
ABSTRACT
The COVID-19 pandemic caused by SARS-CoV-2 has started in December 2019 in Wuhan, China, and become a global health problem. The SARS-COV-2 main protease (Mpro) play a crucial role in the multiplication and control of virus activity. Therefore, we assume that the inhibition of these enzymes inhibits viral replication. To determine potential inhibitors of this protease among existing chemical libraries, we have carried out a virtual screening of 300 antiviral molecules obtained by the ASINEX database. Nine molecules were chosen based on their stability with the target (Mpro), and the molecular docking study was carried out against the same target. These molecules showed higher binding affinities than the positive control to the active site of the same target (Mpro) and exhibit interactions with the amino acids responsible for the inhibitory activity of this enzyme (Cys145 and His 41). Then, these molecules were selected and chosen as potential inhibitors of SARS-Cov-2.
Highlights
Three hundred antiviral compounds obtained from the ASINEX database were subjected to a virtual screening against SARS-COV-2 main protease Mpro.
Nine molecules were selected and were subjected to molecular docking against the same target to evaluate their stability and the mode of binding inside the active site.
The nine molecules show favourable interactions with the two amino acids Cys145 and His41, which suggests that these molecules could have competitive inhibitory activity. against the Mpro target main protease
The graphical abstract was created using BioRender.com
GRAPHICAL ABSTRACT
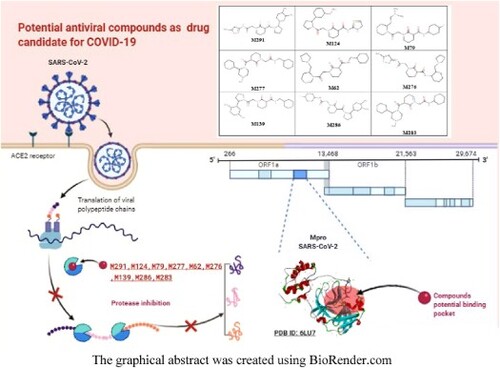
1. Introduction
The end of the year 2019 is marked by the apparition and spread of a zoonotic virulent coronavirus named SARS-CoV-2Footnote1 in Wuhan China [Citation1]. This virus rapidly transformed into a global pandemic (COVID-19 pandemic) [Citation2]. This virus affects not only public health but also the world's global economy. Until 29 June 2020, the COVID-19 pandemic has affected more than 200 countries, with more than 10 million confirmed cases and more than 499 K victims (COVID-19 Situation Reports, s. d.)
Nowadays, no effective treatment or vaccine against this virus has been found, which is a serious global health problem [Citation3]. Several treatment strategies have shown the potential effectiveness of certain drugs already existing on the market such as Hydroxychloroquine, Remdesivir, Famotidine and Ivermectin [Citation4,Citation5]. So the search for new potential effective molecules among the already existing drugs can be a solution to this global health emergency. It is therefore interesting to carry out a high-throughput virtual screening approach based on the structure of the targeted S- protein, to determine potential inhibitors.
Coronaviruses (CoV)Footnote2 have a positive sense, Single-stranded RNA encapsulated in a membrane envelope. The viral membrane is strewn with spiked glycoprotein which gives the coronaviruses their crown-like appearance. Four classes of coronaviruses are known as an alpha, beta, gamma, and delta class. The three most recent coronaviruses, Severe Acute Respiratory Syndrome Virus (SARS-CoV-1), Middle East Respiratory Syndrome Virus (MERS-CoV), and the new causative agent of COVID-19 (SARS-CoV-2) belong to the beta-coronavirus class [Citation6]. Like SARS-CoV-1 and MERS-CoV,Footnote3 SARS-CoV-2 is characterized by a genome that codes for various structural proteins, including advanced glycosylated spike – protein (S), which acts as a major trigger of host immune responses. This spike – protein (S) is involved in the invasion of host cells by SARS-CoV-1 and SARS-CoV-2 via the binding to a receptor protein called angiotensin 2 converting enzyme (ACE2)Footnote4 which codes for several structural proteins [Citation7], including the advanced glycosylated spike – protein (S) which acts as a major inducer of the host's immune responses. This spike – protein is involved in the invasion of host cells by SARS-CoV-1 and SARS-CoV-2 via the binding to a receptor protein called angiotensin 2 converting enzyme (ACE2). Two cysteine proteases in CoVs, the chymotrypsin-like protease (Mpro) and papain-like protease (PLpro),Footnote5 are involved in the maturation of the functional proteins necessary in the process of viral replication [Citation8] The main protease (Mpro) also known as 3C-type protease (3CLpro).Footnote6 Mpro is composed of three-domains (domains I to III) [Citation9]. Their active site comprises a catalytic cysteine Cys145 and His41 that forms at least three hydrogen bonding interactions with neighbouring residues [Citation10]. The SARS-CoV main protease Mpro7Footnote7 has a dyadic non-canonical Cys-His located in the cleavage between domains I and II [Citation11]. The Cys145 and His41 form a catalytic dyad in contrast to serine proteinases and other cysteine proteinases, which have a catalytic triad [Citation11]. The protease inhibition treatment is the most important recent advance in the treatment of viral infection, protease activity can be inhibited by antiviral drugs, which bind to the catalytic site of the main protease Mpro and block the synthesis of infectious virions [Citation12]. The high-resolution crystal structure of CoV-2-SARS main protease Mpro in combination with a covalently bound inhibitor (N3 ligand) has been published recently in the PDB database [Citation13], as soon as it appeared in the protein bank, it gave researchers an opportunity to develop and design potential inhibitors against SARS-CoV-2. This main protease Mpro can be qualified, until now, the most important targets against COVID19. For this reason, our study aimed to implement a virtual structure-based screening approach to identify antiviral drugs as potential treatments for COVID-19. Our strategy focused on identifying small molecules among 300 molecules available on the ASINEX database (http://www.asinex.com/libraries-html/) [Citation14], that may have potential anti-protease activity against SARS CoV-2 main protease Mpro.
2. Materials and methods
2.1. Virtual screening
2.1.1. Chemical database
A series of 300 molecules were obtained from the ASINEX database [Citation14], using the antiviral filter while applying advanced search on the database to select the molecules that have antiviral activity. These molecules have been downloaded in a PDB format file.
2.1.2. Preparation of the target
The three-dimensional (3D) structure of the target (PDB code: 6LU7, resolution 2.16 Å) of the SARS-CoV-2 Mpro [Citation15] was downloaded from the Protein Database (PDB) [Citation13], and prepared by Discovery Studio 2020 [Citation16], we have removed the water molecules that surround the structure, we have eliminated the native ligand (The inhibitor N3), then we determined the coordinates (X, Y and Z) of the active site, at the end we added the polar hydrogen to this structure. The 6LU7 structure was extracted in PDB format and saved for inclusion as a target in the virtual screening.
2.1.3. Virtual screening
The high-speed virtual screening was carried out using the iGEMDOCK programme (Generic Evolution Method for Docking) [Citation12]. To identify new potential hits to inhibit SARS-Cov-2 main protease the Mpro, in silico screening of 300 compounds were performed with the SARS-Cov-2 main protease PDB code (6LU7), the screening score which is based on total energy calculations (Total Energy = VdWFootnote8 + HBondFootnote9 + Electrostatic)Footnote10, was calculated using iGEMDOCK v2.1.[Citation12].
The standard parameters used for screening were as follows (population size = 300, generations = 70 and number of solutions = 2). The results obtained based on the energy were analyzed and 9 potential inhibitors were selected based on the stability (Table ).
Table 1. Interaction results between the SARS-Cov-2 main protease Mpro, and the selected potential ligands.
3. Molecular docking
3.1. Docking process
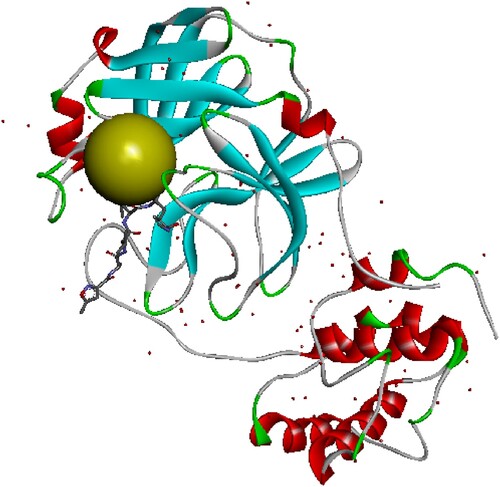
The nine selected molecules according to the virtual screening were used to perform molecular docking against the same target. Autodock vina [Citation17], and MGL Tools programmes [Citation18] were used with the default parameters. Grid box is at 1 angstrom with the active site coordinates (x= −9.378 y = 20.384 z = 68.898) (Figure ).
Figure 1. Structure of the protein SARS-COV-2 main protease: the green sphere represents position of the allosteric active site, the coordinates of the active site are x=−9.378 y = 20.384 z = 68.898.
3.2. Visualization and analysis of results
The results were visualized by the Discovery studio visualizer [Citation16] and we analyzed the results of the molecules which showed an interesting docking score and compared their positioning inside the active site. We also compared the type of interactions established by each molecule inside the active site.
4. Results and discussion
4.1. Virtual screening
Virtual screening is considered an interesting approach for drug discovery, development and research of new drugs. The availability of the three-dimensional crystal structure of the SARS-CoV-2 main protease Mpro, allows us, using this approach, to predict the preferred orientation of the ligand in the receptor cavity, and then to calculate the binding affinity between the ligand and the targeted protein. In this context aimed at exploring the affinity of the ligand–protein complex, over 300 drugs were screened based on their total binding energy and amino acid position. In this step we have selected nine molecules which have a higher stability and affinity than the reference ligand N3 (binding energy less than −6.1 kcal / mol) Among the nine selected compounds, five molecules (M291, M124, M79, M277, M62) showed the lowest total binding energy (less than −100 kcal. mol−1). Furthermore, the drugs M291 and M124 showed higher Vander Waal bonding affinity with amino acids; cysteine, and histidine of protease Mpro. As we have already mentioned all these molecules reported to be antiviral compounds in the ASINEX database.
4.2. Docking results
The nine compounds with higher affinity selected after the virtual screening approach were docked into the active site of the SARS-CoV-2 main protease Mpro. On the basis of their score values, these molecules were scored and ranked. Drugs with a docking score between −8.4 and −7.0 are considered interesting compounds that can be proposed as potential candidates to inhibit the SARS-CoV-2 main protease Mpro. The nine candidates with the best docking score are presented in Table . In this work, we considered the N3 ligand co-crystallized in the 6LU7 structure as a positive control for the binding and screening of 9 antiviral compounds performed on the 6LU7 structure of SARS-CoV-2 main protease Mpro [Citation15], the binding energy of the N3 ligand is −6.1 kcal/mol.
The analysis of the obtained results show that the nine top-ranked candidates (M291, M283, M286, M276, M139, M124, M277, M79, M62) by screening, all showed better binding affinities to the active site of Mpro compared to the N3 ligand (a range of docking score between −8.4 and −7 kcal / mol). However, M291, M283, M286, And M276 showed a higher docking score and show several interactions with histidine 41 and cysteine 145 (Table ). We have visualized the positioning of all these molecules inside the active site and the interactions established between them and the protein SARS-CoV-2 main protease Mpro. Figures show the visualization of docking results and the types of interactions with the amino acids inside the active site.
Figure 2. 2D diagram of interactions, and positioning of the molecule M276 inside the active site of SARS-CoV-2 main protease.
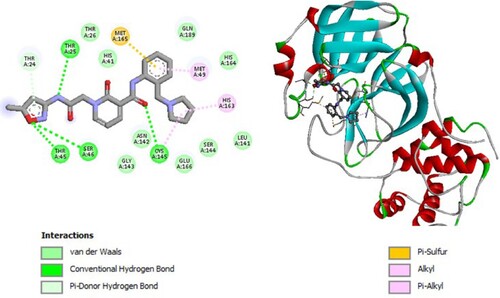
Figure 3. 2D diagram of interactions, and positioning of the molecule M79 inside the active site of SARS-CoV-2 main protease.
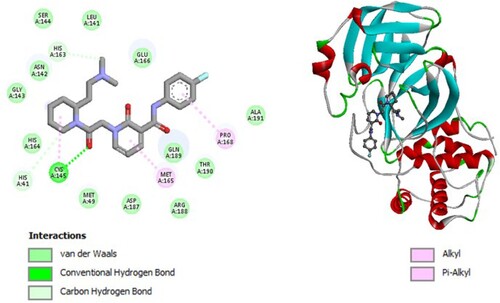
Figure 4. 2D diagram of interactions, and positioning of the molecule M124 inside the active site of SARS-CoV-2 main protease.
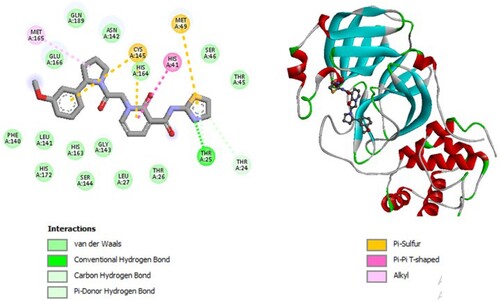
Figure 5. 2D diagram of interactions, and positioning of the molecule M139 inside the active site of SARS-CoV-2 main protease.
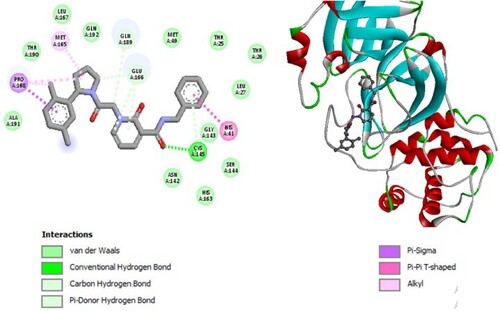
Figure 6. 2D diagram of interactions, and positioning of the molecule M277 inside the active site of SARS-CoV-2 main protease.
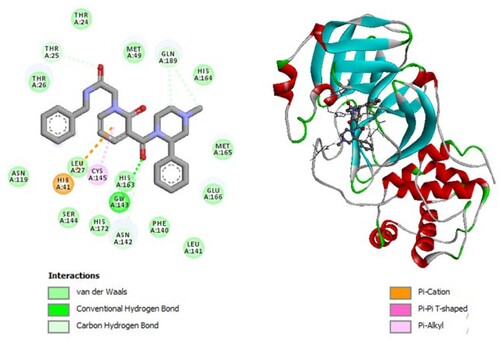
Figure 7. 2D diagram of interactions, and positioning of the molecule M291 inside the active site of SARS-CoV-2 main protease.
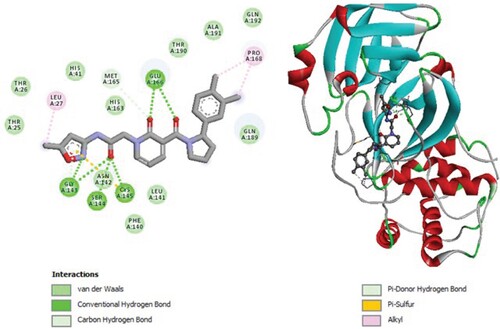
Figure 8. 2D diagram of interactions, and positioning of the molecule M283 inside the active site of SARS-CoV-2 main protease.
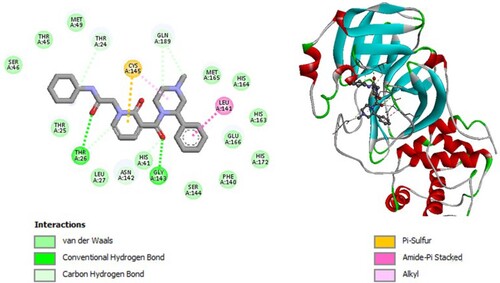
Figure 9. 2D diagram of interactions, and positioning of the molecule M286 inside the active site of SARS-CoV-2 main protease.
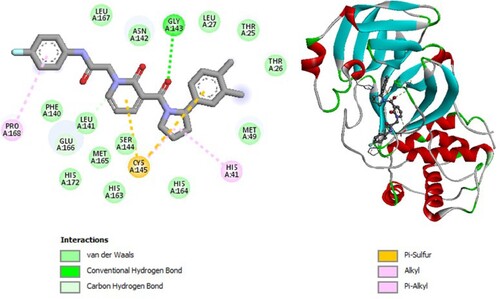
Figure 10. 2D diagram of interactions, and positioning of the molecule M62 inside the active site of SARS-CoV-2 main protease.
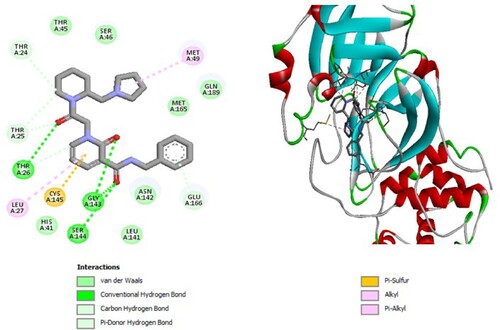
Molecular binding between the selected compound M276 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and hydrophobic interactions as well as van der Waals, π -sulfur and π -alkyl bonds with the residues of the binding pocket to the substrate of Mpro. A van der Waals interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 via hydrogen bonding.
Molecular binding between the selected M79 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals and π -alkyl bonds with the residues of the binding pocket to the substrate of Mpro. A hydrophobic interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 through van der Waals interaction.
Molecular binding between the selected M124 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals and π -alkyl bonds and π -T-shaped with the residues of the binding pocket to the substrate of Mpro. A π -T-shaped interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 through π – Sulfur interaction.
Molecular binding between the selected M139 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals and π -alkyl bonds, π -Sigma and π -T-shaped with the residues of the binding pocket to the substrate of Mpro. A π -T-shaped interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 through van der Waals interaction.
Molecular binding between the selected M277 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals, π -alkyl bonds, π – Cation, and π – π T-shaped with the residues of the binding pocket to the substrate of Mpro. A π – Cation interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 by π -alkyl.
Molecular binding between the selected M291 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals, alkyl bonds and π – Sulfur and carbon–hydrogen bonds with the residues of the binding pocket to the substrate of Mpro. A Van der Waals interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 by a hydrogen bond.
Molecular binding between the selected M283 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals, alkyl bonds, amid- π Stacked, π -Sulfur and Carbon Hydrogen bonds with the residues of the binding pocket to the substrate of Mpro. A van der Waals interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 through π-Sulfur.
Molecular binding between the selected M286 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals, π - alkyl bonds, alkyl bonds, carbon–hydrogen bonds and Pi-Sulfur with the residues of the binding pocket to the substrate of Mpro. A π -Alkyl interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 through π -Sulfur interaction.
Molecular binding between the selected M62 and the residues of the SARS-CoV-2 Mpro substrate-binding pocket (Figure ) have established multiple hydrogen bonds and a hydrophobic interaction as well as van der Waals, π - alkyl bonds, alkyl bonds, carbon–hydrogen bonds, π -donor hydrogen bonds, π -sulfur and π T-shaped interactions with the residues of the binding pocket to the substrate of Mpro. A Van der Waals interaction with catalytic His41 has been observed and it interacts with catalytic Cys145 through π -sulfur interaction.
The molecules (M291, M283, M286, M276, M139, M124, M277, M79, M62) showed an effect against the main COVID19 protease Mpro based on docking scores, ligand efficiency, interaction of different types with the two amino acids of interest (Cys145 and His 41).
After performing the molecular docking, the results showing the score and the types of interactions established between the two amino acids (Cys145 and His41) and our nine molecules are listed below in Table .
Table 2. Docking results showing the best lay conformation ordered by their binding affinities and the types of interaction established with amino acids Cys145 and His41.
5. Discussion
Over the last two years, important results have been obtained with a new class of antivirals protease inhibitors (PIs)Footnote11 [Citation19,Citation20].Footnote12 These inhibitors are characterized by significant antiviral activity, having a particular interest in the current therapeutic approach to the treatment of HIV [Citation21] as well as in the treatment of hepatitis C [Citation22,Citation23].
The 3CLpro known as main protease Mpro is a significant target for designing anti-SARS inhibitors. In SARS COV-1 as in SARS – COV-2 this enzyme is important for viral replication [Citation24]. Crystal structures of SARS-CoV-2 main protease Mpro had been determined to facilitate the structural and functional investigation of this protease. The binding site of SARS-CoV-2 main protease Mpro was found to be mostly located in a hydrophobic cleft and contains Cys145 and His41 [Citation25]. These two amino acid residues interfere together in the active site of the protease to constitute a catalytic (base-nucleophilic) dyad. It has been reported that the inhibition of the SARS-COV Mpro enzyme is mainly due to an irreversible covalent bond by Cys145, or by reversible interaction with Cys145 and His41. Cys145 plays the role of a nucleophile [Citation11] and His41 polarizes and de-protonates the nucleophile (Cys145) to increase its reactivity. Cys145 is attacked by the inhibitor by forming an intermediate complex before releasing the enzyme or forming directly an irreversible complex.
In the current work, we obtained nine molecules M291, M283, M286, M276, M139, M124, M277, M79, M62 that show all interactions (Table ) with the two amino acids of interest (Cys145 and His 41), these results are in agreement with previous docking studies of other molecules with the same target [Citation8,Citation26–30]. Furthermore, these candidates selected based on their screening score. The amino acids of the SARS-CoV-2 substrate-binding pocket have established multiple hydrogen bonding and hydrophobic interaction with the amino acids of the Mpro substrate-binding pocket, as well as interactions with the His41 catalyst in all nine ligands.
In this work, it should be noted that all of the studied candidates interacted with the catalyst residues (Cys145 and His41) in the SARS-Cov-2 Mpro substrate binding site. our results suggest that these molecules could have an inhibitory activity against SARS-COV-2 main protease Mpro by forming temporary complexes (reversibly). Therefore, SARS-COV-2 main protease Mpro is a cysteine protease, in which these molecules M291, M124, M79, M277, M62, M276, M139, M286, and M283 can play the role of an competitive inhibitors and consequently, they might be a drug candidate against COVID-19.
Coronavirus proteases share the same active site [Citation31], therefore, these nine candidates may have potential inhibitory activity on other coronavirus proteases as well. As these results appear to be very encouraging, further biological test are needed to check the inhibitory activity of these compounds against the major SARS-Cov-2 protease.
6. Conclusion
In this work, we have carried out a virtual screening of 300 antiviral molecules available in the ASINEX database. Nine molecules were chosen based on their stability against SARS-CoV-2 main protease target and Molecular Docking was carried out on them, to highlight the different interactions and positioning of each one in the active site domain. Docking results have shown that all of these selected molecules (M291, M283, M286, M276, M139, M124, M277, M79, M62) exhibit interactions with the two amino acids responsible for the inhibitory activity of this enzyme (Cys145 and His 41), which suggests that these molecules could have an inhibitory effect on this protein and may be candidates for an in vitro study against SARS-COV-2 main protease and consequently the inhibition of replication of SARS-COV2 responsible for COVID-19 disease. An experimental study is necessary to confirm these results.
Author’s contributions statement
All authors confirmed they have contributed to the intellectual content of this paper and have met the following requirements: significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; drafting or revising the article for intellectual content; and final approval of the published article.
Below is the participation of author’s by task
Moujane Soumia, Zaki Hanane and Moualij Benaissa concept, design and perform the study; Moujane Soumia, Zaki Hanane, Bouachrine Mohammed and Benlyas Mohamed develop the methodology; Moujane Soumia, Zaki Hanane, Moualij Benaissa and Benlyas Mohamed write the original draft; Moujane Soumia, Zaki Hanane and Bouachrine Mohammed validate the data obtained; Moujane Soumia, Zegzouti Filali Younes, Alem Chakib and Benlyas Mohamed analyse and validate the data; all authors revise, edit and approve the submitted article; Bouachrine Mohammed and Benlyas Mohamed supervise the study.
Acknowledgments
We would like to thank Rihab Benlyas for critical reading the paper and revising the spelling. S. Moujane is a recipient of a grant from the University of Moulay Ismail, 2020.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes
Note: V: Van der Waals is the interaction types, M and S are the main chain and a side chain.
Note: His 41: Histidine (41), Cys145: Cysteine (145).
1 SARS-CoV-2: Severe acute respiratory syndrome coronavirus.
2 CoV: Coronaviruses.
3 MERS-CoV: Middle East respiratory syndrome coronavirus.
4 ACE2: Angiotensin-converting enzyme 2.
5 PLpro: Papain-like protease PLpro.
6 3CLpro: The viral enzyme 3-chymotrypsin-like cysteine protease.
7 Mpro: Main protease.
8 VdW: Van der Waals.
9 HBond: Hydrogen bond.
10 Electrostatic bond.
11 PIs: Protease inhibitors.
12 Protease inhibitors.
References
- Lu H, Stratton CW, Tang Y. Outbreak of pneumonia of unknown etiology in Wuhan, China: the mystery and the miracle. J. Med. Virol. 2020;92:401–402.
- Yuen K-S, Ye Z-W, Fung S-Y, et al. SARS-CoV-2 and COVID-19: the most important research questions. Cell Biosci. 2020;10(40):1–5.
- Cunningham AC, Goh HP, Koh D. Treatment of COVID-19: old tricks for new challenges. Crit Care. 2020;24(91):1–2.
- Panoutsopoulos AA. Known drugs and small molecules in the battle for COVID-19 treatment. Genes Dis. 2020. doi:10.1016/j.gendis.2020.06.007.
- Ojha PK, Kar S, Krishna JG, et al. Therapeutics for COVID-19: from computation to practices—where we are, where we are heading to. Mol. Divers. 2020. doi:10.1007/s11030-020-10134-x.
- Liu C, Zhou Q, Li Y, et al. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6:315–331.
- Du L, et al. The spike protein of SARS-CoV–a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009;7:226–236.
- Liang J, et al. Interaction of the prototypical α-ketoamide inhibitor with the SARS-CoV-2 main protease active site in silico: molecular dynamic simulations highlight the stability of the ligand-protein complex. Comput. Biol. Chem. 2020;87. doi:10.1016/j.compbiolchem.2020.107292.
- Bacha U, Barrila J, Velazquez-Campoy A, et al. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906–4912.
- Yin J, et al. A Mechanistic View of enzyme inhibition and Peptide Hydrolysis in the active site of the SARS-CoV 3C-like Peptidase. J. Mol. Biol. 2007;371:1060–1074.
- Anand K, Ziebuhr J, Wadhwani P, et al. Coronavirus main Proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767.
- Hsu K-C, Chen Y-F, Lin S-R, et al. iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinformatics. 2011;12(S33):1–11.
- Berman HM, et al. The protein data bank. Acta Crystallogr. D Biol. Crystallogr. 2002;58:899–907.
- ASINEX Ltd. Asinex.com – Asinex Focused Libraries, Screening compounds. Available from: http://www.asinex.com/.
- Jin Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020. doi:10.1038/s41586-020-2223-y.
- Dassault Systèmes BIOVIA. BIOVIA Discovery Studio. (Dassault Systèmes, 2020).
- Morris GM, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791.
- Sanner MF. Python: a programming language for software integration and development. J. Mol. Graph. Model. 1999;17:57–61.
- Kneller DW, Agniswamy J, Ghosh AK, et al. Potent antiviral HIV-1 protease inhibitor combats highly drug resistant mutant PR20. Biochem. Biophys. Res. Commun. 2019;519:61–66.
- Shi Y, et al. Identification of two antiviral inhibitors targeting 3C-like serine/3C-like protease of porcine reproductive and respiratory syndrome virus and porcine epidemic diarrhea virus. Vet. Microbiol. 2018;213:114–122.
- Pawar S, et al. Structural studies of antiviral inhibitor with HIV-1 protease bearing drug resistant substitutions of V32I, I47 V and V82I. Biochem. Biophys. Res. Commun. 2019;514:974–978.
- Taylor JG, et al. Discovery of the pan-genotypic hepatitis C virus NS3/4A protease inhibitor voxilaprevir (GS-9857): A component of Vosevi®. Bioorg. Med. Chem. Lett. 2019;29:2428–2436.
- Wypych RM, LaPlante SR, White PW, et al. Structure-thermodynamics-relationships of hepatitis C viral NS3 protease inhibitors. Eur. J. Med. Chem. 2020;192. doi:10.1016/j.ejmech.2020.112195.
- Wu C. et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020. doi:10.1016/j.apsb.2020.02.008.
- Yu R, Chen L, Lan R, et al. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int J Antimicrob Agents. 2020. doi:10.1016/j.ijantimicag.2020.106012.
- Gurung AB, Ali MA, Lee J, et al. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sci. 2020;255. doi:10.1016/j.lfs.2020.117831.
- Hall DC, Ji H-F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020. doi:10.1016/j.tmaid.2020.101646.
- Khodadadi E, et al. Study of combining virtual screening and antiviral treatments of the sars-CoV-2 (Covid-19). Microb. Pathog. 2020;146. doi:10.1016/j.micpath.2020.104241.
- Macchiagodena M, Pagliai M, Procacci P. Identification of potential binders of the main protease 3CLpro of the COVID-19 via structure-based ligand design and molecular modeling. Chem. Phys. Lett. 2020;750; doi:10.1016/j.cplett.2020.137489.
- Mirza MU, Froeyen M. Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020. doi:10.1016/j.jpha.2020.04.008.
- Báez-Santos YM, St. John SE, Mesecar AD. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115:21–38.