
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
COVID-19 pandemic has poses urgent health challenge, and this project aims to identify potential inhibitors to combat this virus. We screened 198 bioactive compounds from five selected medicinal plants previously reported to be antiviral against SARS-CoV-2 protease and two co-receptors followed by molecular dynamics simulations. From the screened compounds, Astragalin demonstrated very strong molecular interactions with the molecular docking binding energies −8.5, −8.0, −7.6 kcal/mol for 6LU7, 6LZG, and 6VXX proteins of SARS-CoV-2, respectively. Hydrogen bonding interaction with the active site catalytic residue HIS-41 or CYS-145 of the main protease SARS-CoV-2 was observed. Binding free energies (ΔGbind) from MM-GBSA after 50 ns MD simulations showed that Astragalin has the highest energy of −33.00 and −34.89 kcal/mol in complex with the main protease and spike glycoprotein of SARS-CoV-2, respectively. The study identifies Astragalin as a better inhibitor for the inactivation of COVID-19 and should be pursued as a potential drug candidate for this virus.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has recently emerged in December 2019 in Wuhan, China [Citation1] causing millions of death worldwide. Common symptoms of SARS-CoV-2 infection are fever, dry cough, breathing difficulties (dyspnoea), diarrhoea, headache, pneumonia, and ventilator support. Disease onset may cause progressive respiratory failure owing to alveolar damage and even death [Citation1,Citation2]. The World Health Organization (WHO) declared it as pandemic on 11 March 2020 because of its fast-growing human-to-human transmission and reported cases in nearly every region [Citation3,Citation4]. So far, there is no effective cure for the treatment of the virus, thus the need to identify potent drugs/vaccines.
SARS-CoV-2 is an enveloped spherical or pleomorphic particle containing single-stranded (positive-sense) RNA paired with a capsid nucleoprotein containing a matrix protein. The envelope contains glycoprotein in the form of clubs with a genome of nearly 30,000 bases belonging to the genus betacoronavirus (β-CoV). The viral genome encodes four structural proteins, the envelope (E), the nucleocapsid (N), the membrane (M), and the spike (S) proteins [Citation5,Citation6]. The spike glycoprotein attaches the virion to the host cell membrane in two functional units, S1 and S2. S1 domain of SARS-CoV-2 spike glycoprotein potentially interacts with the human host cell targets such as (ACE2, cyclophilins, CD26, Ezrin, and other cell adhesion factors) a key immune-regulatory factor for hijacking and virulence [Citation7,Citation8]. SARS-CoV-2 attach to the host cells through Spike protein and human Angiotensin-Converting Enzyme 2 (hACE2) receptor interaction.
For the virus receptor (spike protein) to bind to its cellular ligand (ACE2), activation by transmembrane protease TMPRSS2 is needed [Citation5]. One way to control the infection is to seek protease inhibitors to prevent the S-protein of SARS-CoV-2 cleavage to the S1 domain. Inhibiting the ACE2 receptor is likely to induce a conformational change of ACE2, therefore blocking the interaction between spike protein and ACE2 receptor.
Also, since SARS-CoV-2 is a single-stranded RNA virus, SARS-CoV-2 3-chymotrypsin-like protease (3CLpro) also called main protease (Mpro) is a key CoV enzyme, which plays a critical role in facilitating viral replication and transcription, making it an attractive drug target for the virus [Citation9]. Despite this, some protease inhibitors, and antiviral agents namely Chloroquine, Remdesivir and Hydroxychloroquine have been reported to be highly effective in the control of COVID-19 infection in in vitro under investigation as agents to treat COVID-19 [Citation10–13].
Medicinal plants are important sources of structurally novel compounds that functions as a lead for the development of novel drugs [Citation14]. Herbal medicine has the benefits of being stable at room temperature, non-injectable, and with no serious side effects [Citation15]. The herbal preparation also can be used as a health drink, health food or dietary supplement, to prevent immune system impairment [Citation15], which is needed for the recovery from SARS-CoV-2 infection [Citation16].
The plant Ageratum conyzoides has been specified for the treatment of “craw-craw” better known as rashes, pneumonia by rubbing it on the patient's chest, diarrhoea, wound healing and skin diseases, infectious and mental diseases as well as headaches and dyspnoea [Citation17]. Andrographis paniculata (Burm. F.) Nees herb is well explored therapeutically and effectively used as an immunostimulant, for asthma, influenza, respiratory tract infections, anti-pathogenic bacteria, anti-HIV, antimalarial, anti-snake venom, fever among others [Citation18,Citation19]. Andrographolide showed good inhibition of CHIKV infection, and it reduces the virus production by approximately 3log10 with EC50 of 77 µM without cytotoxicity [Citation20,Citation21].
Azadirachta indica (neem) has proven to be multifunctional and correctly stated as “Tree for Solving Global Problems”. Neem has been extensively used conventionally as a remedy for many diseases. Studies have identified active compounds from different part of neem plant as antimalarial, anti-inflammatory, anti-hyperglycaemic, anti-mutagenic, anti-ulcer, anti-carcinogenic, anti-oxidant, anti-viral, and immune-modulator drugs [Citation22–24].
Momordica charantia (Bitter gourd or Bitter melon) with similar pharmacological importance of A. indica has been widely used traditionally as unorthodox medicines for numerous sicknesses such as antitumor, anti-diabetic, anthelmintic, antimalarial, and antiviral agents [Citation25]. Studies have confirmed that the protein of M. charantia has an effective and wide antiviral activity against different subtypes of influenza A virus [Citation26] and Dengue virus [Citation27,Citation28].
Phyllanthus amarus is a widely distributed tropical medicinal plant, which belongs to the family Euphorbiaceae. The compounds extract of P. amarus show a wide spectrum of therapeutic activities such as antibacterial, antiplasmodial, antiviral, antimalarial, anti-inflammatory, anticancer, anti-microbial, anti-diabetic, anti-oxidant, hypolipidemic, nephroprotective, hepatoprotective, and diuretic properties [Citation25,Citation29–32]. Methanol extract of Phyllanthus species was tested for anti-HIV-1 reverse transcriptase (RT) mechanism using the HIV-RT assay by inhibition of the HIV-1 RT enzyme based on their IC50 values [Citation28]. Some suggest that P. amarus may be helpful in the cure of hepatitis B virus infection [Citation29].
Molecular docking can predict the ligand–protein interactions by recognizing the apt active sites in the protein, getting the best geometry of the ligand–receptor complex, and computing the interaction energy for various ligands to design additional effective ligands.
Owing to the numerous activities reported for these class of compounds, we pursue a virtual screening against receptors of SARS-CoV-2 to determine the inhibitory activity of the bioactive constituents of Ageratum conyzoides, Andrographis paniculata (Burm. F.) Nees, Azadirachta indica, Momordica charantia, and Phyllanthus amarus, which have been used for folk medicine for various diseases.
To discover lead compounds for clinical use, the aim of this article is to indicate the drug candidate from medicinal plants that can act as an antiviral, protease inhibiting agents, and RNA interfering agents for SARS-CoV-2. We initiated virtual screening of compounds, molecular docking, and molecular dynamics to single out new drug leads that target the main protease (MPro) and Spike glycoproteins (S) of SARS-CoV-2.
2. Materials and methods
2.1. Ligand preparation
Two hundred and eight (208) compounds (i.e. 198 extracted compounds from 5 selected plants and 10 extra compounds) were obtained from PubChem [Citation33], in SDF format. Some of these ligands are essentially bioactive compounds in medicinal plants and information on their bioactivities were retrieved from Dr. Duke's Phytochemical and Ethnobotanical Database [Citation34]. The ligands were opened in Spartan ’14 version 1.1.4 and converted to a 3D structure. The ligands were energetically minimized and geometrically optimized using the Semi-Empirical method with a basis set of PM3. Drug-like properties were calculated using Lipinski's rule of five [Citation35] and Veber's standards [Citation36] for oral bioavailability of drug candidates. The considered Lipinski's filters mass (≤500 g/mol), clogP (≤5), Hydrogen bond acceptors (≤5), hydrogen bond donors (≤10), Molar refractivity (), and polar surface area (≤140 Å2) [Citation35,Citation37]. Adherence with Lipinski's rule of five was calculated using SWISSADME prediction [Citation38].
2.2. Receptor preparation
The crystal structure of SARS-CoV-2, COVID-19 3CLpro/Mpro (PDB ID: 6LU7) [Citation9] and spike glycoproteins (PDB ID: 6VXX, 6LZG) [Citation39,Citation40] were retrieved from the RCSB Protein Data Bank [Citation41] in PDB format. All water molecules and heteroatoms were removed from the crystal structures using Discovery Studio software v. 2020 and then saved in PDB format for further analysis. For the spike glycoprotein (6VXX), alpha – chain (A chain) was used for the docking study. This is because there are multiple alignments of the amino acid sequences, this means, they have identical residue.
2.2.1. Determination of active sites
The amino-acids in the active site of the receptor was determined as it was reported in the study of the proteins [Citation9,Citation39,Citation40]. The determination of the amino acids in the active site was used to analyse the docking evaluation results. Meanwhile, a three–dimensional grid box map was made at maximum on MPro (6LU7) and on the Spike glycoproteins (6LZG and 6VXX), to allow the ligand to be docked to all parts of the receptor.
2.3. Molecular docking
The prepared receptors (PDB ID: 6LU7, 6LZG, 6VXX) were treated, i.e. removal of water molecules, ligand, and heteroatoms from the receptor, with the use of the Discovery Studio programme. AutoDock version 4.2.6 was used for protein optimization and the addition of the polar hydrogen group. Virtual screening using automated docking involves the preparation of the receptor (comprises of assigning Gasteiger charges and Kollman charges to all atoms and assigning AD4 forms to protein structure atoms) prior to docking [Citation42], ligands, grid box, and the algorithm. With PyRx and AutoDock Vina [Citation43], the correct grid box size was evaluated for the possible binding site of the ligands, and the receptor grid centre was placed on the receptor's active site residue. AutoDock 4.2.6, Pymol, and Discovery Studio were used for the docking analyses. Analysis of the docked complexes was done to obtain compounds with better binding energy (BE) and ligand's inhibitory constant (Ki) with the target protein. Analysis of the structural complex of the ligand–protein interaction was done using Discovery Studio 2020 programme.
2.4. Molecular dynamics simulations
The top three categories of compounds based on their binding affinities in complexation with the main protease and the two spike glycoproteins of SARS-CoV-2 from the docking studies were subjected to molecular dynamics (MD) simulations. A similar MD protocol in our previous studies [Citation44] was applied in this work. The simulations were conducted on the GPU version of the AMBER 14 software package. The molecular geometries of the ligands and proteins were reproduced using the general AMBER force field (GAFF) and AMBER force field 99SB, respectively. The ligand–protein complexes were neutralized with the required counter ions. Thereafter, the complexes were solvated with TIP3P water box of 12 Å. The solvated parameters (“top and crd)” for MD simulations were saved. The complexes were minimized using steepest descent and conjugated gradient minimizations for 2500 and 10,000 steps respectively to eliminate atomic clashes. Stepwise heating of the complexes was applied until 300 K and relaxation at the equilibration stage before the actual production. A 50 ns MD simulations were performed; trajectories analysis was extracted on the CPPTRAJ module of AMBER 14.
3. Results and discussion
3.1. Docking score
The binding affinities of all the compounds tested against the main protease and the two spike proteins are presented in Table . Based on the docking score, it is obvious that Astragalin, Isoquercitrin, Momordicin, Friedelin, Nelfinavir, Nimbaflavone, Momordicoside-I, Kaempferol, Gedunin, and Meldenin interacted well with the main protease and Spike glycoprotein of SARS-CoV-2 than Remdesivir and Chloroquine.
Table 1. Docking score result for SARS–CoV-2 receptors binding with the potential inhibitors.
Thirty-three active compounds in medicinal plants used as an antioxidant, antiviral, and antimalarial, which passed the Lipinski thumb rule alongside seven other compounds were considered for this study. In total, we docked two hundred and eight (208) compounds. Eight out of the forty-one compounds showed a good binding affinity (ΔG ≤ −7.8) with the Main protease (6LU7); these compounds are Astragalin, Isoquercitrin, Momordicin, Friedelin, Nelfinavir, Nimbaflavone, Momordicoside-I, and Kaempferol.
It was also found that thirty-four compounds have a strong interaction (ΔG ≤ −8.0) with the spike glycoprotein (6LZG); these are Ivermectin, Lectin, Beta-amyrin, Quercetin, Diosgenin, Multiflorenol, Nelfinavir, Gallocatechin, Nimolicinol, Epoxyazadiradione, Friedelin, Taraxerol, Cucurbitacin-B, Nimbolin-B, Momordicoside-I, Astragalin, Azadiradione, Nimbinin, Neoandrographolide, Nimolinone, Momordicin, Remdesivir, Cucurbitacin-K, Quercitrin, Gamma-cadinene, Myricetin, Lopinavir, Gedunin, 5-hydroxy-7,8,2′-trimethoxyflavone, Nimbaflavone, Meliantriol, Ascorbigen, Epiazadiradione, and Isoquercitrin. Besides, seventeen compounds were found to have a strong interaction (ΔG ≤ −7.6) with the spike glycoprotein (6VXX); these are Ivermectin, Friedelin, Diosgenin, Nimolicinol, Multiflorenol, Meliantriol, Taraxerol, Momordicoside-I, Azadiradione, Beta-amyrin, Nimolinone, Nimbaflavone, Epoxyazadiradione, Momordicin, Astragalin, Nimbinin, and Epiazadiradione. The key residue(s) for each ligand interaction with the SARS-CoV-2 receptors through hydrogen bond interaction and their respective distances were presented in Table S2. Hydrogen bonding interactions are essential for proteins, as they provide the organization for distinct folding and the selectivity that supports molecular recognition in the ligand–protein interface [Citation45].
3.2. Post molecular dynamics simulations
The post MD analysis such as root mean square deviation fluctuation (RMSF), root mean square deviation (RMSD), and free binding energy were investigated. For clarity, the analysis in this section will be focused on the main protease while the results for the spike proteins will be provided in the supplementary information. The compounds simulated are Astragalin (Astra), Kaempferol (Kaem), and Nimbaflavone (Nimb) in complex with the main protease of the SARS-CoV-2.
3.3. Binding free energy calculations
The association of ligand–protein interaction is quantified using the MM/GBSA method [Citation46,Citation47]. The Generalized Born (GB) solvation model [Citation48] is used to compute the electrostatic component of the solvation binding free energies. Estimation of the binding free energies are extracted from the equations below
(1)
(1)
(2)
(2)
(3)
(3)
(4)
(4)
(5)
(5)
Where ,
,
is the energy of the complex, protein and ligand respectively. The energy components such as
is the molecular mechanics energy of the system,
, electrostatic energy,
, and van der Waals term,
. Also
, solvation energy which is divided into the polar (
) and non-polar (
) contributions.
, is the non-polar contribution to solvation-free energy that is linearly dependent on the solvent accessible surface area (SASA).
3.4. System stability and flexibility
RMSD value for the heavy backbone atoms was calculated for the 50 ns simulation for all the systems to validate their stabilities. The RMSD plots are represented in Figure with average values of 1.85, 1.25, and 0.82 Å for Astra-6LU7, Kaem-6LU7, and Nimb-6LU7, respectively. The small average RMSD values of the complexes indicate that the simulations were already converged [Citation49].
Figure 1. RMSD plots for the studied complexes.
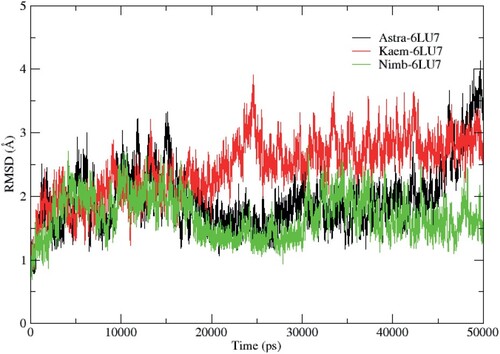
It can be observed that the systems were equilibrated after 5 ns, although some deviations were seen during the simulation period. A stable protein structure is judged with a corresponding smaller deviation [Citation50]. The residual flexibility of the studied proteins on binding with the ligands was evaluated using the RMSF parameter. The RMSF (Figure ) of the α-Carbon backbone for the systems showed a similar evolution. However, the Kaem-6LU7 system displayed more flexible residues at residue number 152–155 in comparison to Astra-6LU7 and Nimb-6LU7. In general, different protein regions showed varying levels of flexibility, which impacted the binding interactions with the ligands as represented on Table .
Figure 2. RMSF plots for the α-Carbon backbone of the main protease for three systems.
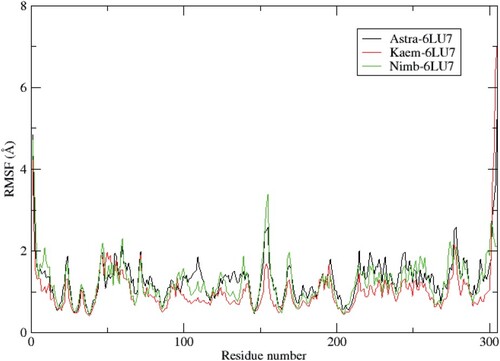
Table 2. The calculated binding free energies and their relative components for the complexes using MM-GBSA method obtained from the last 1000 frames of 50 ns. The energy components are in kcal/mol.
3.5. Binding free energy calculations
To evaluate the binding free energy of the ligand–protein complexes, we employed the MM-GBSA method [Citation51,Citation52] and they were generated from the last 1000 frames of 50 ns. An in silico prediction of ligand–protein binding free energy has been reported to be the main objective of structure-based drug design [Citation53]. The binding free energies for the systems are presented in Table 5 with the accompanying energy components. According to the calculated results presented in Table , ΔGbind for Astragalin, Nimbaflavone, and Kaempferol in complex with the main protease of SARS-Cov-2 were −33.32, −31.86, and −25.35 kcal/mol respectively. The results for the Astra – 6LU7 complex demonstrate larger binding free energies compared to Nimb – 6LU7 and Kaem – 6LU7 with a difference of 1 and 8 kcal/cal respectively. Binding free energies for the spike proteins (6LZG and 6VXX) are presented on Tables S3 and S4.
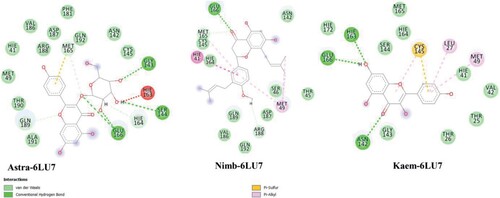
The relative energy contributions for the systems are consistent with their binding free energies with Astra – 6LU7 having the highest increases in electrostatic energy when binding in both gas, polar and non-polar phases and a favourable change in van der Waals energy when binding, the change in electrostatic energy compensates significantly. Important residual interactions were observed as presented in Figure for the systems. Hydrogen bonding interactions were seen for GLU166, GLY143, SER144, ASN142, there is interaction with the catalytic residue CYS145 (Figure ).
Figure 3. Ligand-protein interactions resulting from MM-GBSA calculations.
4. Conclusion
Using various in silico methods, we have identified some potential antiviral inhibitors, which can serve as a starting point to stop the replication of SARS-CoV-2. These inhibitors were derived from known medicinal plants. The use of virtual screening and molecular dynamics simulations were utilized for this study. Top lead compounds (n = 41) showed that many of the compounds interact with the active site residues of SARS-CoV-2 from the virtual screening protocol. Molecular dynamics simulations of the top 3 compounds were performed, Astragalin, Nimbaflavone, and Kaempferol in complex with the main protease of SARS-CoV-2. The results of the study revealed that Astragalin and Nimbaflavone in complex with the main protease of SARS-Cov-2 showed the best binding free energies ΔGbind of −33.32 and −31.86 kcal/mol, respectively. Important residual interactions with the active region of the receptors were observed, such as Cys 145. It has been reported that Astragalin has antiviral activity and has underlying anti-H1N1 influenza virus activity [Citation53]. Studies have demonstrated that Astragalin functions as an effective lead compound antagonizing oxidative stress induced by endotoxin, which leads to airway dysfunction and inflammation [Citation54]. The study which utilized state-of-the-art virtual screening and molecular dynamics simulations are crucial techniques required for drug design and identification of new inhibitors.
Supplemental Material
Download MS Word (749.8 KB)Acknowledgement
The authors are grateful for the computational resources provided by CHPC (www.chpc.ac.za).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Notes
Where, ΔEvdw van der Waals energy, ΔEele electrostatic energy, ΔGgas gas-phase free energy, ΔGpolar polar free energy, ΔGnon-polar non-polar free energy, ΔGsolvation solvation free energy. ΔGbind total binding free energy.
References
- Chan JFW, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi:10.1016/S0140-6736(20)30154-9.
- Zhou P, Lou Yang X, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi:10.1038/s41586-020-2012-7.
- WHO, Coronavirus (COVID-19) events as they happen, https://www.who.int/emergencies/diseases/novel-coronavirus-2019/Events-as-They-Happen. (2020) https://www.who.int/emergencies/diseases/novel-cor. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen (accessed May 18, 2020).
- (World Health Organisation), Coronavirus (COVID-19) events as they happen, (n.d.). https://www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen (accessed June 30, 2020).
- Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect. 2020: 0–4. doi:10.1016/j.jmii.2020.03.022.
- Niranjani Iyer, Decoding the SARS-CoV-2 Genomes – Diagnostic Testing, (2020). https://blogs.3ds.com/biovia/decoding-the-sars-cov-2-genomes-diagnostic-testing/?utm_medium=email&utm_source=promotional&utm_campaign=bioviaeflashsendinwaves_OP53851&utm_content=2020-05-biovia-newsletter-en_DM960160 (accessed May 21, 2020).
- Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9:601–604. doi:10.1080/22221751.2020.1739565.
- Habibzadeh P, Stoneman EK. The novel coronavirus: a bird’s eye view. Int J Occup Environ Med. 2020;11:65–71. doi:10.15171/ijoem.2020.1921.
- Jin Z, Du X, Xu Y, et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020. doi:10.1038/s41586-020-2223-y.
- Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi:10.1038/s41422-020-0282-0.
- Moore N. Chloroquine for COVID-19 infection. Drug Saf. 2020;43:393–394. doi:10.1007/s40264-020-00933-4.
- Strodel B, Jülich F, Olubiyi OO, et al. High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2 Mechanistic study View project Sickle cell disease (SCD) drug discovery View project High throughput virtual screening for M pro inhibitors A PREPR, Researchgate.Net. (2020). doi:10.20944/preprints202004.0161.v1.
- Tallei T, Tumilaar S, Niode N, et al. Potential of plant bioactive compounds as SARS-CoV-2 main protease (Mpro) and spike (S) glycoprotein inhibitors: a molecular docking study. Preprints. 2020: 1–18. doi:10.20944/preprints202004.0102.v1.
- Okereke SC, Ijeh II, Arunsi UO. Determination of bioactive constituents of Rauwolfia vomitoria Afzel (Asofeyeje) roots using gas chromatography-mass spectrometry (GC-MS) and Fourier transform infrared spectrometry (FT-IR). African J Pharm Pharmacol. 2017;11:25–31. doi:10.5897/ajpp2016.4712.
- Ganjhu RK, Mudgal PP, Maity H, et al. Herbal plants and plant preparations as remedial approach for viral diseases. VirusDisease. 2015;26:225–236. doi:10.1007/s13337-015-0276-6.
- Ali I, Alharbi OML. COVID-19: disease, management, treatment, and social impact. Sci Total Environ. 2020;728:138861. doi:10.1016/j.scitotenv.2020.138861.
- Okunade AL. Ageratum conyzoides L. (Asteraceae). Fitoterapia. 2002;73:1–16. doi:10.1016/S0367-326X(01)00364-1.
- Churiyah, Pongtuluran OB, Rofaani E, et al. Antiviral and immunostimulant activities of andrographis paniculata. HAYATI J Biosci. 2015;22:67–72. doi:10.4308/hjb.22.2.67.
- Benoy GK, Animesh DK, Aninda M, et al. An overview on andrographis paniculata (burm. F.) Nees. Int J Res Ayurveda Pharm. 2012;3:752–760. doi:10.7897/2277-4343.03610.
- Tang LIC, Ling APK, Koh RY, et al. Screening of anti-dengue activity in methanolic extracts of medicinal plants. BMC Complement Altern Med. 2012;12:3. doi:10.1186/1472-6882-12-3.
- Wintachai P, Kaur P, Lee RCH, et al. Activity of andrographolide against chikungunya virus infection. Sci Rep. 2015;5:1–14. doi:10.1038/srep14179.
- Dubey S, Kashyap P. Azadirachta indica: a plant with versatile potential. Rajiv Gandhi Univ Heal Sci J Pharm Sci. 2014;4:39–46. doi:10.5530/rjps.2014.2.2.
- Emmanuel O, Agbaji AS, Williams IS, et al. An overview of Neem, Azadirachta indica research in Nigeria. Niger J Sci. 2015;49:1–9.
- Ahmad A, Javed MR, Rao AQ, et al. Designing and screening of universal drug from neem (Azadirachta indica) and standard drug chemicals against influenza virus nucleoprotein. BMC Complement Altern Med. 2016;16:1–8. doi:10.1186/s12906-016-1469-2.
- Bharti Sarin AM, Verma N, PedroMartín J. An overview of important ethnomedicinal herbs of phyllanthus species: present status and future prospects. Sci World J. 2014;2014:12. doi:10.1155/2014/839172.
- Pongthanapisith V, Ikuta K, Puthavathana P, et al. Antiviral protein of Momordica charantia L. inhibits different subtypes of influenza A. Evidence-Based Complement Altern Med. 2013;2013; doi:10.1155/2013/729081.
- Tang LIC, Ling APK, Voon KGL. Screening of anti-dengue activity in methanolic extracts of medicinal plants Background Methods Plant materials and extraction. BMC Complement Altern Med. 2014: 1–13.
- Salehi B, Anil Kumar NV, Şener B, et al. Medicinal plants used in the treatment of human immunodeficiency virus. Int J Mol Sci. 2018;19. doi:10.3390/ijms19051459.
- Yeh SF, Hong CY, Huang YL, et al. Effect of an extract from Phyllanthus amarus on hepatitis B surface antigen gene expression in human hepatoma cells. Antiviral Res. 1993;20:185–192. doi:10.1016/0166-3542(93)90019-F.
- Londhe JS, Devasagayam TPA, Foo LY, et al. Antioxidant activity of some polyphenol constituents of the medicinal plant Phyllanthus amarus Linn. Redox Rep. 2008;13:199–207. doi:10.1179/135100008X308984.
- Yeo SG, Song JH, Hong EH, et al. Antiviral effects of Phyllanthus urinaria containing corilagin against human enterovirus 71 and Coxsackievirus A16 in vitro. Arch Pharm Res. 2015;38:193–202. doi:10.1007/s12272-014-0390-9.
- Patel JR, Tripathi P, Sharma V, et al. Phyllanthus amarus: ethnomedicinal uses, phytochemistry and pharmacology: a review. J Ethnopharmacol. 2011;138:286–313. doi:10.1016/j.jep.2011.09.040.
- PubChem, (n.d.). https://pubchem.ncbi.nlm.nih.gov/ (accessed June 27, 2020).
- R. Quarcoo, A. Court, C. Division, Search list, (2014) 2013–2015. https://phytochem.nal.usda.gov/phytochem/search (accessed June 6, 2020).
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17. doi:10.1016/j.addr.2012.09.019.
- Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi:10.1021/jm020017n.
- Giménez BG, Santos MS, Ferrarini M, et al. Evaluation of blockbuster drugs under the rule-of-five. Pharmazie. 2010;65:148–152. doi:10.1691/ph.2010.9733.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:1–13. doi:10.1038/srep42717.
- Walls AC, Park YJ, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–292.e6. doi:10.1016/j.cell.2020.02.058.
- Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020: 1–11. doi:10.1016/j.cell.2020.03.045.
- Protein Data Bank, RCSB PDB: Homepage, Rcsb Pdb. (2019) 1. https://www.rcsb.org/ (accessed June 30, 2020).
- Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges. Tetrahedron. 1980;36:3219–3228. doi:10.1016/0040-4020(80)80168-2.
- Allouche A. Software news and updates Gabedit — a graphical user interface for computational chemistry softwares. J Comput Chem. 2012;32:174–182. doi:10.1002/jcc.
- Sabe VT, Tolufashe GF, Ibeji CU, et al. Identification of potent L,D-transpeptidase 5 inhibitors for Mycobacterium tuberculosis as potential anti-TB leads: virtual screening and molecular dynamics simulations. J Mol Model. 2019;25; doi:10.1007/s00894-019-4196-z.
- Baker EN, Hubbard RE. Hydrogen bonding in globular proteins. Prog Biophys Mol Biol. 1984;44:97–179. doi:10.1016/0079-6107(84)90007-5.
- Srinivasan J, Cheatham TE, Cieplak P, et al. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J Am Chem Soc. 1998;120:9401–9409. doi:10.1021/ja981844+.
- Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi:10.1021/ar000033j.
- Gohlke H, Case DA, Biology M, et al. Converging free energy estimates: MM-PB (GB) SA studies on the protein – protein complex Ras – Raf. J Comput Chem. 2003: 238–250.
- Liu K, Watanabe E, Kokubo H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J Comput Aided Mol Des. 2017;31:201–211. doi:10.1007/s10822-016-0005-2.
- Aier I, Varadwaj PK, Raj U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci Rep. 2016;6:1–10. doi:10.1038/srep34984.
- Sun H, Li Y, Tian S, et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys. 2014;16:16719–16729. doi:10.1039/c4cp01388c.
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov. 2015;10:449–461. doi:10.1517/17460441.2015.1032936.
- Cournia Z, Allen B, Sherman W. Relative binding free energy calculations in drug discovery: recent advances and practical considerations. J Chem Inf Model. 2017;57:2911–2937. doi:10.1021/acs.jcim.7b00564.
- Cho IH, Gong JH, Kang MK, et al. Astragalin inhibits airway eotaxin-1 induction and epithelial apoptosis through modulating oxidative stress-responsive MAPK signaling. BMC Pulm Med. 2014;14:1–11. doi:10.1186/1471-2466-14-122.