
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In the current study, optical and nonlinear optical (NLO) properties of supersalt Al(BH4)3 doped boron nitride (BN) were studied by employing density functional theory (DFT) at the MPW1PW91/6-31 G (d,p) level of theory. HOMO–LUMO energy gap of 7.48 eV was found for pure BN which is reduced to 4.85 eV after doping. Supersalt doping has shown better results on the NLO properties including a decrease in the excitation energy, change in vertical ionization energies, increase in oscillator strength, and increase in dipole moment. Doped molecules have lesser transition energies (ΔE) that lead to higher hyperpolarizability. Electron density difference map was plotted which describes the distribution of electrons of doped compounds. The enhanced values of αiso and βstatic are observed which increases the NLO properties. Overall, the results have shown that supersalt doped boron nitride is an effective approach for designing efficient nonlinear material.
1. Introduction
Nonlinear optical (NLO) effects were being proposed around 1930s [Citation1], which deal with the interaction of LASER with the matter. Engrossed study on NLO began in 1961 and led to the second harmonic generation shortly after the contriving of the first LASER in 1960 [Citation2]. Because of its usage in different types of optoelectronic devices, biosensors, LASER, LED, photovoltaics, dynamic holography, [Citation3] characteristics of molecules and polymers have sparked the interest of both experimental and theoretical chemists [Citation4–15]. Optical devices have begun to retrieve electrical gadgets in a variety of applications during the last few decades [Citation16].
The NLO substances are broadly classified into four categories that are inorganic [Citation17], organic [Citation18], hybrid materials [Citation19], and organometallics [Citation20]. Among these, organic NLO materials perform better as compared to inorganic materials due to their structural diversity flexibility, and elevated NLO coefficient. Some species with excess electrons also exhibit a strong NLO response, where the excess electrons are responsible for augmenting the first hyperpolarizability () [Citation21, Citation22].
BN materials are the most exciting inorganic materials. B12N12, B16N16, B18N18, B22N22, and B28N28 are stable structures of BN [Citation23]. Two major forms of BN are cubic and hexagonal boron nitride. Cubic form (c-BN), shows excellent wear resistance, good chemical inertness, and high thermal conductivity. The hexagonal form is thermally stable, used as heat resistant and electrical insulators [Citation24]. B12N12 is most stable because of its small size. Density functional theory (DFT) based treatment at visualizing applicability of B12N12 like useful storage medium of encapsulating hydrogen in the molecular state. Particulars about potential worth of B12N12 cage corresponds to optimal hydrogen storage; enlighted with analysis of stability of H2-bounded B12N12 cage cluster with stance of aromaticity[Citation23,Citation25].
Lai-sheng et al. reported very first actual evidence of superhalogens in comparison to theoretical data via a series of (photoelectron spectra) [Citation26]. These anions are counterbalanced by metal cations which are super alkalis. The combination of these two will form supersalts like CsAuF6, KMnO4, and KMnCl3. [Citation27, Citation28]. Supersalts show resemblance to alkali halides which are salts.
Borohydrides are a special class of complex hydrides that possess larger volumetric and gravimetric densities, these materials face hydrogen desorption over 85°C. Al(BH4)3 is strong pyrophorous liquid. Al(BH4)3 has planar trigonal geometry having double hydrogen bridge. Al(BH4)3 shows a gravimetric density of 16.8 wt % and this also releases hydrogen at low temperature. This is a non-polar compound and these neutral colored crystals melts a round −64.5°C, its vapour tension at 0° is 119.5 mm, and its boiling point (extrapolated) is 44.5°C. It is rapidly hydrolyzed at ordinary temperature and produces aluminium hydroxide, hydrogen and boric acid. It rapidly interacts with hydrogen chloride, at even -80°C, to produce diborane,aluminium chloride and hydrogen [Citation29].
In present work, effects of supersalt doping on linear, nonlinear and electrical properties of BN were examined. Significant changes in optical and electronic properties were examined after doping. We attempted to clarify several aspects, such as the impact on the bandgap, changes in geometry after doping, and modifications in the energy gap of HOMO-LUMO doped compounds.
2. Computational details
Density functional theory using MPW1PW91 [Citation30] level of theory being used to do all calculations because this is utmost reliable method accompanying Gaussian 09 [Citation31]. Geometries and molecular orbital images were visualized using GaussView 6.0 [Citation32]. The MPWIPW91/6-31G* level of theory is the best approach to calculating transition energies and dipole moment of the studied molecular systems. The vertical ionization energy (VIE), energy gap among HOMO and LUMO, ultraviolet–visible (UV-Vis) absorption spectra of optimized substances, alongwith difference in dipole moment, all were calculated using computational methods. HOMO–LUMO energy gap is calculated by equation [Citation33].
(1)
(1)
Where EH and EL are energies of HOMO and LUMO.
The vertical ionization energies (EVI) [Citation34] are defined as.
(2)
(2)
E(X+) represents energy of cation of supersalt doped boron nitride (SS@BN)+ as well as E(X) is the energy of the neutral SS@BN when neutral geometry of SS@BN being optimized. (DOS) of boron nitride and doped boron nitride calculated by MPW1PW91/6-31G (d,p) level of theory, their images were drawn by using PyMOlyze software [Citation35].
3. Results and discussion
3.1. Geometrical parameters
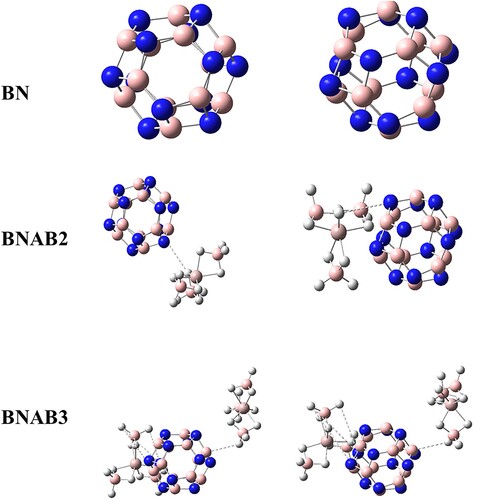
BN is used for pure boron nitride, BNAB2 is used for supersalt which is single doped and BNAB3 represents supersalt which is doped twice with boron nitride. In BNAB2, supersalt is placed in such a way that the 16th N atom of BN is hexagonal with 2 rings and also cubical with two rings. In BNAB3, supersalt is placed in such a way that the 16th N atom of BN is hexagonal with 2 rings and also cubical with two rings. Super salt interacted with (BN) was explored by applying altered combinations that are described below: the super salt (Al(BH4)3) was placed on a single side and then placed on the double side and the following combinations were produced depicted in Figure .
Figure 1. Optimized geometries of BN, BNAB2, and BNAB3 front view in left side view in right.
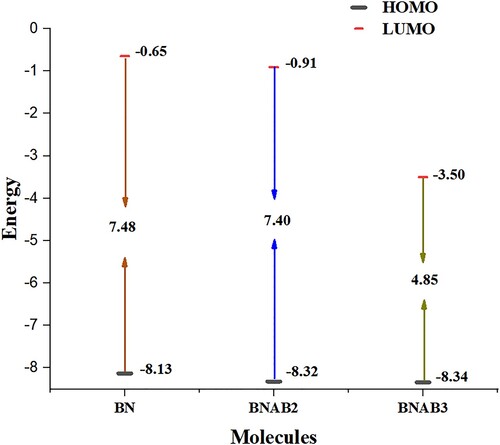
The vertical ionization energy of pure boron nitride (BN) is 8.13 eV. And the vertical ionization energy of BNAB2 is 8.32 eV and BNAB3 has vertical ionization energy of 8.34 eV (Table ).
Table 1. HOMO, LUMO energies of observed molecules, their band gaps, and VIE in eV.
3.2. Electronic properties
Frontier molecular orbitals (FMO) that include the highest molecular orbital (HOMO) and lowest molecular orbital (LUMO) help inunderstanding optical as well as electronic properties [Citation36]. It also provides particulars regarding transfer of charge [Citation37]. The outcomes related to FMO assessment at MPW1PW91 functional through 6-31G (d,p) basis set. HOMO of boron nitride (BN) shows the energy of −8.13 eV that increases to −0.65 eV when it enters the excited state showing the band gap between them of 7.48 eV. During excitation, electron density moves from BN to supersalt Al(BH4)3. Similar to that, in BNAB2, energy values of HOMO and LUMO are −8.32 eV and −0.91 eV, and they show a bandgap of 7.40 eV, which is less than the bandgap of BN. The lowest band gap is shown by BNAB3 which is 4.83 eV it shows the value of HOMO as −8.33 eV and the value of LUMO is −3.50 eV. Upon arranging these molecules in the ascending order of bandgap energy values, this trend was observed BNAB3 < BNAB2 <BN. This order shows that this molecule BNAB3 shows the lowest bandgap; upon excitation, it will help the electron transfer from HOMO to LUMO. When boron nitride was double doped with supersalt, some new energy levels were created in this process. The lower values of Eg make the excitation of electrons feasible, lesser Eg is related to lower kinetic stability because it helps to exclude the electrons from lower energy HOMO towards higher energy LUMO; due to these energy levels, major changes were noticed in Eg of BNAB3. BN in its pure form is an insulator, after doping with supersalt Al(BH4)3, the HOMO–LUMO energy gap has reduced, and BN is converted into a semi-conductor. This alleviation in HOMO LUMO will increase its conducting effects. Figure . Shows the FMOs of all 3 molecules. The red colour depicts the positive part of the orbital and the green colour depicts the negative part of an orbital. At MPW1PW91/6-31G level of theory calculated energies of HOMO & LUMO. Figure
Figure 2. FMOs of pure BN, BNAB2, and BNAB3.
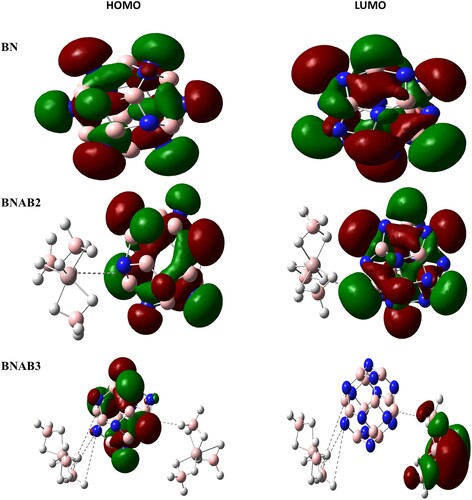
Figure 3. Energy difference in (eV) of HOMO LUMO for BN, BNAB2, and BNAB3.
Polarizability which is observed for pure boron nitride is 141.37 au. When pure boron nitride is doped with supersalt then its polarizability is increased. Observed polarizability for αiso for BN, BNAB2, and BNAB3 is 141.37, 213.58, and 288.40 au, sequentially. Polarizability values of αiso are in the increasing order like BN < BNAB2< BNAB3 and βstatic increases in the order of BN < BNAB2< BNAB3.
3.3. Nonlinear optical properties
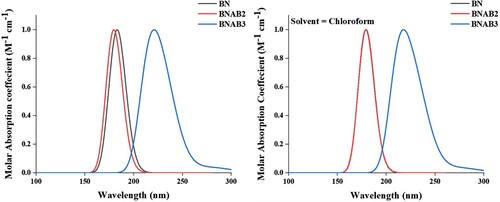
The first hyperpolarizability which is a 3×3×3 matrix depicts intrinsic molecular hyperpolarizability due to lack of resonating effect. f has no dimensional quantity, its portrays probabilities of emission and absorption of electromagnetic radiations among transitions within different energy levels of the atom. ΔE represents energy gap among HOMO and LUMO in eV showing percent transition of charge from HOMO to LUMO. λmax embodies the maximum absorption for complexes about visible and ultraviolet light. Observed results indicated that pure BN showed a linear behaviour with 141.37 au value of linear isotropic polarizability with negligible first hyperpolarizability value. The nonlinear optical response of BN in pure form is very low (0.0014 au) that increases via doping as in BNAB2, and BNAB3. The NLO response of the doped complexes enhances to 373.47, and 383.30 au respectively (Table ).
Table 2. The polarizability αiso, βstatic in (a.u) for BN, BNAB2, and BNAB3.
The analysis of the two-level method is performed to study the variation of hyperpolarizability βo, also responsible for this change. The estimations are done to evaluate values for these factors such as a change in dipole moment (δµ), ΔE, and the strength of oscillation fo in the excited state of molecules. Relation between βo along these factors is given by the following equation.
(3)
(3)
The oscillator strength is fo, the critical excited state's excitation energy is the distinction among excited as well as ground state is denoted by Δμ. fo is inversely proportional to
E3, that describes third power of excitation energy, βo is directly proportional to Δμ. In this case, BNAB3 shows less energy gap among LUMO, and HOMO shows maximum chances of excitation; HOMO towards LUMO that are 69%.
3.4. Uv–Vis spectral properties
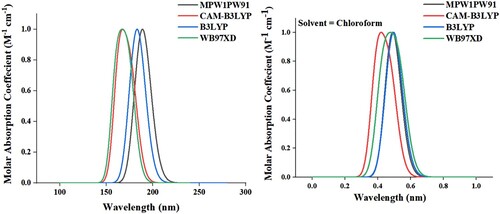
Considering the absorption coefficient and optical properties of boron nitride evaluated using chloroform as a solvent by using B3LYP [Citation38], CAM-B3LYP [Citation39], MPW1PW91, and WB97XD [Citation40] functionals. From Figure , CAM-B3LYP and WB97XD show absorption in the middle of 130–190 nm. B3LYP and WB97XD show absorption in the range of 200–230. The figure depicts that the MPW1PW91 level of theory shows better absorption. All of the calculations of BN, BNAB2, and BNAB3 were calculated using MPW1PW91 functional. In the liquid phase, chloroform was used as a solvent to calculate the absorption coefficient. Due to the presence of chloroform, the absorption band moves towards a longer wavelength i.e. shows redshift because of stabilization of delocalized electrons. DFT approach was used to optimize designed compounds, and their λmax values in solvent as well as gas phase as reported in Tables and .
Figure 4. Calculated αiso and βstatic for BN and doped BN.
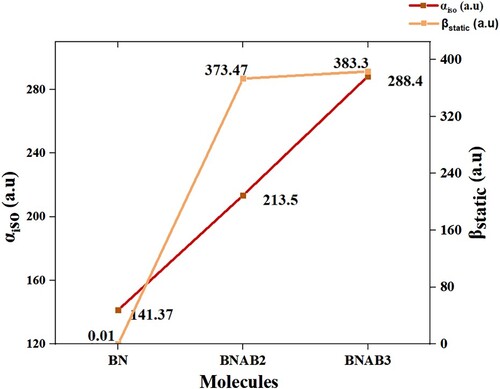
Table 3. Calculated maximum wavelength (λmax), first excited state oscillator strength (ƒo), as well as transition character of considerate molecules in the gas phase.
Table 4. Calculated maximum wavelength (λmax), oscillator strength (fo), as well as transition character of considerate molecules in the solvent phase.
BNAB3 shows the maximum λmax around 220 nm amongst other compounds.. All charge transfer and absorption properties relate to the HOMO–LUMO energy bandgap. When the energy band gap is lower charge transferring and absorption will be higher. On the contrary, these compounds show lesser absorption in gas phase. Absorption profile of each molecule in gaseous and solvent phases are depicted Figure .
Figure 5. UV visible absorption graphs using different functionals for BN as well as doped BN in Chloroform solvent (right) and gaseous phase (left).
3.5. Density of states (DOS) analysis
DOS analysis is accomplished for studying densities electronic structure to support the results of FMOs. DOS of BN and doped BN with supersalt Al(BH4)3 were observed by MPW1PW91 functional using 6-31G (d,p) level of theory, their outcomes are depicted in Figure . Various coloured peaks showed electronic densities of BN, supersalt doped BN. DOS graphs show negative and positive energy values in eV that further depicts bonding and antibonding orbitals that are involved in the distribution of charge; zero depicts the non-bonding interaction of orbitals. Fermi levels for pure BN is −4.39 eV, for BNAB2 is −4.615 eV and for BNAB3 is −5.915 eV. Fermi levels are calculated by this formula:
(4)
(4)
Figure 6. UV visible absorption graphs for BN as well as doped BN in Chloroform solvent (right) and gaseous phase (left).
3.6. Dipole moment
Dipole moment can discard the intermolecular disorders as it determines the dissolution of substance within the organic solvent. Molecules that have crystalline geometry possess elevated values of dipole moment as well as shows solubility in organic solvents in comparison with molecules that show less dipole moment [Citation41]. Dipole moment as well as solubility are interlinked [Citation42]. The dipole moment of BN and doped boron nitride is performed at MPW1PW91 functional and shown in Table (Figures –).
Figure 7. DOS spectra of BN, BNAB2, and BNAB3.
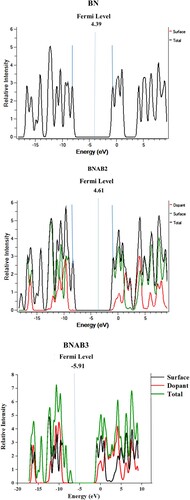
Figure 8. Observed dipole moment (μ) of BN, BNAB2, and BNAB3.
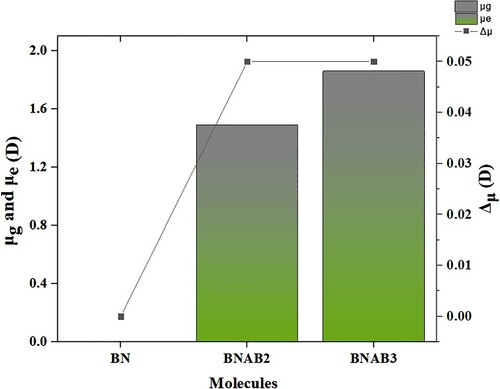
Table 5. Observed dipole moments in the excited state (μe), ground state (μg), and difference among excited state as well as ground state (Δμ) in Debye for observed molecules.
Table 6. Computed HOMO (EH), LUMO energy (EL) of investigated molecules and their band gap (Egap).
The value of dipole moment for BN, BNAB2, and BNAB3 in the gaseous phase are 0.00, 1.44, and 1.81 D respectively. And when chloroform was used as a solvent calculated values are 0.00, 1.49, and 1.86 D respectively. In the ground state, descending order of dipole moment is, BNAB3> BNAB2> BN. It increased from the ground state to the excited state due to the greater ionization potential of electrons in chloroform as a solvent. The results have shown that BNAB3 has higher values of dipole moment because the interactions between dopant and surface are very strong, it shows better NLO properties and it made BNAB3 more versatile.
3.7. The electron density difference map (EDDM)
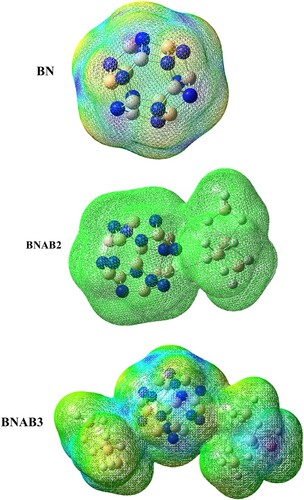
The electron density difference map describes rearrangement of electrons in pure and doped boron nitride. The map of doped boron nitride is obtained by evaluating the contrast between the excited state as well as ground state. Ground states as well as excited states of complexes were estimated through the very same MPW1PW91/6-31G level of theory. Regions related to nucleophilic as well as electrophilic reactions are highlighted with different colours to differentiate them. Figure shows sea-green colour shows lessening of electronic charge and purple colour shows an excess of electrons. These studies also revealed the non-covalent bonding interaction among the molecules. It can also be estimated from the analysis that charge shifting occurred among the boron nitride and supersalt. It was difficult to calculate the difference among low energy orbitals (HOMO) and high-energy orbitals (LUMO). EDDM analysis was carried out to take into account the distribution of electrons among molecule, it transitioned through excitement to a ground state [Citation43] (Table ).
Figure 9. EDDM of pure S, BNAB2, and BNAB3.
The covalent characteristics of the bond which is present between different atoms of molecules are well explained by Infrared spectroscopy (IR). IR spectra of BN, as well as doped boron nitride, are well studied using DFT calculations by applying MPW1PW91/6-31 G level of theory. Molecules in IR region in the range of 400-4000 cm−1. IR of BN shows various peaks around 1446, 1224, 1000, and 807 cm−1. Figures
Figure 10. Calculated EH, EL, and Eg of BN, BNAB2, and BNAB3.
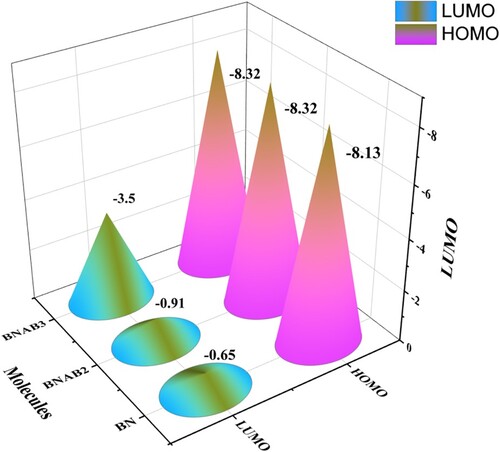
Figure 11. IR spectra of BN, BNAB2, and BNAB3.
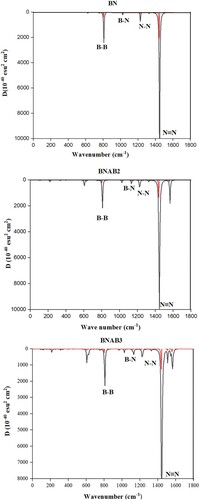
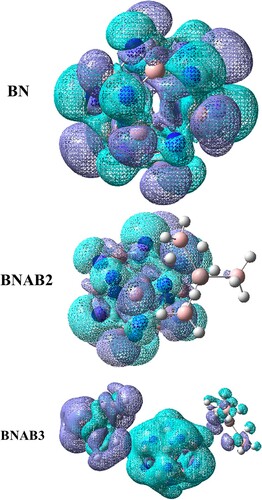
Figure 12. TDM pictures for BN, BNAB2, and BNAB3.

Figure 13. Molecular electrostatic potential of BN, BNAB2, and BNAB3.
3.8. Transition density matrix (TDM)
The charge distribution, electronic excitation, and charge fluctuations during excitation were all acknowledged using transition density matrix assessment [Citation44]. The transition density matrix has been known for describing exciton effects [Citation45]. A transition density matrix may be depicted as; a spatial map that reveals electron–hole pairs are being produced and to which dimensional ranges they expand for a given electronic excitation process linking the excited state ψn to the ground state . The contribution of hydrogen atoms was ignored due to their insignificant involvement in transitions. The X-axis in TDM image depicts number of atoms of distinct material, and y-axis on right side shows the range of electron density. Using MPW1PW91/ 6-31G (d, p) level of theory, the distribution of charge is studied well. In BNAB3 the charge density is uniformly distributed all over the molecule. All 3 molecules have shown diagonal and off-diagonal properties, which shows the better separation of charges among all molecules.
3.9. Molecular electrostatic potential (MSEP)
Molecular electrostatic potential a projection map calculated about supersalt doped boron nitride was calculated using MPWIPW91/6-31G method. It helps in calculating physicochemical properties and molecular structure. 3D visual is shown by this method to apprehend the electrostatic potential effect. The green colour shows neutral electrostatic potential. The blue colour shows the electron poor region which is electron-deficient [Citation46].
4. Conclusion
We have conducted a thorough examination of supersalt Al(BH4)3 doped with boron nitride (BN) to examine the optical and NLO properties using density functional theory (DFT). HOMO–LUMO gap is reduced when BN is doped with Al(BH4)3. BNAB3 shows the lowest energy gap of 4.83 eV. BNAB3 has shown the higher values of λmax. When the most stable geometry of the complex was selected, their DOS, EDDM and FMO analysis give information regarding its charge distribution. The thermodynamic stability of pure as well as doped compounds has been evaluated with the aid of vertical ionization potential. The doped system shows extraordinary higher values of the first hyperpolarizability. This study will provide a unique perspective for the design of novel NLO substances with useful applications in wavelength conversion.
Acknowledgment
The authors are acknowledged for the technical support from the University of Agriculture, Faisalabad. We thank Dr. Khurshid Ayub for the additional resources from the COMSATS University, Abbottabad Campus, Pakistan.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Ward J, Miller C. Measurements of nonlinear optical polarizabilities for twelve small molecules. Phys Rev A. 1979;19(2):826.
- Menzel R. Photonics: linear and nonlinear interactions of laser light and matter. Springer Berlin, Heidelberg: Springer Science & Business Media; 2013.
- Liang F, et al. Molecular construction using (C3N3O3) 3–anions: analysis and prospect for inorganic metal cyanurates nonlinear optical materials. Cryst Growth Des. 2017;17(7):4015–4020.
- Di Bella S, et al. Synthesis, characterization, optical spectroscopic, electronic structure, and second-order nonlinear optical (NLO) properties of a novel class of donor− acceptor bis (salicylaldiminato) nickel (II) Schiff base NLO chromophores. J Am Chem Soc. 1997;119(40):9550–9557.
- Zahid S, et al. Tuning the optoelectronic properties of triphenylamine (TPA) based small molecules by modifying central core for photovoltaic applications. J Mol Model. 2021;27(9):1–14.
- ul Ain Q, et al. Designing of benzodithiophene acridine based Donor materials with favorable photovoltaic parameters for efficient organic solar cell. Computational and Theoretical Chemistry. 2021;1200:113238.
- Shehzad RA, et al. Exploring the optoelectronic and third-order nonlinear optical susceptibility of cross-shaped molecules: insights from molecule to material level. J Mol Model. 2021;27(1):1–10.
- Shehzad RA, et al. Electro-Optical and Charge Transport Properties of Chalcone Derivatives Using a Dual Approach from Molecule to Material Level Simulations. Computational and Theoretical Chemistry. 2021;1203:113349.
- Rasool A, et al. Designing of benzodithiophene (BDT) based non-fullerene small molecules with favorable optoelectronic properties for proficient organic solar cells. Computational and Theoretical Chemistry. 2021;1203:113359.
- Naseem Z, et al. Theoretical investigation of supramolecular hydrogen-bonded choline chloride-based deep eutectic solvents using density functional theory. Chem Phys Lett. 2021;769:138427.
- Naeem N, et al. Molecular Engineering Strategy of Naphthalimide Based Small Donor Molecules for High Performance Organic Solar Cells. Computational and Theoretical Chemistry. 2021;1204:113416.
- Khan AU, et al. DFT study of superhalogen (AlF4) doped boron nitride for tuning their nonlinear optical properties. Optik (Stuttg). 2021;231:166464.
- Khan AU, et al. DFT study of superhalogen and superalkali doped graphitic carbon nitride and its non-linear optical properties. RSC Adv. 2021;11(14):7779–7789.
- Ishaq M, et al. DFT study of superhalogen-doped borophene with enhanced nonlinear optical properties. J Mol Model. 2021;27(6):1–11.
- Fatima R, et al. Exploring the potential of tetraazaacene derivatives as photovoltaic materials with enhanced photovoltaic parameters. Int J Quantum Chem. 2022;122:e26817.
- Chen C, et al. Nonlinear optical borate crystals: Principals and applications. Weinheim Wiley-VCH-Verl: John Wiley & Sons; 2012.
- Champagne B, et al. Nonlinear optical molecular switches as selective cation sensors. J Am Chem Soc. 2012;134(19):8101–8103.
- Gao F-W, Xu H-L, Su Z-M. The inner-induced effects of YCN in C 76 on the structures and nonlinear optical properties. J Mol Model. 2016;22(8):1–7.
- Wang Y-F, et al. Hopping of Li@ AR (AR= benzene and naphthalene) between electride and lithium salt configurations brings considerably different first hyperpolarizabilities: Candidate for high-performance nonlinear optical molecular switches. J Phys Chem C. 2019;123(39):24248–24254.
- Janjua MRSA, et al. Prediction of remarkably large second-order nonlinear optical properties of organoimido-substituted hexamolybdates. J Phys Chem A. 2009;113(15):3576–3587.
- Zhong R-L, et al. Role of excess electrons in nonlinear optical response. J Phys Chem Lett. 2015;6(4):612–619.
- Shafiq S, et al. DFT study of OLi3 and MgF3 Doped Boron Nitride with Enhanced Nonlinear Optical Behavior. J Mol Struct. 2021;1251:131934.
- Moon WH, Son MS, Hwang HJ. Theoretical study on structure of boron nitride fullerenes. Appl Surf Sci. 2007;253(17):7078–7081.
- Raza Ayub A, et al. Super alkali (OLi3) doped boron nitride with enhanced nonlinear optical behavior. Journal of Nonlinear Optical Physics & Materials. 2020;29(01n02):2050004.
- Li J, He T, Yang G. An all-purpose building block: B 12 N 12 fullerene. Nanoscale. 2012;4(5):1665–1670.
- Graudejus O, et al. Room Temperature Syntheses of AuF6-and PtF6-Salts, Ag+ AuF6-, Ag2+ PtF62-, and Ag2+ PdF62-, and an Estimate for E (MF6-)[M= Pt, Pd]. Inorg Chem. 1999;38(10):2503–2509.
- Gutsev GL, et al. Origin of the unusual stability of MnO4−. Chem Phys Lett. 1999;312(5-6):598–605.
- Wu MM, et al. Manganese-based magnetic superhalogens. Angew Chem. 2011;123(11):2616–2620.
- Schlesinger H, Sanderson RT, Burg A. Metallo borohydrides. I. Aluminum borohydride. J Am Chem Soc. 1940;62(12):3421–3425.
- Adamo C, Barone V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J Chem Phys. 1998;108(2):664–675.
- Frisch M, et al. gaussian 09, Revision d. 01, Gaussian. Inc. Wallingford (CT); 2009. 201.
- Dennington R, et al. Semichem Inc. Shawnee Mission KS, GaussView, Version; 2009. 5.
- Shehzad RA, et al. Designing of benzothiazole based non-fullerene acceptor (NFA) molecules for highly efficient organic solar cells. Computational and Theoretical Chemistry. 2020;1181:112833.
- Shehzad RA, et al. Enhanced linear and nonlinear optical response of superhalogen (Al7) doped graphitic carbon nitride (g-C3N4). Optik (Stuttg). 2021;226:165923.
- Tenderholt A. (2019). PyMOlyze: a program to analyze quantum chemistry calculations, version 2.0.
- Ans M, et al. Tuning opto-electronic properties of alkoxy-induced based electron acceptors in infrared region for high performance organic solar cells. J Mol Liq. 2020;298:111963.
- Adeel M, et al. Exploration of CH⋯ F & CF⋯ H mediated supramolecular arrangements into fluorinated terphenyls and theoretical prediction of their third-order nonlinear optical response. RSC Adv. 2021;11(14):7766–7778.
- Civalleri B, et al. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm. 2008;10(4):405–410.
- Yanai T, Tew DP, Handy NC. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett. 2004;393(1-3):51–57.
- Chai J-D, Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys Chem Chem Phys. 2008;10(44):6615–6620.
- Manzoor F, et al. Theoretical calculations of the optical and electronic properties of dithienosilole-and dithiophene-based donor materials for organic solar cells. ChemistrySelect. 2018;3(5):1593–1601.
- Sutradhar T, Misra A. Role of Electron-Donating and Electron-Withdrawing Groups in Tuning the Optoelectronic Properties of Difluoroboron–Napthyridine Analogues. J Phys Chem A. 2018;122(16):4111–4120.
- Muhammad S, et al. Benchmark study of the linear and nonlinear optical polarizabilities in proto-type NLO molecule of para-nitroaniline. Journal of Theoretical and Computational Chemistry. 2019;18(06):1950030.
- Li Y, Ullrich C. Time-dependent transition density matrix. Chem Phys. 2011;391(1):157–163.
- McWeeny R. Some recent advances in density matrix theory. Rev Mod Phys. 1960;32(2):335.
- Medjahed S, et al. Computational study of molecular electrostatic potential, drug likeness screening and structure-activity/property relationships of thiazolidine-2, 4-dione derivatives. Journal of Bionanoscience. 2016;10(2):118–126.