
Abstract
A theoretical assessment of the effects of the OH, NH2 and Br substituents on the properties of 2,2′-bipyridine has been carried out using density functional theory approach. The substituents were found to alter both electronic and structural properties of substituted 2,2′-bipyridine with the inductive and resonance effects playing significant roles. For the relative stability study, an order of ortho > para > meta was predicted for each set of the 2,2′-bipyridine derivatives with the ortho conformers being more stable by 4.62, 6.54 and 2.18 kcal/mol than the meta counterparts of the hydroxo, amino and bromo configurations respectively. On the part of electronic property, the electron donating / withdrawing tendency of the substituents was crucial in making electrons available to the bipyridine system. A general reduction in HOMO – LUMO energy gap was noticed upon substitution with the NH2 substituent having greater effect. Furthermore, the calculated reactivity parameters of the 2,2′-bipyridine were altered by substitution with –OH and –NH2 substituents causing reduction in ionization potential (IP) and absolute electronegativity (χ) while the –Br substituent did otherwise. For the solubility simulation, a more negative ΔGsolution was predicted for the hydroxo and amino derivatives at the para position, but reverse was the case for the bromo counterpart. In the case of the optical property, the range separated Time-dependent (LC-TDDFT) calculation revealed that the shifts in absorption λmax caused by the substitution is both substituent and position dependent.
1. Introduction
2,2′-Bipyridine is a N-donor organic molecule that consists of two pyridine rings linked together by single C–C bond [Citation1,Citation2]. The presence of lone pairs of electrons on both nitrogen atoms makes 2,2′-bipyridine basic and as such, functions as a bidentate ligand by donating these lone pairs electrons to a transition metal [Citation3]. Bipyridines have a long history of being used in forming a variety of transition metal complexes for both catalysis and material applications, [Citation4–9] but their chemistry is less studied compared to phosphine [Citation10]. 2,2′-Bipyridine was found to give excellent results when used as a ligand in homogenous catalytic transfer hydrogenation of acetophenone [Citation3]. In another report, its derivatives were applied in different catalytic reactions ranging from C–H borylation reaction to alkyl halides and CO2 reduction [Citation11–16] as well as in photocatalysis [Citation17–19].
It is expected that the electronic properties of 2,2′-bipyridine should be altered upon substitution [Citation20–24]. Electron donating substituents (EDS) are believed to increase the electron density in the 2,2′-bipyridine system which in turn aids the electron donating ability of 2,2′-bipyridine ligand to the transition metal. On the other hand, electron withdrawing substituents (EWS) which are deactivating agents are expected to behave otherwise [Citation24]. However, the aftermath of the substitution on the resultant bipyridine properties is governed by two factors which are inductive and resonance effects. These two factors compete, and as a result making it difficult to predict a definite effect that a particular substituent will have on bipyridine system [Citation25,Citation26].
Nevertheless, the use of a specific ligand for catalyst design is a subject of compatibility with the metal centre [Citation3]. A right combination gives a highly efficient catalytic system. Thus, screening of bipyridine derivatives (BPD) prior to their use as ligands in catalytic system would be considered essential. However, the scanty information about the effects of substituents on the properties of 2,2′-bipyridine makes the screening challenging [Citation4]. For this study, –OH, –NH2 and –Br substituents have been chosen to examine their effects on the properties of bipyridine. Both –OH and –NH2 are known for their excellent nucleophilic properties with the latter having a strong mesomeric effect, whereas, –Br is a good leaving group [Citation27,Citation28]. Their presence is believed to influence the organic reactions undertaken by the BPD [Citation22–24,Citation29]. Hence, investigating the overall effects that these substituents have on the properties of the resulting BPD would be a significant contribution to the ever dynamic field of catalysis where the bipyridine complexes are important catalysts. Moreover, this task can be effectively undertaken theoretically using Density Functional Theory (DFT) which has proved to be a useful tool in understanding catalysis at molecular level [Citation3]. It is on this premise that we intend to study the effect of –OH, –NH2 (EDS) and –Br (EWS) on the structural and electronic properties of 2,2′-bipyridine using DFT approach.
2. Computational details
The structures of all the 2,2′-bipyridine derivatives were fully optimized by means of GAUSSIAN 09 program [Citation30], and the nature of convergence was verified via frequency calculation which in return predicted positive value for all the calculated frequencies. Then the reactivity parameters such as ionization potential (IP), electron affinity (EA), absolute electronegativity (χ) and absolute hardness (η) were computed using the method outlined in the literature [Citation31,Citation32]. Furthermore, natural bond orbital (NBO) calculation was carried out to establish the hybridization of heteroatom N [Citation33], whereas the Frontier molecular orbitals for HOMO and LUMO energy levels were generated from formatted check file of the optimized structures, using a range separated functional LC-BLYP [Citation34,Citation35]. The absorption property of the bipyridine derivatives was computed via the same range separated functional with the use of linear response formalism of Time-dependent (TDDFT) calculation approach. However, the other calculations were carried out through ab initio density functional theory (DFT) by adopting the B3LYP method [Citation36,Citation37]. Basis set of 6-311++g(d,p) was used for non-metals, while SDD was used for Rh metal with the inclusion of empirical dispersion term gd3 [Citation38] for the metal complex calculations in order to correct for the London dispersion interactions. .
Figure 1. Structure of 2,2′-bipyridine.
3. Results and discussion
3.1 Stability
Molecules have ability to exist in either singlet or triplet state based on the arrangement of electrons in the orbital. As a result, it is always preferred to establish the more stable state at the onset of any study. For this reason, the energy gap between the singlet and triplet states for the 2,2′-bipyridines were determined using their zero-point electronic energy and the results are presented in Table . The energy gaps were predicted to be within the same range and their positive values affirm the singlet states to be of lower energy, thereby making them more stable than the corresponding triplet states. The lower stability of triplet state molecules compared to that of singlet, is understood to be due to the presence of two unpaired electrons of the same type that strongly repel each other. Another property worthy of examining is the relative energy, which determines the stability of one configuration relative to the others. These values were obtained from zero-point corrected electronic energies for the fully optimized configurations of the three set of bipyridine derivatives. The results obtained for hydroxo derivatives of bipyridine as listed in Table , affirms the ortho-substituted configurations as the most stable, followed by the para (1.99 kcal/mol), while the meta counterpart (4.62 kcal/mol) was the least stable conformer [Citation39–41]. The stability of ortho configuration over para and meta could be attributed to the electron donating ability of –OH species, which in principle shows activating effect in the order of ortho > para > meta. This sequence is actually in consonant with the result obtained for the relative stability shown in Table . For the amino substituent which is also an electron donating species, the same trend of relative stability of configurations was found for the derivatives (Table ), with ortho conformer being the most stable while meta was the least. The heteroatom N in the pyridyl ring is more electronegative than the carbon, as a result, pulls electrons towards itself which in turn destabilizes the ring system. However, the lone pairs of electrons present on oxygen and nitrogen of –OH and –NH2 respectively are resonantly donated to the pyridyl ring. Since the electrons donated from the substituents stabilize the ring system, substitution at the ortho position is more inductive-ly favoured than any other position, thereby making the ortho configuration most stable. In a similar version to the EDS, the relative stability for the configurations of bromo derivatives was predicted to follow the same order (ortho > para > meta). Though, bromine is an EWS, but as a member of halogen group, its order of deactivating effect is ortho > para > meta. This behaviour of –Br can be attributed to its electronegativity which makes it withdraw electrons from pyridyl ring whereas its lone electron pairs are in turn resonantly donated to the ring. Overall, the resonant donation of electrons is inductively favoured at the ortho position and this makes the process more active at this position than the others due to its closeness to the heteroatom N.
Table 1. Energy gap (ES-T) in eV between the singlet and triplet states of bipyridine and its derivatives.
Table 2. Relative energies (kcal/mol) for the bipyridine derivatives.
Analysis of the result suggests the stability of bipyridine system to be controlled by the nitrogen atom, hence, the understanding of its hybridization state via the exploration of natural bond orbital (NBO) result is essential. Furthermore, the heteroatom N hybridization state could be substantiated by examining its surrounding bond parameters in accordance with the atom numbering given in Scheme 1. The results obtained as presented in Table shows that the predicted bond angles for C1-N2-C3 are very close to 120o which is the required bond angle for sp2 hybridization. On the other hand, NBO result indicates that the activating substituents (–OH and –NH2) pushed the hybridization of heteroatom N towards sp2 whereas –Br, a deactivating type did otherwise at the ortho, which is the most active position.
Scheme 1. Atom numbering of 2,2′-bipyridine derivative.
Table 3. Selected geometrical parameters and N-hybridization for bipyridine and its derivatives.
3.2 Molecular geometry
The structural property of a molecule is predicated upon its molecular geometry which influences its inherent properties ranging from reactivity to biological activity [Citation42,Citation43]. This essential property was examined via the optimization calculation, of which the bond lengths and the bond angles; the important parameters for geometrical specification were extracted as listed in Table . The analysis of these results shows that the bond lengths associated with the substituent bearing pyridine part of the derivatives changed with respect to the position of the substituent whereas those of the unsubstituted part were less sensitive as they remained nearly constant. This observation signals that the presence of substituent does affect the geometrical property of the derivatives and also that, this effect is both positional and species-wise dependent. For the C–N bond lengths, C3–N2 seems to be more sensitive to the position of the substituent than the C1–N2 counterpart as distinct bond distances were predicted for different positions in each of the derivatives. On the part of C–C bond lengths, C3–C4 were calculated to be longer when the substituents were at the ortho position (140.4, 141.0 and 139.7 pm for –OH, –NH2, and –Br derivatives respectively) than the others, with the para position of the –OH and –NH2 derivatives giving rise to the distance shorter than that recorded for the parent bipyridine. However, an opposite trend was predicted for the C4–C5 of –OH and –NH2 conformers. As expected, the bond distances between the substituent and the carbon to which it is attached (C–O and C–N for –OH and –NH2 derivatives respectively) at different positions were found to be sensitive to the activity of the substituent. The EDS (–OH and –NH2) which are more active at ortho and para positions, gave rise to shorter bond length (136.1 pm for –OH and 138.3 pm for –NH2) than the meta position (136.5 pm for –OH and 139.1 pm for –NH2). Another important parameter worth considering is the bond angle at the carbon bearing the substituent, as it gives insight into the carbon hybridization. For the hydroxo derivatives, bond angles 123.7o (N2–C3–C4), 118.8o (C4–C5–C6) and 118.2o (C3–C4–C5) were predicted for the ortho, para and meta positions respectively. Similar trend (ortho > para > meta) was obtained for the bond angles of both amino and bromo derivatives. This result suggests that the hybridization of the substituent bearing carbon at ortho position is approximately sp2 as the bond angle requirement for this type of hybridization is 120o. However, the decrease in the bond angle as we move towards meta position is an indication of the carbon hybridization tending towards sp3. In addition, the higher H1–N–H2 (115.2o) predicted for amino group at ortho position gives the N greater sp2 character than either para or meta position with lower bond angle.
Table 4. Bond parameters for 2,2′-bipyridine and its derivatives.
3.3 Reactivity parameters
A key factor in the choice of ligands for complexation is HOMO–LUMO energy gap, as this determines the reactive nature of ligands. The lower this gap is for a ligand, the more reactive it would be and as such, this factor is sometime used for ligands screening. In this study, the HOMO-LUMO energy gap prediction task was undertaken using range separated LC-BLYP, a functional which had been shown to give minimized error in the La valence excitation [Citation44,Citation45]. The results obtained from this calculation are highlighted in Table . It is clearly shown in the Table that substitution caused reduction in the HOMO–LUMO energy gaps of the resulting derivatives, except for the para substituted bromo bipyridine. Comparing the substituents, each of the amino derivatives was found to have greater effect on the energy gap than their corresponding hydroxo and bromo counterparts. This feat makes the amino derivatives more reactive. Moreover, the substitution at the meta position resulted in a greater energy gap reduction than the other positions for all the substituents. This observation is in consonant with the relative energies for which the meta derivatives were predicted to be less stable than the other configurations. The effect of the substituents on the electronic properties of bipyridine system was further investigated, via Frontier molecular orbitals showing the distribution of HOMO and LUMO for 2,2′-bipyridine and its derivatives, and the results are presented in Figure . As observed, the electron clouds were evenly distributed over both carbons and the π-electrons in the ring for the HOMOs of all the bipyridines, except for the ortho and para amino derivatives where the pyridyl ring without substituent showed deficiency in electron density. On the part of the LUMO, both the atoms and the π system of the bipyridine ring and those of the derivatives with EDS were characterized with highly insufficient electron clouds. On the contrary, the LUMOs of bromo derivatives were identified with even distribution of electron cloud over the atoms and π electrons in the ring system while their bromine substituent was electron deficient.
Figure 2. Frontier molecular orbitals for 2,2′-bipyridine and its derivatives at LC-BLYP theory.
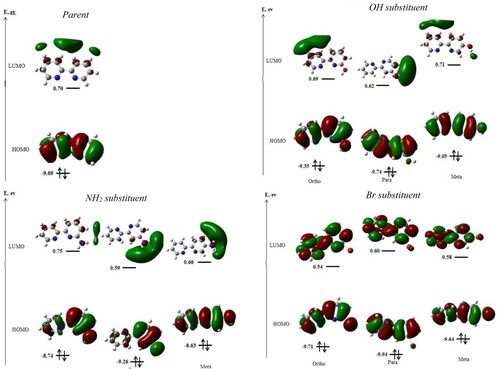
Table 5. EHOMO-LUMO gap (eV) for bipyridine and its derivatives.
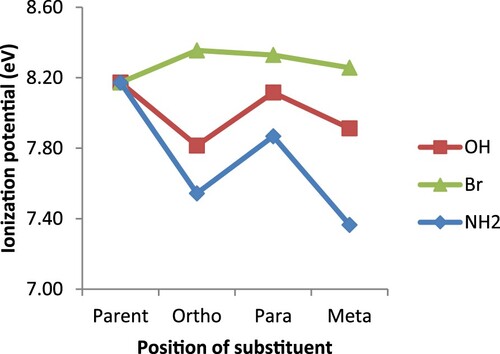
The other electronic properties that determine the reactivity of a ligand are ionization potential (IP), electron affinity (EA), absolute electronegativity as well as absolute hardness. Both absolute electronegativity (χ) and absolute hardness (η) are known to be important parameters in catalyst design, as they are essential in assessing polarity of M-L bonds [Citation33]. On the other hand, ionization potential reveals the basic nature, stability as well as the conductivity of a substance. The lower the IP value, the more basic and conductive, but less stable a substance is [Citation33,Citation46–48]. The result presented in Figure shows that both hydroxyl and amino substituents caused reduction in IP value of the resultant derivatives at all positions. This observation could be attributed to the electron donating ability of the substituents which in return populates the bipyridine system with electrons, thereby making them more basic, conductive and reactive. On the contrary, bromine substituent which is a deactivating agent increased the IP value by withdrawing electrons from the bipyridine system and as such, making the resultant derivatives less basic and conductive, but more stable. Careful analysis of the result, reveals the influence of inductive effect on this reactivity parameter. For the hydroxyl substituent which is an electron donating specie, electrons are more availed to bipyridine system at ortho position than others that are farther from heteroatom N. This eventually makes the ortho derivative have lower IP than other positions. On the other hand, the bromine which is an EWS caused greater increase in IP value at ortho position than meta or para, because of its closeness to the heteroatom N.
Figure 3. Ionization potential for 2,2′-bipyridine and its derivatives.
Another important reactivity parameter examined in this study is electron affinity, which denotes the acidic nature of a ligand. This parameter also, indicates the conductive nature as well as the stability of a substance. The greater its value, the more acidic but less conductive a substance is [Citation33,Citation46,Citation49–51]. In this regard, the higher EA values recorded for the ortho substituted derivatives in Figure , is an indication of their being more acidic but less conductive than the para and meta counterparts. This result is consistent with the report by Guha and his co-workers [Citation33] for which a species can be both more basic and acidic than its counterparts at the same time. In addition, the order of EA (ortho > para > meta) is the same as that of configurations’ relative stability earlier discussed. The highly acidic nature predicted for the bromo derivative is due to the deactivating property of bromine substituent which eventually makes bipyridine electron deficient. On the part of the absolute electronegativity, parent 2,2′-bipyridine seems to be more electronegative than both amino and hydroxo derivatives as presented in Figure . This could be understood from the fact that both –NH2 and –OH are activating agents and as such, making the bipyridine system electron rich; a feat that eventually decreases its ability to attract electrons. For the bromine substituents, its electron withdrawing ability greatly contributed to the higher electronegativity value recorded for bromo derivatives over the parent 2,2′-bipyridine. The other interesting property often considered in catalysis is the absolute hardness. It is an important parameter in the design of an efficient catalytic system, based on the principle that hard ligands go with hard metals while the soft ones go with soft metals [Citation52]. The results represented in Figure shows that the substituted bipyridines are generally softer than the parent and as such they would be much more compatible with the soft metal systems than the parent 2,2′-bipyridine.
Figure 4 Electron affinity for 2,2′-bipyridine and its derivatives.
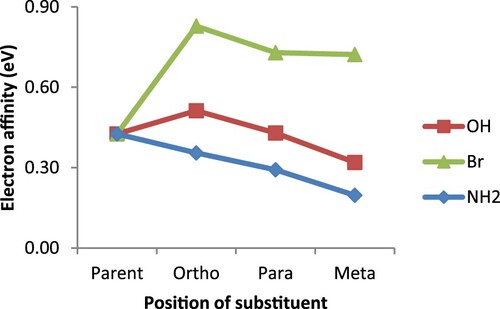
Figure 5 Absolute electronegativity for 2,2′-bipyridine and its derivatives.
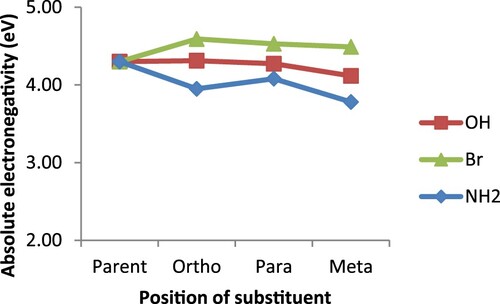
Figure 6 Absolute hardness for 2,2′-bipyridine and its derivatives.
3.4 Solubility
The applicability of compounds in both industry and research is predicated upon the ease of their solubility in common solvents for which solute-solvent interaction plays a significant role. This interaction is usually predicted via Polarizable Continuum Model (PCM) adopting the integral equation formalism for which the calculated change in Gibbs free energy of the solution (ΔGsolution) can be correlated to the degree of solubility. The result presented in Table , highlights the ΔGsolution predicted for the interaction between the bipyridines understudied and the selected solvents. The negative values obtained for the ΔGsolution is an indication that the interaction is an exergonic process. Besides, the solubility of parent and the derivatives was predicted to increase with the solvent polarity as reflected by the increasing negative value of ΔGsolution moving from the diethyl ether, the least polar to the water which is the most polar among the solvents. The ortho derivatives of –OH and –Br showed similar energy of interaction with the parent. Moreover, the order of interaction for the –OH and –NH2 derivatives, was predicted as ortho < meta < para while that for the –Br was para < meta < ortho. It is worth noting that the EDS (–OH and –NH2) generally improved the solubility of bipyridine in the selected solvents with greater effect being recorded at para and meta positions. Steric hindrance could be seen playing a significant role in the solute-solvent interaction, as the ortho substitution could not significantly improve the solubility. In contrary, the derivatives’ solubility was appreciably enhanced by the para and meta substitution, for which the EDS are far from the hetero nitrogen atom.
Table 6. Calculated ΔGsolution (kcal mol−1) for the interaction between bipyridines and solvents.
3.5 Calculated absorption spectra
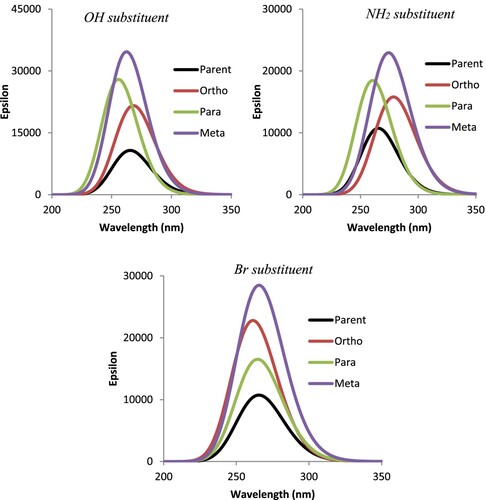
Absorption is one of the important optical properties of a compound arising from its interaction with electromagnetic radiation. Moreover, the absorption maximum wavelength (λmax) which is an inherent property of a compound owing to its functional group or the nature and the type of it bonds is environmentally dependent, and as such changes upon interacting with the solvents. Furthermore, solvent polarity together with its hydrogen bonding ability is believed to play significant role in the bathochromic (red shift) or hypsochromic (blue shift) shift of the compound’s λmax upon dissolution [Citation53,Citation54]. As a way of examining the nature of shift in λmax caused by the understudied substituents on bipyridine upon substitution, a long-range-corrected Time-dependent (LC-TDDFT) calculation was carried out using Gaussian 09 [Citation30]. The choice of this range-separated functional is based on its ability to give results that assent to experimental data [Citation34,Citation35]. As depicted in Figure , the absorption of the parent bipyridene predicted at 263 nm could be attributed to π-π* transition owing to the conjugated π electron system in both pyridyl rings. Upon substitution, the absorption energy red shifted at ortho and meta positions, but blue shifted at para for both –OH and –NH2 (EDS). The bathochromic shift observed at ortho and meta could be due to the inductive effect as the EDS are closer to the heteroatom N and as such, able to easily compensate for the electron withdrawing tendency of the heteroatom N and at the same time, increasing the electron density in π system. This eventually brings the energy of molecular orbital closer, thus leading to the longer absorption wavelength recorded at the two positions. However, the bromine substituent (EWS) was found to have less significant effect on the absorption property of bipyridine as both parent and derivatives have comparable λmax (Figure ).
Figure 7. Effect of substituent substitution on calculated absorption spectra of bipyridines.
The effect of solvent interaction on the derivatives λmax is another important parameter worth considering. This was examined via Time-dependent (TD) DFT calculation using linear response (LS) in conjunction with PCM model but benchmarked with the LC-TDDFT. The results presented in Figure 1S show loss of excitation energy by the ortho derivative of hydroxobipyridine in the TDDFT calculation when compared with that of benchmark LC-TDDFT. This disparity in excitation energy is due to the incorrect Physics of formulation of B3LYP for which both short and long ranges were not separated as established by Wong and co-workers [Citation34,Citation35]. Nevertheless, the trend in the λmax shift predicted by the TDDFT calculation, for the ortho hydroxo derivative is consistent with that of the benchmark calculated at LC-TDDFT for all the corresponding solvents, as shown in Figure 1S. Moreover, the TDDFT results depicted in Figures 1S–3S showed a general red shift in λmax for the selected solvents with the extent of shift increasing with the solvent polarity. In a nutshell, the observed increasing λmax shift with the solvent polarity suggests more stabilization in the excited due to solvation. This behaviour actually leads to the decrease in Franck-Condon energy with increasing solvent polarity and as such resulting to longer λmax [Citation54].
3.6 Metal complexes
Carbonyl complexes of rhodium (I) with chelating bidentate ligands have been reported to have high reactivity and catalytic activity [Citation55]. Moreover, the effect of these ligands on the of complex metal-carbonyl interaction, which informs their sigma donating ability could be understood via Dewar-Chatt-Duncanson model [Citation56]. Also, using spectroscopic approach, the frequency associated with C = O stretching vibrational mode could be used as a measure for ligand sigma donating ability. In order to examine this property together with other important complex properties, the resulting 2,2′–bipyridine derivatives were theoretically used to form rhodium (I) carbonyl complexes. The structures of these complexes are shown in Scheme 2, while their relative stabilities are presented in Table . The result showed that the complexes formed from ortho derivatives were the most stable for the hydroxyl and amino derivatives whereas the most stable configuration predicted for the bromo derivative was the para. Steric hindrance due to the presence of substituent at ortho position is expected to make the resulting complex less stable, but the ability of –OH and –NH2 to have hydrogen bonding interaction with the chlorine ligand overrides the hindrance and thus makes the complexes of hydroxyl and amino bipyridine derivatives more stable at ortho position than any other position. On the part of the bromo derivatives, the steric effect was much more pronounced at the ortho than other positions and as such, made it the least stable (Table ).
Scheme 2. Predicted structure for bipyridine derivatives ([Rh(CO)Cl(Bipy-X)]).
![Scheme 2. Predicted structure for bipyridine derivatives ([Rh(CO)Cl(Bipy-X)]).](/cms/asset/722a951a-531f-479a-b033-d0dd00141913/tusc_a_1843872_f0009_ob.jpg)
Table 7. Relative energies (kcal/mol) for the Rh complexes [Rh(CO)Cl(Bipy-X)].
Another important parameter worth considering is the complex structural property. Following the structural optimization, all the complexes were predicted to exhibit square planar geometry around rhodium atoms as shown in Scheme 2. As presented in Table , both Rh-N and Rh-C bond lengths were found to follow the same trend, with the complexes of ortho derivatives having shortest bond length in the respective set. Using Dewar-Chatt-Duncanson model, the shorter the Rh-C bond length the greater the π back-bonding effect [Citation56]. Hence, the π back-donation would be more pronounced at ortho than other positions. Although, the C = O bond length is used as a measure for σ donation tendency of ligands, but the result depicted in Table shows that it not affected by substitution as the similar C = O bond lengths were calculated for both complexes of parent and substituted bipyridines. This suggests that the nature of these substituents as well as their position is insensitive to σ donation tendency of biypridine system. This assertion is further supported by the calculated frequencies for the C = O stretching mode (Table ), which were predicted to be approximately the same (≈ 2060cm−1) for all the complexes.
Table 8. Selected bond lengths and calculated C = O frequencies of Rhodium complexes of bipyridine and its derivatives.
4. Conclusion
In this study, the effects of OH, NH2 and Br substituents on the properties of 2,2′-bipyridine have been documented. The singlet ground state derivatives of the 2,2′-bipyridine were found to be more stable than the triplet state counterparts for all the substituents. Furthermore, the relative energies computation revealed that substitution at the ortho position resulted in a more stable configuration than either para or meta. For the electronic property, the inductive and resonance effects together with the electron donating / withdrawing ability of the substituents played significant role. Substitution of the parent 2,2′-bipyridine resulted in reduced HOMO–LUMO energy gaps with the EDS having greater effect than the EWS, while the extent of the reduction in position-wise followed meta > ortho > para sequence. In addition, OH and NH2 substituents caused reduction in the ionization potentials of the resultant bipyridine systems while the Br counterpart did otherwise. Furthermore, the calculated absolute electronegativity of 2,2′-bipyridine was enhanced by the Br substituent but reduced by the OH and NH2. As for the solubility simulation, substitution at the para position resulted in more soluble hydroxo and amino BPDs but less soluble bromo derivative. Regarding the optical property, a red shift in absorption energy was predicted for the substitution at ortho and meta positions by EDS.
Supplemental Material
Download MS Word (529 KB)Acknowledgements
The authors gratefully acknowledge the support from the Islamic University of Madinah, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Merritt LL, Schroeder ED. The crystal structure of 2,2’-bypiridine. Acta Cryst. 1956;9:801–804.
- Kaes C, Katz A, Hosseini MW. Bipyridine: the most widely used ligand. A review of molecules comprising at least two 2,2’-bipyridine units. Chem Rev 2000;100:3553–3590.
- Popoola SA, Jaseer EA, Al-Saadi AA, et al. Iridium complexes as catalysts in the hydrogen transfer of isopropanol to acetophenone: ligand effects and DFT studies. Inorg Chim Acta. 2015;436:146–151.
- Bednářová E, Dračínský M, Malatinec Š, et al. Synthesis of a Bolm’s 2,2'-bipyridine ligand analogue and its applications. Adv Synth Catal. 2018;360:2869–2878.
- Puntus LN. Photophysical processes in ‘supramolecular balls’ formed by lanthanide chloride with 2,2′-bipyridine. Helv Chim Acta. 2009;92:2552–2564.
- Hotze ACG, van der Geer EPL, Kooijman H, et al. Characterization by NMR spectroscopy, X-ray analysis and cytotoxic activity of the ruthenium(II) compounds [RuL3](PF6)2 (L = 2-Phenylazopyridine or o-Tolylazopyridine) and [RuL'2L'’](PF6)2 (L’, L'’ = 2-Phenylazopyridine, 2,2'-bipyridine). Eur J Inorg Chem. 2005: 2648–2657.
- Jiang P, Yan L, Zhang X, et al. Electrogenerated chemiluminescence sensor based on tris(2,2’-bipyridine)ruthenium(II)-immobilized natural clay and ionic liquid. Electroanal. 2010;22:204–208.
- Penner A, Schröder T, Braun T, et al. Synthesis, structure, and reactivity of rhodium bipyridine compounds: formation of a RhII hydrido cluster and a RhIII peroxido complex. Eur J Inorg Chem. 2009: 4464–4470.
- Norris MR, Concepcion JJ, Glasson CRK, et al. Synthesis of phosphonic acid derivatized bipyridine ligands and their ruthenium complexes. Inorg Chem. 2013;52:12492–12501.
- Buonomo JA, Everson DA, Weix DJ. Substituted 2, 2′-bipyridines by nickel catalysis: 4, 4′-di-tert-butyl-2, 2′-bipyridine. Synth (Stuttg). 2013;45:3099–3102.
- Shrestha R, Dorn SCM, Weix DJ. Nickel-catalyzed reductive conjugate addition to enones via allylnickel intermediates. J Am Chem Soc. 2013;135:751–762.
- Everson DA, Shrestha R, Weix DJ. Nickel-catalyzed reductive cross-coupling of aryl halides with alkyl halides. J Am Chem Soc. 2010;132:920–921.
- Ishiyama T, Takagi J, Ishida K, et al. Mild iridium-catalyzed borylation of arenes. high turnover numbers, room temperature reactions, and isolation of a potential intermediate. J Am Chem Soc. 2002;124:390–391.
- Mkhalid IAI, Barnard JH, Marder TB, et al. C-H activation for the construction of C-B bonds. Chem Rev. 2010;110:890–931.
- Smieja JM, Sampson MD, Grice KA, et al. Manganese as a substitute for rhenium in CO2 reduction catalysts: the importance of acids. Inorg Chem. 2013;52:2484–2491.
- Wang S, Qian Q, Gong H. Nickel-catalyzed reductive coupling of aryl halides with secondary alkyl bromides and allylic acetate. Org Lett. 2012;14:3352–3355.
- Shih HW, Vander Wal MN, Grange RL, et al. Enantioselective α-benzylation of aldehydes via photoredox organocatalysis. J Am Chem Soc. 2010;132:13600–13603.
- Jayasundara CRK, Unold JM, Oppenheimer J, et al. A catalytic borylation/dehalogenation route to o– fluoro arylboronates. Org Lett. 2014;16:6072–6075.
- Slinker JD, Gorodetsky AA, Lowry MS, et al. Efficient yellow electroluminescence from a single layer of a cyclometalated iridium complex. J Am Chem Soc. 2004;126:2763–2767.
- Rittmeyer SP, Groß A. Structural and electronic properties of oligo- and polythiophenes modified by substituents. Beilstein J Nanotechnol. 2012;3:909–919.
- Inokuma Y, Yoon ZS, Kim D, et al., meso-Aryl-substituted subporphyrins: Synthesis, structures, and large substituent effects on their electronic properties. J Am Chem Soc. 2007;129:4747–4761.
- McGrath KK, Jang K, Robins KA, et al. Substituent effect on the electronic properties and morphologies of self-assembling bisphenazine derivatives. Chem Eur J. 2009;15:4070–4077.
- Sahu H, Panda AN. Computational study on the effect of substituents on the structural and electronic properties of thiophene−pyrrole-based π–conjugated oligomers. Macromol. 2013;46:844–855.
- Vaschetto ME, Retamal BA. Substituents effect on the electronic properties of aniline and oligoanilines. J Phys Chem A. 1997;101:6945–6950.
- Reynolds WF, Hamer GK. Concerning the relative importance of inductive effects and polar field effects on 19F chemical shifts in aromatic derivatives. J Am Chem Soc. 1976;23:7296–7299.
- Clark DT, Fairweather DJ. The electronic effect of the methyl and methoxyl groups partial rate factors for nitration of some substituted naphthalenes. Tetrahedron. 1969;25:5525–5533.
- Godfrey M, Murrell JN. Substituent effects on the electronic spectra of aromatic hydrocarbons. III. An analysis of the spectra of amino- and nitrobenzenes in terms of localized-orbital model. Proc R Soc Lond A. 1964;278:71–90.
- Smith MB, March J. March’s advanced organic chemistry. 6th Ed. Hoboken (NJ): John Wiley & Sons, Inc; 2007.
- Meyer TD, Steyaert I, Hemelsoet K, et al. Halochromic properties of sulfonphthaleine dyes in a textile environment: The influence of substituents. Dyes Pigm. 2016;124:249–257.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision A.02. Wallingford (CT): Gaussian, Inc.; 2009.
- Pearson RG. Absolute electronegativity and hardness: application to inorganic chemistry. Inorg. Chem. 1988;27:734–740.
- Foresman JB, Frisch A. Exploring chemistry with electronic structure methods. 2nd ed. Pittsburgh (PA): Gaussian, Inc; 1996.
- Guha AK, Das C, Phukan AK. Heterocyclic carbenes of diverse flexibility: a theoretical insight. J Organomet Chem. 2011;696:586–593.
- Wong BM, Piacenza M, Sala FD. Absorption and fluorescence properties of oligothiophene biomarkers from long-range-corrected time-dependent density functional theory. Phys Chem Chem Phys. 2009;11:4498–4508.
- Raeber AE, Wong BM. The importance of short- and long-range exchange on various excited state properties of DNA monomers, stacked complexes, and Watson-crick pairs. J Chem Theory Comput. 2015;11:2199–2209.
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098–3100.
- Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789.
- Grimme S, Antony J, Ehrlich S, et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132:154104.
- Onawole AT, Halim MA, Ullah N, et al. Structural, spectroscopic and docking properties of resorcinol, its -OD isotopomer and dianion derivative: a comparative study. Struct Chem. 2018;29:403–414.
- Mary YS, Varghese HT, Panicker CY, et al. Molecular conformational analysis, vibrational spectra, NBO, NLO, HOMO–LUMO and molecular docking studies of ethyl 3-(E)-(anthracen- 9-yl) prop-2-enoate based on density functional theory calculations, Spectrochim. Acta Part A Mol Biomol Spectrosc. 2015;150:533–542.
- Popoola SA, Al-Saadi AA. Spectroscopic and theoretical evaluation of the metal-olefin interaction in di-µ-chlorobis (1,5-cyclooctadiene) complexes of Ir and Rh Vib. Spectrosc. 2016;86:109–123.
- McMurry JE. Organic Chemistry. 3rd ed. Belmont: Wadsworth; 1992.
- Cotton FA, Wilkinson G, Murillo CA, et al. Advanced Inorganic Chemistry. 6th ed. New York: Wiley-Interscience; 1999.
- Peach MJG, Tellgren EI, Sałek P, et al. Structural and electronic properties of polyacetylene and polyyne from hybrid and Coulomb-attenuated density functionals. J Phys Chem A. 2007;111:11930–11935.
- Wong BM, Hsieh TH. Optoelectronic and excitonic properties of oligoacenes: substantial improvements from range-separated time-dependent density functional theory. J Chem Theory Comput. 2010;6:3704–3712.
- Liang Z, Zhang Y, Souri M, et al. Influence of dopant size and electron affinity on the electrical conductivity and thermoelectric properties of a series of conjugated polymers. J Mater Chem A. 2018;6:16495–16505.
- Kim Y, Morita K. Thermal conductivity of Molten Li2O–B2O3 and K2O–B2O3 systems. J Am Ceram Soc. 2015;98:3996–4002.
- Klein A, Körber C, Wachau A, et al. Surface potentials of magnetron sputtered transparent conducting oxides. Thin Solid Films. 2009;518:1197–1203.
- Chang R. Chemistry. 10th ed. Mc Graw Hill; 2010.
- Faustov VI, Egorov MP, Nefedov OM, et al. Ab initio G2 and DFT calculations on electron affinity of cyclopentadiene, silole, germole and their 2,3,4,5-tetraphenyl substituted analogs: structure, stability and EPR parameters of the radical anions. Phys Chem Chem Phys. 2000;2:4293–4297.
- Martin RB. Metal stabilities correlate with electron affinity rather than hardness or softness. Inorg Chim Acta. 1998;283:30–36.
- Crabtree RH. The organometallic chemistry of the transition metals. 4th ed. Hoboken (NJ): John Wiley & Sons; 2009.
- Popoola SA. Spectroscopic study of 2-methylindole and 3-methylindole: solvents interactions and DFT studies, Spectrochim. Acta Part A Mol Biomol Spectrosc. 2018;189:578–585.
- Lakowicz JR. Principle of fluorescence spectroscopy. 2nd ed. New York: Kluwer Academic/ Premium Publishers; 1999.
- Das P, Konwar D, Sengupta P, et al. Rhodium(I) carbonyl complexes of triphenylphosphine chalcogenides and their catalytic activity. Transit Met Chem. 2000;25:426–429.
- Bistoni G, Rampino S, Scafuri N, et al. How π back-donation quantitatively controls the CO stretching response in classical and non-classical metal carbonyl complexes. Chem Sci. 2016;7:1174–1184.