locusts (the hugest for at least 25 years). KSA has become with Yemen, Ethiopia, Eritrea, Sudan and Somalia in the central region of the present outbreak of S. gregaria. The invasion of locusts to many cities including Mecca and Al-Madinah has been recorded over the summer of 2019. During the last quarter of 2019, locust numbers have persisted in activity on the coastal plains near Jizan. Hoppers continued developing to adults and oviposition has started [Citation3]. At the start of 2020, huge numbers of swarms have been detected in Somalia and Ethiopia. They continue spreading to Uganda, Kenya and Sudan. Swarms have been shaping in Yemen, Saudi Arabia, Iran, Pakistan and India [Citation4].
The importance of bacterial load studies of the desert locust, S. gregaria, rely on its consumption as alternative protein source, on its ability to form swarms and to serve as mechanical vector of pathogens, as well. The Food and Agriculture Organization (FAO) reported that approximately 2.5 billion humans consume insects as alternative protein source worldwide [Citation5]. The desert locust, S. gregaria, is one of the most consumable insects in African countries such as Zambia, South Africa, Cameroon, Congo, Botswana, Tanzania, Sudan, Uganda, Ethiopia, Kenya, Sierra Leone, Morocco, Guinea, Lesotho, Mauritania, Somalia, Eritrea, Guinea Bissau [Citation6]. In addition, the desert locust is one of the most-feared insects among farmers. It was reported early that S. gregaria is the most economically important locust of approximately 20 locust species. This is due to its vast area of distribution during both recession and plague phases [Citation7]. Crop loss of 80–100% and more than 500 million dollars were the average costs of controlling the 2003–2005 locust plague [Citation2,Citation8]. In the last decade, grasshopper model was successfully used as an experimental model for evaluating vertebrate brain uptake by investigating blood–brain barrier (BBB) permeability. This is very important in development and assessment of drugs against disorders of central nervous system [Citation9]. The economic importance and ecological contribution of locusts call for a superior perception on the structure of their microbial communities.
Much has been published on characterization and function of microbiota in animal physiology, ecology and development. Studies on insect-microbiota relationships are very important to understand the role of symbiosis and mutualism in many interactions. Herbivore-microbiomes model has attracted researchers to work on, especially, beneficial insects like honey bee which have a high environmental impact and high economic value [Citation10]. In general, due to the little attention paid to study microbiome of orthopteran hosts, this model is still poorly understood [Citation11]. The edibility, vast mobility and environmental impact of locusts necessitate a special consideration to study the dangers of microorganisms of edible insects [Citation12]. Microbiota may affect host development, host ecology, host defense against microbes and mechanical distribution of pathogens, as well [Citation13,Citation14]. Like most insects, locusts could play a role in mechanical transmission of their bacterial load in addition to eating our food. Regarding the biodiversity of insects, we are in bad need to learn more about insect-microbiota interaction. Therefore, the main objective of this study was to identify the cultivable bacterial community associated to the desert locust, S. gregaria, collected from KSA.
Materials and methods
Locust collection
Sixty adults (30 males and 30 females) of the desert locust S. gregaria were, randomly, collected from moderate density populations between November 2018 and February 2019. Collection were done from Mecca and Al-Madinah, KSA. Collected locusts were divided alive into two groups. Locusts from both sampled locations were equally distributed in the two groups. These were subdivided into males and females and kept in sterile containers. In order to exclude contamination by food, plant material was soaked in 2% potassium permanganate for about five minutes, rinsed under sterile water running from a tank for five minutes and then dried inside laminar flow hood before administration. Locusts were starved for two days before experiments have been executed. No direct contact between the collectors and locusts until furtherly processed. Locust samples were processed for bacterial cultures within one week from collection date.
Isolation of external bacteria
In a sterile conditions, starved locusts (15 males and 15 females) were anesthetized by freezing at 0°C until stop mobility. Each individual locust was then divided into three regions: head, thorax and abdomen. This step was carried out using frozen chilling plate inside the freezing area (0°C) to keep exuding of the intestinal contents from being mixed with external bacteria. Additionally, freezing conditions provided enough time for careful sealing of body regions. Each region was immediately sealed by making a thin film of mineral-based sealer (BioRoot™ RCS). Sealing material was purchased from Septodont company (Cat. #: 01E0300, USA). With such sealing, we excluded external contamination by gut and other internal tissue. Two millilitres of sterile phosphate buffered saline (PBS) were added to each body region in a test tube and samples were thoroughly shaken for 2 min. Tubes of the same body region were pooled and a fixed volume (0.1 mL) of each body region collective tube was spread separately on 20 cm diameter plates containing nutrient agar (NA) media (0.5% peptone, 0.3% yeast extract, 1.5% agar, 0.5% NaCl, dH2O, pH = 6.8 at 25°C) using a sterilised spreader inside laminar flow hood [Citation15]. NA was purchased from Merck company (Cat. #: 70148, Germany). Plates were incubated at 30°C for 24–48 h and then examined for bacterial identification. Colonies were differentiated according to their morphology including colour, shape, margins, elevation, and texture. Colonies were purified and stored separately in 5% glycerol at −80°C.
Isolation of internal bacteria
In a sterile conditions, locusts (15 males and 15 females) were anesthetized by freezing at 0°C until stop mobility. Five washes with 70% ethanol, two washes with PBS, followed by three washes with sterile distilled water were carried out for surface sterilisation of the insect samples. Surface-sterilised samples were centrifuged for 5–10 s at 1,000 rpm to get rid of excess ethanol. Surface-sterilised individual locust was then divided into three regions: head, thorax and abdomen. Each region was homogenised in a suitable amount of sterile Ringer’s solution (7.2 g NaCl, 0.37 g KCl, 0.17 g CaCl2, in 1 L dH2O, pH = 7.3–7.4). The homogenate was centrifuged at 1000 rpm to remove cell debris. Suspensions of the same body region were pooled. A fixed volume (0.1 mL) of each body region was spread on 20 cm diameter plates containing NA media using a sterilised spreader. All steps of the isolation procedure were carried out inside laminar flow hood [Citation15]. Plates were incubated at 30°C for 24–48 h and then examined for bacterial identification. Colonies were differentiated on the basis of morphology including colour, shape, margins, elevation, and texture. Each colony was purified and stored in 5% glycerol at −80°C.
Isolation of bacteria from rearing environment
As the locusts were kept in sterile containers until being processed, bacterial swaps were taken from containers and from fecal mass of locust, too. Suitable amount of sterile PBS were added to cotton of the swaps in a test tube and samples were thoroughly shaken for 2 min. All tubes were pooled, passed through cotton mesh to remove debris and 0.1 mL of the collective filtrate was spread on agar plate using a sterilised spreader. Procedure was completed as described above.
Genomic DNA purification
As visually identical bacterial colonies may actually contained more than one bacterial species, 2 or 3 colonies of the same morphology were sent for molecular studies. Total bacterial DNA of each isolate was purified using mericon DNA Bacteria Kit (QIAGEN, GmbH, Germany), as described by the supplier’s protocol. Purified DNAs were transferred into new tubes and stored at −20°C until processed. Concentration of DNA was determined at OD260 and purity of DNA was assessed by measuring OD260/OD280 and OD260/OD230 ratios. DNA was considered pure when 260/280 ratio is at ∼1.8 and 260/230 ratio is at ∼2.0.
Oligonucleotides and PCR amplification of 16S rRNA gene
Primers used in this study were designed to amplify 500 bp fragment from 16S rRNA gene [Citation16]. A master mix of PCR (2.5 μl PCR buffer, 1.5 mM MgCl2, 200 μM dNTPs, 1 U Taq DNA polymerase (AmpliTaq, Perkin-Elmer)) was prepared within a cabinet of biosafety for all reactions. PCR tube contains 7.5 µl master mix, 2.5 µl of 10 pmol forward primer (5′-TGGAGAGTTTGATCCTGGCTCAG-3′), 2.5 µl of 10 pmol reverse primer (5′-TACCGCGGCTGCTGGCAC-3′), 4 µl of DNA (40 ng), and 8.5 µl distilled water to reach a 25 µl final volume. A no-template PCR tube was used as negative control. PCR amplification was carried out in a thermocycler ABI GeneAmp 9700 (Applied Biosystems, USA), which was programmed for one cycle at 94°C for 5 min (initial denaturation), followed by 40 cycles of denaturation for 1 min at 94°C, annealing for 1 min at 55°C, extension for 1 min at 72°C, and a final extension for 10 min at 72°C. PCR products were displayed on 1.5% agarose gel and photographed utilising gel documentation system. Molecular size of PCR products was determined in relation to 1 Kb ladder (MBI, Fermentas, USA) as a marker. On a UV-transilluminator, the specified bands were cut from the gel with least amount of gel fraction. DNAs were eluted and purified using QIAGEN gel extraction kit (QIAGEN, GmbH, Germany) as described in the manufacturer’s manual.
DNA sequencing of bacterial 16S rRNA gene
For each bacterial isolate, two positive PCR products were sent for sequencing (to exclude PCR errors certainly) using the specific 16S rRNA gene forward and reverse primers. Sequencing was accomplished using ABI PRISMTM 3100 DNA sequencer (Applied Biosystems) and Big Dye terminator sequencing kit (Version 3.1, Applied Biosystems, USA).
Sequence analyses and phylogeny
The sequences of the 9 bacterial isolates obtained from both 16S rRNA primers were first edited to readable sequences. These were subjected to analysis by the Ribosomal Database Project (RDP) Classifier (https://rdp.cme.msu.edu/classifier/classifier.jsp) to assign the putative genus of each isolate. A preliminary phylogenetic tree was then built to position each isolate in its specific genus. Sequences of the type species of the genera suggested by RDP Classifier were downloaded to be used in deduction of phylogenetic relationship. Our sequences were then subjected to multiple alignment with other similar sequences using Clustal Omega and manually refined using BioEdit 7.2.5 (https://www.mybiosoftware.com/bioedit-7-0-9-biological-sequence-alignment-editor.html). Alignments were manually edited where necessary. All nucleotide sequences of the bacterial isolates have been submitted to GenBank for accession numbers. BLAST searches of each sequence with similar sequences were done using NCBI database (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Phylogenetic trees were constructed using Neighbor Joining (NJ) method and MrBayes 3.2.6 software (http://www.phylogeny.fr/one_task.cgi?task_type=mrbayes). Each bacterial genus was then designated to its species using the inference of phylogenetic trees and measures of sequence similarity. Additionally, Oxidase Test was carried out to differentiate between two confused genera in the case of the eighth isolate [Citation15]. In brief, fresh alive bacteria were cultivated on tryptone soy agar plates (soy peptone, casein peptone, NaCl, agar, pH 7.3). Plates were incubated at 37°C for 24–48 h. After bacterial growth, 2–3 drops of N,N-dimethyl-p-phenylenediamine (DMPD) have been added to each one of the two organisms. Development of violet to purple colour within 10–30 S indicate positive test.
Results
The main objective of our study was to identify the cultivable bacterial community associated to the desert locust, S. gregaria, collected from KSA. This goal was achieved by colony and cell characterization of the different bacterial isolates. Molecular identification and morphological data were combined to have a clear complementary conclusion.
Morphological characteristics of bacteria
Seven opaque, one translucent and one iridescent colonies were identified. Eight pigmented and one unpigmented colonies have been characterized. Eight convex and one raised colonies were observed. Out of the 9 circular and entire-margined colonies, 6 medium-sized and 3 small-sized colonies were described (Table ). In addition, 5 Gram -ve, 3 Gram + ve and one variable Gram’s stain bacterial cells were recognised. Out of the nine bacterial strains, 6 bacilli, 2 cocci and one coccobacilli bacterial cells were identified (Table ).
Table 1. Colony and Cell Characteristics of bacterial isolates from adult S. gregaria.
Molecular identification of 9 pure colonies were done using 16S rRNA sequences (Tables and ). Combination of the morphological and molecular data resulted in confirmation of 9 different bacterial genera belonging to 5 families (Enterococcaceae, Lactobacillaceae, Bacillaceae, Staphylococcaceae and Enterobacteriaceae), 3 orders (Lactobacillales, Bacillales and Enterobacteriales), 2 classes (Bacilli and Gammaproteobacteria) and 2 phyla (Firmicutes and Proteobacteria). Out of the 9, 5 genera belong to order Enterobacteriales, class Gammaproteobacteria and phylum Proteobacteria (55.56%), 2 genera belong to order Lactobacillales, class Bacilli and phylum Firmicutes (22.2%) and 2 genera belong to order Bacillales, class Bacilli and phylum Firmicutes (22.2%). Bacterial genera identified in this work and their taxonomic analysis are listed in Table (2). We included the most probable species of the confirmed genera, whenever more likely (Table ).
Table 2. A key-table showing the main information of the bacterial isolates using RDP Classifier analysis.
Table 3. Summarised similarity measures of the identified bacterial species using 16S rRNA gene.
PCR identification
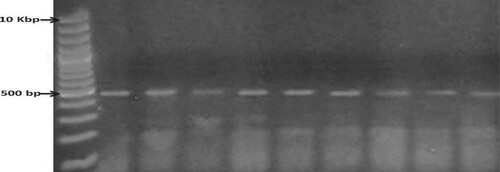
In the present study, the primer set of 16S rRNA gene was successfully used to amplify 500 bp fragments (Figure ). Bands of approximately 500 bp in relation to marker size, were eluted and two positive amplicons of each isolate were submitted to direct sequencing using the same amplifying primers. Readable sequences were analyzed and used for identification of the isolate (Table ).
Figure 1. PCR amplification of 16S rRNA of 9 bacterial isolates. Lane M: 10 kb DNA marker. Lanes 1–9: The nine bacterial isolates. The amplicon size is 500 bp.
Sequence alignment
Firstly, sequences of the two positive amplicons of each isolate were aligned to ensure the correctness of sequence. Meanwhile, the intraspecific identity of nine corrected sequences was examined using Clustal Omega to ensure the number of different isolates. Sequence analyses of our 16S rRNA sequences revealed that 9 bacterial genera were isolated from the different body regions of both male and female S. gregaria. The RDP Classifier succeeded to confirm 8 out of 9 genera. The ninth genus was suggested as Plesiomonas or Shigella using RDP Classifier (Table ). In addition, BLAST search did not succeed to specify one of the two genera. It is worthy to mention that the unclassified genus exhibited the least identity 94.17%, query coverage 59% and maximum score 183 with both Shigella and Plesiomonas (Figure ). In order to differentiate between Plesiomonas or Shigella genera, Oxidase Test was carried out. The unclassified isolate was oxidase positive which indicated that it may be Plesiomonas spp. This genus should be confirmed by more experimental laborious work. Thus, we preferred to designate it as unclassified bacterium.
Figure 2. BLAST scores of 16S rRNA gene of the isolate number 8 in comparison to 16S rRNA gene of Plesiomonas and Shigella (Acc# X60418 and NR_104826, respectively).

To specify each of the eight confirmed genera to the most probable species, 16S rRNA genes of the type species and all available 16S rRNA genes of the genus have been downloaded. Each genus was aligned to its corresponding sequences using Clustal Omega software. A distance-based phylogenetic trees were created. A genus has placed to its closest probable species on the basis of the inference of phylogenetic trees and measures of sequence similarity (Figure and Tables and ). Maximum score, percentage of query coverage and percentage identity were the most weighted items to have a pointer to the closest species (Table ). Our bacterial sequences were separately submitted to FASTA (https://www.genome.jp/tools/fasta/) and BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi?) searches to reveal their identity with previously published sequences. Our sequences showed similarities of 94.17% to 99%, query coverages of 59% to 88% and maximum scores of 183–555 in comparison to corresponding sequences in GenBank databases. Out of the 8 bacterial genera, five genera created less than 98% similarity with their related genera. E. faecalis, L. paracasei and S. epidermidis exhibited higher similarity measures (Table ).
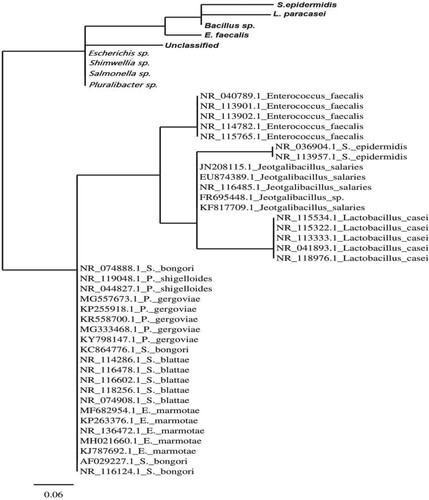
Figure 3. Distance-based phylogenetic tree of 47 sequences of 16S rRNA gene using Joining (NJ) method and MrBayes 3.1.2 software.
Molecular phylogeny
Phylogenetic tree was generated by 47 sequences using Neighbor Joining (NJ) method and MrBayes 3.1.2 software. Tree’s topology showed two major divisions. Division I collects our nine sequences in four sister clades. Meanwhile, division II includes all other sequences in five species-collective sister clades. Overlap of species was observed between S. bongori, P. shigelloides, P. gregoviae, S. blattae and E. marmotae (Figure ).
Isolation of bacteria from body regions of adult S. gregaria
A total of 9 bacterial genera were isolated from adult S. gregaria (Tables and ). Out of 9 bacterial genera, 5 genera (55.56%) were isolated from both males and females (Salmonella, Escherichia, Bacillus, unclassified and Enterococcus), 3 genera (33.33%) were isolated from males (Shimwellia, Pluralibacter and Lactobacillus), and one genus (11.11%) was isolated from females (Staphylococcus). Regarding body regions of adult S. gregaria, two genera (22.22%) were isolated from heads of both males and females (Salmonella, externally and Escherichia, internally) and one genus (11.11%) from heads of males, internally (Shimwellia). One genus (11.11%) was isolated from thoraces of both males and females, internally (Bacillus) and one genus (11.11%) from thoraces of females, internally (Staphylococcus). Two genera (22.22%) (unclassified, externally and Enterococcus, internally) were isolated from abdomens of both males and females and two genera (22.22%) (Pluralibacter and Lactobacillus) from abdomens of males, internally (Tables and ).
Table 4. Cultivable bacteria of different body regions of adult male S. gregaria.
Table 5. Cultivable bacteria of different body regions of adult female S. gregaria.
Isolation of bacteria from rearing environment
Four bacterial isolates were identified from the bacterial swaps of rearing environment of S. gregaria. These isolates were identified as four genera: unclassified, Enterococcus, Staphylococcus and Salmonella (Table ).
Table 6. Cultivable bacteria isolated from rearing environment of adult female S. gregaria.
Discussion
The importance of studying S. gregaria-microbiota interactions rely on the fact that this insect has a great impact on the heath of people who consumed insects as food. In addition, this insect could disseminate pathogens mechanically as its swarms are able to fly up to 150 Km/ day in searching for food [Citation17]. That is why we threw the light on internal and external bacterial genera of S. gregaria. Additionally, the reason for dividing insect body into head, thorax and abdomen is that some locust-eating people only eat the abdomen without the rest of the body.
Out of the nine genera, 7 genera were isolated internally and 2 genera were isolated from the external surface of insect. Regarding the sex, 8 and 6 bacterial genera were isolated from male and female, respectively. Concerning body region, male abdomen was the most loaded region (4 bacterial genera). Meanwhile, male thorax was the least loaded region (one genus of bacteria). Generally, no persistent species was isolated from all body regions. Regarding the rearing environment of S. gregaria, the four identified genera had already isolated from locust’s body regions. No additional genera were identified from the rearing environment. These results confirmed the hypothesis that locust bodies have contaminated their rearing environment with bacteria via direct contact. Previous studies on S. gregaria-bacteria association concentrated on the gut bacteria. Earlier studies described the isolation of Enterobacter liquefaciens, Enterobacter cloacae, Escherichia coli, Klebsiella pneumoniae, Pantoea (Enterobacter) agglomerans, Enterococcus casseliflavus, Acinetobacter sp., Serratia marcescens, Streptococci and some Gram-positive Cocci from the gut of S. gregaria [Citation11, Citation17–21]. Enterobacter agglomerans was reported to be 10–100-folds more abundant than Streptococci [Citation18]. Additionally, 293 bacterial taxa were isolated from the alimentary canals of six different grasshopper species. Firmicutes, Actinobacteria and Proteobacteria phyla were conserved in all tested grasshoppers [Citation21]. Conclusively, our study presented 5 bacterial genera (Lactobacillus and Pluralibacter from abdomen, Bacillus and Staphylococcus from thorax and Shimwellia from head) that were not isolated in previous studies. Possibly, the gut bacteria were acquired with the locust’s food. Furthermore, this study isolated two bacterial genera from the external surface of S. gregaria (unclassified from abdomen and Salmonella from head). It is generally believed that the gut flora of an insect is acquired from its environment [Citation21].
Several previous studies compare culture-dependent and molecular methods to identify bacteria. The faster, easier, more accurate and deeper method is very crucial, especially, in health and healthcare issues [Citation22–24]. However, the culture-dependent methods are intensively laborious, time-consuming and require additional microbiological tests to differentiate phenotypically similar species [Citation24]. On the other hand, molecular markers were successfully used for classification and identification of many microbial strains [Citation24,Citation25]. Recently, the positive and negative points of bacterial identification by molecular and other techniques have been reviewed, intensively [Citation25–27].
The present investigation relied on combining different methodologies to identify bacteria at species level. Cell and colony’s morphological characterization succeeded in differentiating isolates. Sequencing, sequence analyses and phylogenetic inference of 16S rRNA gene succeeded in definite identification of the isolates at genus level. RDP Classifier succeeded in assigning genera. Phylogenetic inference, multiple alignment and BLAST search succeeded in positioning genera to their most probable species in only 3 genera out of 9. On the other hand, morphological and molecular did not succeed to assign the isolate 8, even at genus level (Plesiomonas/ Shigella). We designated this isolated as unclassified. Standard biochemical and more confirmatory tests are recommended in the case of isolate 8, where genus is not resolved by the previous methodologies.
Our sequences displayed significant similarities of 94.17–99% with other sequences in GenBank. Out of 9, six of our sequences generated identities less than 98%. The general rule of thumb stated that: “Less than 97% sequence identity with the closest relative species is an indication of novel species status” [Citation28]. Relying on this rule, only one new genus was isolated in the present study (isolate 8 displayed 94.17% identity with Plesiomonas). In addition, the 97% similarity rule was recently updated [Citation29,Citation30]. Edgar [Citation29] assessed taxonomy-sequence identity association (using one locus) and found a reduction of prediction accuracy to 50% whenever the identity became 97% [Citation29]. In another study, he recommended sequencing of two or more hypervariable loci [Citation30]. A 98.65% sequence similarity was suggested as threshold for differentiating two species of bacteria using full-length 16S rRNA gene [Citation31]. Pearson has suggested that FASTA and BLAST are trustful tools of similarity and could be used to infer homology [Citation32]. He preferred protein and translated DNA sequences to be used in comparison of sequences than using DNA: DNA comparisons. Furthermore, he preferred using bit scores (bits >50) and E-values for inferring homology than similarity percent [Citation32]. Based on the aforementioned updates of the 97% general rule, we have at least 3 isolates confirmed to species level, 5 isolates confirmed to genus level with unconfirmed species and one unconfirmed genus (unclassified).
E. faecalis has been isolated, internally, from male and female abdomens. Meanwhile, L. paracasei and Pluralibacter sp. have been isolated, internally, from male abdomen. Consistent results were presented by Ike [Citation33] who reported that both Enterococcus and Lactobacillus are inhabiting gastrointestinal tracts of a varied group of animals from insects to human [Citation33]. These commensals are beneficial because they are lactic acid-producing bacteria. On the other hand, both genera were involved in endocarditis and other bacteremia when the bacterial species get into the blood [Citation34]. In addition to the isolation of Pluralibacter spp. from the guts of some insects like pink bollworm and fruit flies, it was incriminated in the outbreak of nosocomial bacteremia in Croatia and in other human bacteremia [Citation34].
Bacillus sp. has been isolated, internally, from male and female thoraces. Meanwhile, S. epidermidis has been isolated, internally, from female thorax. Many species of the genus Bacillus have been previously isolated from guts of many insects. Further confirmation to explain the source of this bacterial infections is recommended [e.g. Citation35,Citation36]. S. epidermidis is the first commensal organism on the mucous surfaces and human skin [Citation37]. In addition, S. epidermidis inhibits S. aureus biofilm formation and nasal colonization [Citation38]. The isolation of S. epidermidis from insects was previously reported as a contamination resulting from close association between insects and surgical tools [Citation38].
Escherichia sp. has been isolated, internally, from male and female heads. Meanwhile, Shimwellia sp. has been isolated, internally, from male head. Escherichia was previously isolated from the gut of B. mori [Citation39]. Many Escherichia sp. are well known endosymbionts whereas certain species are pathogens [Citation39]. Shimwellia sp. was first isolated from the hindgut of cockroaches [Citation40]. Shimwellia sp. is non-pathogenic, aerobic and enteric bacterium, having the ability to synthesise commercial products [Citation41–43].
Both Salmonella sp. were isolated as external contaminants of the locust body. S. bongori is considered the cause of salmonellosis, a gastrointestinal disease characterized by cramping and diarrhea and associated with cold-blooded animals [Citation44]. It is generally believed that both internal (gut) and external bacteria, isolated in this study are either environmentally acquired and/ or commensal endosymbionts. Possibly, the internal bacteria were acquired from food and the external bacteria were acquired from contaminated environment.
Conclusion
The present wok described isolation of 9 bacterial species from the different body regions of adult S. gregaria. This study revealed that S. gregaria could harbour 7 bacterial species. Additionally, 2 species were carried on the external body. Touching the sex, the bacterial load of male is higher than female. In general, no bacterial species was found persistent in all body regions. Male abdomen was the most loaded body region (4 bacterial species). We concluded that the bacterial species harboured by S. gregaria may have a symbiotic function (further studies are needed). In addition S. gregaria is involved in mechanical transmission and circulation of the isolated bacteria in nature.
Acknowledgements
Thanks go to the Deanship of Scientific Research at Jouf University for funding this work by the grant number (652/39) and for supporting us to complete this work. Both FHG and AMS contributed this work equally. They designed the study, conducted experimental work, performed data analyses, discussed the results and wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Cressman K. Desert locust assessment, Kingdom of Saudi Arabia, 23 May-1 June 1997. Rome (IT): Food and Agriculture Organization; 1997.
- Belayneh YT. Acridid pest management in the developing world: a challenge to the rural population, a dilemma to the international community. J Orthopt Res. 2005;14:187–196. doi:10.1665/1082-6467(2005)14[187:APMITD]2.0.CO;2.
- Belayneh YT. Emergency Transboundary Outbreak Pests (ETOPs) situation for September with a forecast through mid-November 2019. ETOP Bull. IX- USAID/OFDA, 2019.
- Roussi A. The battle to contain gigantic locust swarms. Nature. 2020;579:330. doi: 10.1038/d41586-020-00725-x
- Fraqueza M, Patarata L. Constraints of HACCP application on edible insect for food and feed, In: Future foods, IntechOpen, 2017.
- Kelemu S, Niassy S, Torto B, et al. African edible insects for food and feed: inventory, diversity, commonalities and contribution to food security. J Insects Food and Feed. 2015;1:103–119. doi: 10.3920/JIFF2014.0016
- Steedman A. Locust Handbook. 3rd ed Chatham: Natural Resources Institute; 1990.
- Brader L, Djibo H, Faye FG, et al. Towards a more effective response to desert locusts and their impacts on food security, livelihoods and poverty. Multilateral evaluation of the 2003–05 Desert Locust Campaign. Food and Agriculture Organisation, Rome, 2006.
- Andersson O, Steen HH, Karin H, et al. The grasshopper: A novel model for assessing vertebrate brain uptake. J Pharmacol Exp Ther. 2013;346(2):211–218. doi:10.1124/jpet.113.205476.
- Engel P, Kwong WK, McFrederick Q, et al. The bee microbiome: impact on bee health and model for evolution and ecology of host-microbe interactions. mBio. 2016;7(2):e02164–15. doi:10.1128/mBio.02164-15.
- Muratore M, Prather C, Sun Y. The gut bacterial communities across six grasshopper species from a coastal tallgrass prairie. PLoS ONE. 2020;15(1):e0228406. doi:10.1371/journal.pone.0228406.
- Van der Spiegel M, Noordam MY, Van der Fels-Klerx HJ. Safety of novel protein sources (insects, microalgae, seaweed, duckweed, and rapeseed) and legislative aspects for their application in food and feed production. Compr Rev Food Sci Food Saf. 2013;12:662–678. doi: 10.1111/1541-4337.12032
- Dillon RJ, Vennard CT, Charnley AK. Gut bacteria produce components of a locust cohesion pheromone. J Appl Microbiol. 2002;92:759–763. doi: 10.1046/j.1365-2672.2002.01581.x
- Dillon RJ, Vennard CT, Buckling A, et al. Diversity of locust gut bacteria protects against pathogen invasion. Ecol Let. 2005;8(12):1291–1298. doi: 10.1111/j.1461-0248.2005.00828.x
- Thiery I, Frachon E. Identification, isolation, culture and preservation of entomopathogenic bacteria. In: Lacey AL, editor. Manual of Techniques in Insect Pathology. IX. London, UK: Academic Press; 1997. p. 55–77. https://doi.org/10.1016/B978-012432555-5/50006-3.
- Hall L, Doerr KA, Wohlfiel SL, et al. Evaluation of the MicroSeq system for identification of mycobacteria by 16S ribosomal DNA sequencing and its integration into a routine clinical mycobacteriology laboratory. J Clin Microbiol. 2003;41:1447–1453. doi: 10.1128/JCM.41.4.1447-1453.2003
- Lavy O, Gophna U, Gefen E, et al. The effect of density-dependent phase on the locust gut bacterial composition. Front Microbiol. 2019;9:3020. doi: 10.3389/fmicb.2018.03020
- Dillon R, Charnley K. Mutualism between the desert locust Schistocerca gregaria and its gut microbiota. Res Microbiol. 2002;153(8):503–509. doi: 10.1016/S0923-2508(02)01361-X
- Dillon R, Webster G, Weightman A, et al. Composition of Acridid gut bacterial communities as revealed by 16S rRNA gene analysis. J Invertebr Pathol. 2008;97:265–272. doi: 10.1016/j.jip.2007.09.010
- Dillon RJ, Webster G, Weightman AJ, et al. Diversity of gut microbiota increases with aging and starvation in the desert locust. Antonie Van Leeuwenhoek. 2010;97:69–77. doi: 10.1007/s10482-009-9389-5
- Eutick ML, O’Brien RW, Slaytor M. Bacteria from the gut of Australian termites. Appl Environ Microbiol. 1978;35:823–828. doi: 10.1128/AEM.35.5.823-828.1978
- Wolcott RD, Cox SB, Dowd SE. Healing and healing rates of chronic wounds in the age of molecular pathogen diagnostics. J Wound Care. 2010;19:272–278.
- Dowd SE, Wolcott RD, Kennedy J, et al. Molecular diagnostics and personalized medicine in wound care: assessment of outcomes. J Wound Care. 2011;20(232):234–239.
- Rhoads DD, Wolcott RD, Sun Y, et al. Comparison of culture and molecular identification of bacteria in chronic wounds. Int J Mol Sci. 2012;13(3):2535–2550. doi: 10.3390/ijms13032535
- Bajinka O, Secka O. Integration of molecular methods into microbiological diagnostics. Appl Microbiol Open Access. 2017;3;130. doi: 10.4172/2471-9315.1000130
- Buszewski B, Rogowska A, Pomastowski P, et al. Identification of microorganisms by modern analytical techniques. J AOAC Int. 2017;100(6):1–16. doi: 10.5740/jaoacint.17-0207
- Rajapaksha P, Elbourne A, Gangadoo S, et al. A review of methods for the detection of pathogenic microorganisms. Analyst. 2018; doi:10.1039/c8an01488d.
- Rosselló-Mora R, Kampfer P. Defining microbial diversity-the species concept for prokaryotic and eukaryotic microorganisms. In: Bull AT. Microbial diversity and bioprospecting. ASM Press, Washington DC; 2004.
- Edgar R. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. Peer J. 2018;6:e4652. doi: 10.7717/peerj.4652
- Edgar R. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics. 2018a;34(14):2371–2375. doi: 10.1093/bioinformatics/bty113
- Kim M, Oh H-S, Park S-C, et al. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Sys Evol Microbiol. 2014;64:346–351. doi: 10.1099/ijs.0.059774-0
- Pearson W. An introduction to sequence similarity (“Homology”) searching. Curr Protoc Bioinformatics. 2013;03:1–8.
- Ike Y. Pathogenicity of Enterococci. Nippon Saikingaku Zasshi. 2017;72(2):189–211. doi: 10.3412/jsb.72.189
- Cannon JP, Lee TA, Bolanos JT, et al. Pathogenic relevance of Lactobacillus: a retrospective review of over 200 cases. Eur J Clin Microbiol Infect Dis. 2005;24:31–40. doi: 10.1007/s10096-004-1253-y
- Lee J, Hwang S, Cho S. Immune tolerance to an intestine-adapted bacteria, Chryseobacterium sp., injected into the hemocoel of Protaetia brevitarsis seulensis. Sci Rep. 2016;6:31722.
- Anand A, Vennison S, Sankar S, et al. Isolation and characterization of bacteria from the gut of Bombyx mori that degrade cellulose, xylan, pectin and starch and their impact on digestion. J Insect Sci. 2010;10:107. doi: 10.1673/031.010.10701
- Otto M. Staphylococcus epidermidis the ‘accidental’ pathogen. Nat Rev Microbiol. 2009;7:555–567. doi: 10.1038/nrmicro2182
- Iwase T, Uehara Y, Shinji H, et al. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010;465:346–349. doi: 10.1038/nature09074
- Liu S, Dong J, Ruiting L, et al. Escherichia marmotae sp. nov., isolated from faeces of Marmota himalayana. Int J Sys Evol Microbiol. 2015;65:2130–2134. doi: 10.1099/ijs.0.000228
- Brzuszkiewicz E, Waschkowitz T, Wiezer A, et al. Complete genome sequence of the B12-producing Shimwellia blattae strain DSM 4481, Isolated from a Cockroach. J Bacteriol. 2012;194(16):4436–4436. doi: 10.1128/JB.00829-12
- Andres S, Wiezer A, Bendfeldt H, et al. Insights into the genome of the enteric bacterium Escherichia blattae: Cobalamin (B12) biosynthesis, B12-dependent reactions, and inactivation of the gene region encoding B12-dependent glycerol dehydratase by a New Mu-like prophage. J Molec Microbiol Biotechnol. 2004;8(3):150–168. doi: 10.1159/000085788
- Acedos M, Ramon A, de la Morena S, et al. Isobutanol production by a recombinant biocatalyst Shimwellia blattae (p424IbPSO): Study of the operational conditions. Biochem Eng J. 2018;133:21–27. doi: 10.1016/j.bej.2018.02.002
- Acedos M, de la Torre I, Santos V, et al. Kinetic modelling of the isobutanol production from glucose using Shimwellia blattae (p424IbPSO) strain: effect of initial substrate concentration. Ind Eng Chem Res. 2019;58(4):1502–1512. https://doi.org/10.1021/acs.iecr.8b05121.
- Tortora GA, Funke BR, Case CL, et al. Microbiology: An introduction. 13th ed. New York, USA: Pearson; 2018.