
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The main mathematical results on the data processing in vibrational spectroscopy are presented. The approaches and algorithms proposed for molecular force field calculations have been constructed on a base of regularizing methods for solving nonlinear ill-posed problems and have been implemented in the software package SPECTRUM. These algorithms were used for constructing the regularized ab initio force fields of important biological molecules including the melatonin and Vitamin B6 derivatives.
1. Introduction
We consider the inverse problems of vibrational spectroscopy related to calculations of the molecular force field parameters on the base of experimental data (mainly obtained from analysis of the molecular vibrational (infrared and Raman) spectra) [Citation1,Citation2]. The molecular force field plays an important role in determining the properties of a molecule, in particular its vibrational spectrum.
The ill-posed character of inverse vibrational problem has led to some degree of subjectivity, related basically to constraints imposed on the solution. Many models of the molecular force fields are known and a great number of force field matrices have been calculated for various series of compound. This lead to situation, in particular, for complicated polyatomic molecules, when a number of proposed force fields are inconsistent due to different criteria for the physical feasibility of solutions used by various investigators and due to the instability (with respect to small perturbations in the input information) of numerical methods used to solve the inverse problem.
To overcome these difficulties we have proposed to apply the stable numerical methods based on the theory of regularization for solving inverse vibrational problem [Citation3,Citation4] based on the formulation of problem and formalization of all possible obvious (and not so obvious) model assumptions concerning the character of force fields which are widely used in vibrational spectroscopy. The principle for choosing a unique solution from the set of solutions have constructed in terms of the closeness of the solution to the given matrix of force constants which satisfy all a priori assumptions concerning the model characteristics of the solution. Within this approach the new statement of inverse vibrational problem has been proposed as a problem of finding the solution which is closest to the definite quantum mechanical result obtained for the molecular force field – the so-called regularized quantum mechanical force field (RQMFF) [Citation5].
In this paper, we describe the results of application of stable methods based on the RQMFF approach to solve inverse problems in vibrational spectroscopy for bulky molecules, e.g. such as biological ones.
2. Molecular force fields and experimental sources of information
The most important sources of knowledge about molecular structure are vibrational spectra (Infrared and Raman [Citation1,Citation2]), which provide information on molecular vibrational frequencies. Vibrational spectra are very sensitive to the small changes in molecular structure. As an example, consider the structures of the most stable equatorial conformations for two biologically important compounds related to the manufacturing of drugs – 5H-dibenz [b,f] azepine (Figure (a)) and 5H-dibenz[a,d]cyclohepten-5-ol (Figure (b)) [Citation6].
Figure 1. Stable conformations of 5H-dibenz[b,f]azepine (a) and 5H-dibenz[a,d]cyclohepten-5-ol (b) molecules.
![Figure 1. Stable conformations of 5H-dibenz[b,f]azepine (a) and 5H-dibenz[a,d]cyclohepten-5-ol (b) molecules.](/cms/asset/caaf7d66-ca7f-4e61-9b3c-737464d17ee9/gipe_a_1797717_f0001_ob.jpg)
The structures of these two molecules are different only in the central seven-atom-cyclic-fragment, where the azepine derivative (a) has the NH functional group, while the cycloheptane derivative (b) contains the CH(OH) group. Nevertheless, the infrared spectra of these compounds are varied widely, especially in the so-called ‘finger-print’ spectral region (500–1500 cm−1) [Citation6] which is usually used to identify unknown or two different organic compounds (Figure ).
Figure 2. Infrared spectra (400–3000 cm−1) of 5H-dibenz [b,f] azepine (top) and 5H-dibenz[a,d]cyclohepten-5-ol (bottom) [Citation6].
![Figure 2. Infrared spectra (400–3000 cm−1) of 5H-dibenz [b,f] azepine (top) and 5H-dibenz[a,d]cyclohepten-5-ol (bottom) [Citation6].](/cms/asset/1b8c3f9c-773f-4baa-9e5e-3ee7256e7868/gipe_a_1797717_f0002_ob.jpg)
This sensitivity of infrared spectra to the small changes in the molecular structure determines their wide scientific and practical applications in many fields of science and industry including the life-science field (biochemistry, biophysics, medicine, plant science, etc.) as the very important instrument for the identification and non-destructive characterization of substances. The additional information about molecular structure can be obtained from the Raman spectra which are complimentary to the IR ones, because the selection rules for the IR and Raman spectra are different [Citation1,Citation2].
A number of inverse problems have arisen in the analysis of spectral data on molecular vibrations. One of the most important is the so-called inverse vibrational problem, the problem of determining parameters of the molecular force field from experimental data, The set of data can include different types of experimental information: vibrational frequencies, isotope frequency shifts, Coriolis constants, centrifugal distortion constants, etc.
The idea of the force field arises when a molecule is considered as a mechanical system of nuclei while all the interactions due to the electrons are included in an effective potential function U(q1,q2, … ,qn) where {q1,q2, … ,qn} denote n = 3N−6 generalized coordinates describing mutual positions of N atomic nuclei of the molecule. Together with the nuclear masses, this function determines the most important properties of a molecule. As is well known (see, e.g. [Citation1,Citation2]), the equilibrium configuration of the molecule satisfies the relation
(1)
(1) where we define coordinates { q1, q2, … , qn} so that in the equilibrium configuration q1 = q2 = … = qn = 0, and the following expansion is valid:
(2)
(2) U0 is a certain constant. The force constants
constitute a positive definite matrix F determining all the molecular characteristics related to the small vibrations. Mathematically, the concept of the force field may be obtained through the adiabatic theory of perturbations with the use of a small parameter related to the ratio of electron mass to the mass of nuclei [Citation1,Citation2]. In a certain approximation the nuclei may be treated as particles moving in the force field determined by the potential energy function (2).
The molecular force field can be used to check validity of various model assumptions commonly used by spectroscopists for approximation of the potential function and to predict the vibrational properties of certain molecules. These applications use the fundamental properties of the force field such as its isotopic invariance and the approximate transferability of specific force constants in a series of related compounds. The accumulation of data on molecular constants, in particular on force constants, helps us not only to predict spectra and thermodynamic properties of compounds not yet investigated but also assists the development of physical models in a theory of molecular structure.
The spectral frequencies ωi (the main type of experimental data on molecular vibrations) are connected with the matrix of force constants F by the eigenvalue equation
(3)
(3) where
is a diagonal matrix consisting of the squares of the molecular normal vibrational frequencies, and G is the kinetic energy matrix in the momentum representation which depends only on nuclear masses and the equilibrium configuration (assumed to be known within specified limits of error). The matrix L determines the form of each normal vibration, i.e. the relative amplitudes of vibrations in terms of classical mechanics. Both G and F matrices are symmetric and positive determined ones.
While Equation (3) is the main source of data determining the force constants, it is evident that (except for diatomic molecules) the n(n+1)/2 parameters of matrix F cannot be determined from n frequencies ω1, … , ωn (a molecule with N atoms has n = 3N−6 vibrations or 3N−5 in a case of linear molecule). As a result, for finding the molecular force fields of polyatomic molecules, we need to apply the certain model assumptions concerning the structure of the matrix F, and to use the additional experimental data.
The model assumptions which are used in the constructing the force constant matrix for the certain molecule are related with the different models of molecular force fields and basically are reduced to the assumptions on the relative values of force constants The additional experimental data can be obtained by using the spectra of isotopic molecular species, because within the approximations considered, the force field of the molecule is independent of the nuclear masses, and hence for the spectra of m molecular isotopic species we have, instead of (3), the system of equations
(4)
(4) where the subscript i = 1,2, … , m indicates the isotopomers of a molecule. Usually, the introduction of isotopomers frequencies leads to a limited number of independent equations in system (4), thus leaving the inverse problem underdetermined.
In the case of so-called high symmetry molecules (with the main symmetry axis order ≥ 3) the important additional information on the molecular force field is provided by the Coriolis constants, ζ, which characterize the vibrational-rotational interaction in the molecule. These constants are determined from rotational structure of vibration-rotational spectra and are connected with matrix F in terms of the eigenvectors L of the problem (3):
(5)
(5) where ζ is a matrix with vector elements consisting of the Coriolis constants, M is a diagonal matrix consisting of the nuclear masses, M is the sum of nuclear masses of the molecule, and A is a matrix with transpose A* connecting the Cartesian displacements of atoms with coordinates q, which can be found from the equilibrium configuration of the molecule. In a similar manner we can write the dependencies of other measured values on the matrix F, such as the mean-square amplitudes of the vibrations (obtained from gas-phase electron diffraction) which may be calculated from the eigenvalues and eigenvectors of (3) [Citation1,Citation2]. The above-mentioned experimental quantities (frequencies of isotopomers, Coriolis constants, mean-square amplitudes), while not so easily obtainable, are very important because their values do not depend on additional unknown parameters of any kind (unlike, e.g. the calculation of intensities in vibrational spectra).
3. Inverse vibrational problem
The inverse vibrational problem is formulated as nonlinear operator equation in finite-dimensional spaces
(6)
(6) where A is an operator completely defined by the certain molecular configuration, vector Λ∈Rm represents the set of available experimental data (vibrational frequencies, etc.) and F is an unknown symmetrical force constant matrix, F∈ Rn(n+1)/2. Experimental data as well as operator A are known with certain errors, that is, instead of A and Λ we are given Ah and Λδ where ||Λ–Λδ|| ≤ δ and h is a numerical estimate of the operator A uncertainty:
(7)
(7)
This problem is an ill-posed problem: it does not satisfy all three well-posedness conditions (existence of solution, its uniqueness and stability to perturbations in input data) [Citation7]. Vibrational frequencies of any molecular structure (except diatomic molecules), can be calculated using an infinite number of force field matrices which result in the same normal vibration frequencies. In the most general formulation the stable solution of problem (7) may be obtained on the base of Tikhonov regularization method [Citation4,Citation5] that allows to include additional information on the solution properties. The main idea is that this solution should be (in chosen metrics) close to an a priori known force constant matrix F0 that is either specified or based on some model assumptions [Citation8–16]. In the framework of this approach, stable solution may be obtained as an extremum Fα of Tikhonov’s functional [Citation5,Citation8–10]
(8)
(8) on the set of constraints D where F0 is some a priori chosen stabilizing force constant matrix. Tikhonov’s regularization technique requires that regularization parameter α should be chosen with account to the input data errors: α = α (h, δ). As a result, among all solutions of Equation (7) compatible with experimental data within a given error level, we obtain matrix Fα(h,δ) that is the closest in the sense of Euclidian norm to a given matrix F0. In our approach we choose the stabilizing matrix F0 as a result of modern quantum mechanical calculations, and thus search for matrix F which is the nearest by the chosen Euclidean norm to the given ab initio F0. The optimized solution is referred to as Regularized Quantum Mechanical Force Field (RQMFF) [Citation16]. The RQMFF is defined as the force constant matrix Fα that (1) is the nearest by a norm to the quantum mechanical matrix F0, (2) is reproduced the experimental data (frequencies, etc.) within the given error level, and (3) is satisfied to the set D – a closed set of a priori constraints/ These constraints can include the next assumptions:
some force constants may be stipulated on a priori grounds to be a zero;
some force constants may be stipulated to satisfy inequalities aij≤ fij≤ bij, where aij, bij – are certain known values;
some force constants may be stipulated to conform to be equal in a series of related molecules (or conformers);
the final solution may be stipulated to conform to the scaled force constant matrix, which may also be considered as a kind of constraint. In this case, set D is specified as:
Quantum mechanical calculations in the vibrational spectroscopy
One of the advantages of the regularizing procedure is the possibility to use any system of generalized coordinates, including redundant system of internal coordinates, and various constraints on the values of force constants. Within the Tikhonov regularization theory, it is well known that the stability and accuracy of the calculated solution Fα can be increased by using
an extended set of experimental data
an improved choice of the stabilizer matrix F0
an improved choice of the constraints set D
So the correct choice of constraint set D is extremely important. Physically stipulated limitations may either decrease a range of possible matrices F, or provide the criteria for selecting a concrete solution from the set of tolerate ones. An incorrect choice of constraints may lead to increasing incompatibility of the inverse problem, eventually resulting in a pseudosolution having no physical meaning.
The last three decades are characterized by the significant advances in quantum mechanical calculations which allow the analysis of rather bulky polyatomic molecules including the biological ones and the prediction of their physicochemical properties. Traditionally, the quantum mechanical calculations are used in molecular spectroscopy for the justification of molecular structure and for the identification and/or an assignment of fundamentals. Results of quantum mechanical calculations on molecular structure and molecular force fields can serve as the additional information in the data processing in vibrational spectroscopy, namely, for the appropriate definition of set D.
Despite the rapid development of the modern quantum mechanical calculations, the processing experimental data remains very important and reliable channel of information on the molecular structure as well as on the intramolecular forces. New applications of ab initio results in structural chemistry and vibrational spectroscopy are connected with using theoretical results as a background in data proccessing and solving inverse problems which are related to the simultaneous determination of molecular geometry and force field parameters on a base of joint use of vibrational spectra, microwave spectra and electron diffraction data. It is the so-called general inverse problem of structural chemistry.
While solving such general inverse problem, experimental infrared and Raman spectra are the main source of information, and the additional information can be obtained from the structural methods which reveal information on the internuclear distances in a molecule. Determined in such way molecular geometrical structure and force field parameters can then be used to predict other properties of the molecules, both existing and those yet to be synthesized.
5. Regularizing procedures for solving the general inverse problem of structural chemistry
As it was mentioned above, the extended set of experimental data on molecular vibrations can include the vibrational spectra of molecular isotopomers, the values of Coriolis constants, the mean square amplitudes etc. A molecular model which is sufficient for the molecular force field analysis is completely described by the following set of parameters:
Equilibrium geometry of a molecule (e.g. a set of interatomic distances, bond angles and dihedral angles);
Force field parameters (harmonic (and when available – anharmonic terms in the expansion of the potential function, usually up to the 3rd or 4th order)).
On the base of these parameters, the effective molecular model has been developed that is capable of predicting both spectroscopic frequencies and electron diffraction patterns [Citation17–19]. To implement an integrated procedure of simultaneous refinement of the force field and equilibrium geometry, a dynamic model of a molecule has been created. Based on the general approximation of small vibrations, the model has been extended to include cubic and quartic anharmonic potential terms for proper description of the large-amplitude motion. Within this approach, a molecule is described using a set of equilibrium geometry parameters R, and a molecular force field F represented by the matrices of quadratic, cubic and possibly quartic force constants defined in the framework of a certain nonlinear system of internal coordinates. Both parameter’s sets R and F can be considered as finite-dimensional vectors.
This model can be used for the extracting simultaneously structure and force field parameters from the experimental data on a base of solving the corresponding inverse problem. It allows to predict experimentally measured values, such as vibrational frequencies ω, electron diffraction intensity M(s), rotational constants {A, B, C} obtained from microwave molecular spectra, etc. All of these values are functions of geometric (R) and force field (F) parameters. With experimental data and parameters represented as elements of normalized finite-dimensional spaces, we can formulate the problem of simultaneous refinement of the force field and equilibrium geometry of the molecule as a system of non-linear equations. The result of such data processing is the Regularized Quantum mechanical Force Field + Regularized Equilibrium Molecular Structure (RQM FF + RES). Examples of such applications are presented in Refs. [Citation17–19]. The solution within this extended model is found as the solution of the system of three nonlinear equations:
(9)
(9) on a set of predefined constraints F∈DF, R∈DR. These three equations are corresponded to three types of experimental information obtained from the vibrational spectra, electron diffraction data and microwave spectra, correspondingly.
The system (9) can be extended to include additional experimental evidence when available (for example, data for isotopic species of a molecule sharing the same force field and equilibrium geometry). Due to the experimental errors, the lack of experimental data and the model limitations, this system of equations (that can be also treated as a finite-dimensional non-linear operator equation) usually fails to define the unique solution, often proves to be incompatible and does not provide stability with respect to the errors of input data. To avoid these unfavourable features characteristic of the ill-posed problems, it is necessary to implement a regularizing algorithm for its solution. We have suggested [Citation20,Citation21] to use a regularizing algorithm based on optimization of the next Tikhonov’s functional
(10)
(10) where in the last (‘stabilizer’) term F0 and R0 represent parameters of ab initio force field and equilibrium geometry, respectively. With the appropriate choice of regularization parameter α (that depends on the experimental errors characterized by some numerical parameter δ), it proves possible to obtain approximations converging to a normal pseudosolution of the system (10) when experimental errors tend to zero. These approximations are obtained as the extremals {Fα(δ), Rα(δ)}of Tikhonov’s functional.
6. Applications to biological molecules
Two main purposes for using a molecular force field can be defined as:
to check validity of various model assumptions commonly used by spectroscopists for approximation of the potential function;
to predict the vibrational properties of certain molecules (including those not yet observed) using fundamental properties of the force field such as the approximate transferability of specific force constants in a series of related compounds.
Investigations of the structures and functions of biological molecules with a help of vibrational spectroscopy and computational chemistry are composed the rapidly growing and developing field rich in revealing, new techniques and experiments. The basic concepts which are established for the small and modest size molecules begin to apply to more and more complex systems, making feasible the structure and physicochemical properties of bulky systems in relation to separate molecule behaviour and disturbance. These applications are also more and more popular in the medical field.
In practice spectroscopists try to use distinct models of force fields which allow to reduce the number of different force constants by the improved choice of the constraints set D. The significant progress in this direction was connected with appearance of very effective approach for obtaining force constants compatible with experimental data based on the so-called Pulay scale factors method which has been originally suggested and implemented for the force fields defined in the internal or symmetry (local symmetry) coordinates [Citation22]. Within this approach the discrepancy is minimized on the set of force constant matrices D such that
(11)
(11) where B is a diagonal matrix of scale factors (n is a number of different scale factors), and F0 is an a priori given matrix. The constraints (11) do not necessarily allow obtaining solution that is compatible with experimental data within experimental errors. Nevertheless, the scaling procedure is very popular and very important procedure for the practicing spectroscopists, due to its simplicity. It has been shown that for many molecular fragments the scale factors (within a given level of quantum-mechanical method) should be approximately constant in a wide range of similar molecules. The force constant scaling factors for different levels of quantum mechanical calculations have been obtained and in the most cases allow to predict experimental frequencies with a reasonable degree of accuracy.
The parameterization (11) does not allow completely avoid the ambiguity of the solution of Equation (7), but it may be realized by using the same regularization scheme on the set D by searching for the matrix F which is the closest to F0, or by searching for the scale matrix B closest to unit matrix. Solution in terms of scale factors is also obtained as an extremum of Tikhonov’s functional [Citation5,Citation16]. Nowadays the scaling in internal coordinates is very popular as very convenient and easy way of approaching calculated frequencies to observed ones
In the last decades, the interest of chemists has been shifted to the different rather bulky molecular systems such as biological molecules, nanostructures. For such molecular systems with hundreds and thousands atoms the number n of standard internal coordinates (n = 3N−6, where N is a number of atoms) is very large and the number of force constants of a molecule (equal to n(n + 1)/2) is also too large. The scaling factors expressed in terms of internal coordinates are the certain stipulation for the correction of results. So we have proposed [Citation23,Citation24] the principally new scheme of scaling procedure in Cartesian coordinates. For rather large molecular systems there can be many specific situations where the 3N Cartesian space is actually more efficient than using other generalized coordinates. In such cases, the choice of the Cartesian coordinates for constructing a force field from fragments remains much simpler procedure than using the internal coordinates.
Quantum-chemical calculations usually provide the force constant matrix in Cartesian coordinates. The additional feature of our approach is developing the principally new technique of the scaling molecular force fields in the Cartesian coordinates (within a frame of general approach of scaling) [Citation23,Citation24] which allows to avoid difficulties with introducing internal coordinates and is beneficial for fitting vibrational spectra of the large biological molecules, associates, polymers and nanostructures where number of atoms exceeds hundreds and thousands, and only moderately accurate quantum chemistry methods may be applied. The scale factors (or scaled force matrices) in the Cartesian coordinates may be then used for the correction of quantum mechanical force fields of large molecules when accurate quantum chemistry calculations are impossible or extremely time-consuming with the goals:
to predict the IR and Raman spectra of the large molecules or clusters when quantum-chemistry calculations are inaccessible. In this case, it is possible to obtain the estimates of vibrational spectra by the synthesis of the force constant matrix from separate blocks of the scaled force constants;
to improve the calculated spectra of the large molecular systems when only a low-level quantum-chemical calculation are possible. In this case, scale factors obtained for the same method for the smaller (model) molecules may be applied to correct the calculated force matrix.
The problem of scaling in the Cartesian coordinates, however, is nontrivial [Citation23,Citation25]. The main problem of scaling force field matrix in the Cartesian coordinates is that the potential energy of a molecule defined by this scaling matrix is not automatically independent of the molecular position and orientation as in the case of internal coordinates. As a result, a physically meaningful force constant matrix should satisfy certain conditions to eliminate translational and vibrational degrees of freedom in the expression of the potential energy. Nevertheless, the certain conditions allowing to find appropriate scale factors using Cartesian coordinates have been formulated and proved [Citation23,Citation24], and included to our software package Spectrum [Citation5,Citation15,Citation21,Citation23–25].
In this approach the force field matrix in Cartesian coordinates is represented as an array of 3 × 3 submatrices corresponding to each atom:
(12)
(12) where N is the number of atoms in a molecule. Independence of potential energy of the translations and rotation of a molecule as a whole leads to the following requirements which were introduced in [Citation23]:
(13)
(13) where 3 × 3 submatrices Vi are defined as
and
are Cartesian components of the i-th atom equilibrium position.
Imposing constraints (13) reduces the rank of matrix F to 3N−6 (or 3N−5 for linear molecules), thus leaving only vibrational degrees of freedom. When scaling procedure (11) is applied to the matrix F in Cartesian coordinates, we may assume that a priori matrix F0 satisfies the requirements (13). However, this does not necessarily mean that the scaled matrix also satisfies these requirements. To ensure that scaled matrix also contains only vibrational degrees of freedom, the scale matrix B should also satisfy certain conditions as it was shown in [Citation23,Citation24]:
Matrix B similarly to force field matrix in Cartesian coordinates (12) consists of the
unit submatrices multiplied by certain factors βij (i, j, = 1, … , N):
The factors βij are satisfied to the next two conditions:
On this way we come to the formulation of inverse vibrational problem in a form (11) where a set of a priori constraints D on the molecular force field includes conditions (14). The solution (a set of scaling factors) can be found by minimization of functional (8). Additionally, a set D can include the constraints such as equality of some off-diagonal factors to zero, in-pair equalities of factors, symmetry constraints etc. similar to constraints for the force constants in internal coordinates [Citation25]. The routine for such calculation is included in our renewed software package SPECTRUM [Citation25]. This approach as well as other models of corrections of ab initio quantum mechanical force fields have been applied to solving the inverse vibrational problem of a series of biological molecules [Citation24–27]. Traditional way in investigations of the bulky molecules is moving from the simplest objects to more complicated ones. Here, we consider only a few examples of results obtained for the rather simple glutamic acid in comparison with results obtained for melatonin, Vitamin B6 derivative, and some other related molecules which have the similar structural fragments.
Glutamic acid is an important representative of α-amino acids that participates in the biosynthesis of proteins and is widely involved to the living beings. The molecule of glutamic acid has 19 atoms (Figure ) and, correspondingly, it has 51 normal vibrations (equal to n = 3N−6 where N is the number of atoms).
Figure 3. Optimized (B3LYP/6–31G*) structure of the most stable cis-isomer of Glutamic acid molecule.
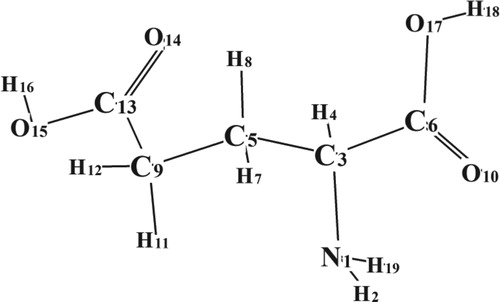
The least number of internal coordinates (including all bond stretchings, band angle deformations and deformations of dihedral angles) which should be introduced for this molecule is equal to n(n+1)/2, so the total number of different force constants in internal coordinates is equal to 1326. Even if some of the force constants in internal coordinates are taken as equal to zero, the total number of force constants remains large and cannot be determined with appropriate exactness from experimental frequencies [Citation28]. Application of Cartesian coordinates allows us to simplify the problem and decrease the number of minimized parameters.
In a case of glutamic acid the total number of different scaling factors in Cartesian coordinates equal to 190 (compare to 1326 different force constants in the internal coordinates). But this general model can be more simplified because the interaction of the far removed atoms of the molecule are very small and corresponding scaling factors can be taken as equal to zero. Here we demonstrate the result of solving the inverse problem of glutamic acid within the simplest model of scaling matrix which included with the least number of minimized parameters. The values of diagonal scaling factors were chosen the same for the identical type of atoms (C, N, O, H). The off-diagonal scaling factors were introduced only for the pairs of chemically connected atoms. Finally, three different diagonal scale factors for the C, N, O atoms and three factors for the different types of hydrogen atoms (in hydroxyl group OH, in amino-group NH2, and participating in CH bonds) have been introduced and, additionally 28 factors for the interatomic interactions, The total number of different minimized factors was equal to 34. Experimental frequencies were taken from Ref. [Citation28], with error level equal to ±5–10 cm−1.
Quantum mechanical calculation were performed with the program GAUSSIAN 09 (Revision D.01) [Citation29] at the B3LYP/6–31G(d) level of theory [Citation30]. Visualization of optimized molecular structures (Figures and ) was carried out with a help of the Chemcraft software [Citation31]. The resulting regularized scaling matrix in Cartesian coordinates of cis-glutamic acid for the B3LYP/6–31G* level of theory is presented in Table .
Table 1. Regularized Cartesian scale factors (B3LYP/6–31G*level of theory) for the cis-isomer of glutamic acid.
The comparison of fitted frequencies with experimental and theoretical frequencies is presented in Table . The results of solving the inverse vibrational problem in Cartesian coordinates demonstrate rather good correspondence between the experimental and observed frequencies of glutamic acid in the ‘finger-print’ region. The scaled Cartesian force constant matrix of glutamic acid allows to approximate the observed vibrational frequencies with reliable accuracy. Results of minimization demonstrate that the difference between values of scaling factors for different types of hydrogen atoms is very small and almost negligible. It seems that it is possible to use simpler model for the analysis of scaling factors in the cases of related compounds with the unified value of scaling factor for all hydrogen atoms. Similar results have been obtained for other organic molecules [Citation24–26].
Table 2. Theoretical (B3LYP/6–31G*), observed [Citation28] and fitted (α = 2.27 × 10−2) vibrational frequencies (in cm−1) of glutamic acid (cis-isomer) in the ‘finger-print’ region.
The regularized quantum mechanical force fields of the series of key biological molecules have been calculated in the similar way for the different levels of theory. Among them there are co-ferments – melatonin and vitamin B6 (the last in the most active methylamine pyridoxal-5′-phosphate Shiff base form, PLP) [Citation24]. Both compounds are participants of the protein metabolism, strong radical scavengers, and can be considered as the markers of important biological processes, and as the basis of physiologically active substances. The general idea of this work is to determine for the first time the spectral ‘fingerprint regions’ for discrimination between possible metabolites of melatonin and Vitamin B6 in isolated state and in biological surrounding. Such regions are necessary for finding ‘spectra-structure’ correlations and development of spectral methods for identification of metabolites. Melatonin can exist as a mixture of at least 18 different conformers and also can form the different molecular clusters (dimers, trimers, etc.) through the intermolecular H-bonds. Quantum mechanical calculations carried out at the different levels of theory allowed us to range all possible conformations of the melatonin molecule by their total energy and determine the most stable conformer in the gaseous state of compound which is presented in Figure (a). The pyridoxal-5′-phosphate methylamine can exist in a form of six isomers corresponded to three enol-, keto- and zwitterionic tautomers of compound [Citation31]. The most stable enolic structure of PLP is shown in Figure (b).
Figure 4. Stable conformations of melatonin (a) and pyridoxal-5′-phosphate methylamine (b).
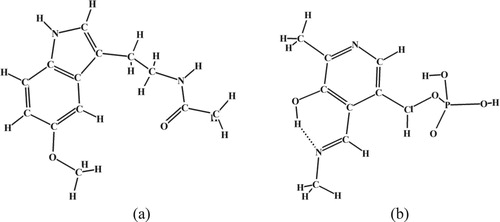
The sets of scaling factors in Cartesian coordinates have been calculated for most stable configurations of melatonin and PLP molecules in gaseous state (Figure ) in the way similar to the glutamic acid. The melatonin molecule have 33 atoms: three ‘heavy’ ones – C, N, O and others are the hydrogen atoms, PLP molecule contains 30 atoms, also 3 types of ‘heavy’ ones (C, N, O) and hydrogen atoms. Inverse problems for melatonin and PLP have been solved in the way similar to glutamic acid using the similar scaling procedure in Cartesian coordinates.
Some regularized diagonal scaling factors in the Cartesian coordinates (for the B3LYP/5–31G* level of theory) of melatonin and PLP are presented in Table in comparison with results for the simple related molecules. The values of the fitted scaling factors in Cartesian coordinates of similar fragments are very close and this result demonstrates the possibility of transferring the scaling factors in Cartesian coordinates between related molecules.
Table 3. Comparison of some scaling factors in Cartesian coordinates (B3LYP/6–31G* level) of melatonin and pyridoxal-5′-phosphate methylamine with corresponding factors of related molecules with similar strcutural fragments [Citation24–27].
7. Computational details
Ab initio and DFT calculations have been performed with the program GAUSSIAN 03 (Revision D.01) [Citation26]. The fully optimized geometries, analytical force constants and harmonic vibrational frequencies of all mentioned above molecular structures have been calculated at the B3LYP/6–31G* level of theory [Citation27,Citation29]. The minima of the potential surface were found by relaxing the geometric parameters with the standard optimization methods. The visualization of optimized molecular structures has been made using Chemcraft (version 1.8) software [Citation31]. Using theoretical force constants as F0 and sets of experimental frequencies collected in [Citation26–28] for molecules presented in Table , the inverse scaling problems was solved and scale factors were found as the extrenal of functional (7). Calculation of scale factors was carried out with the special routine of the software package SPECTRUM [Citation26,Citation32,Citation33] which has been supplied with the additional options mentioned in the previous section. All considered inverse problems have been solved for the single parent molecules without inclusion the experimental data of isotopic species to avoid the incompatibility of inverse problems within harmonic model.
8. Discussion of results and conclusions
Some results of the application of new regularizing algorithms for the fitting quantum mechanical force fields are presented and efficiency of new algorithms for the biological molecules is discussed. Results presented in Table and some other calculations [Citation23–27], which demonstrated good correspondence between experimental and fitted frequencies, support the conclusion of rather good quality of the obtained scale factors in Cartesian coordinates. This shows that the model of correcting theoretical vibrational frequencies by means of scaling Cartesian force matrices is quite reasonable; generally, the results of frequency correction with Cartesian scaling factors are very close to the quality of their correction by scaling force matrices in internal coordinates [Citation34,Citation35]. Quite predictably, scale factors for the Density Functional Theory B3LYP simulations are much closer to unit matrices, and the frequency fitting is considerably better. It should also be noted that diagonal scale factors for similar atoms in considered molecules are close for each theoretical level [Citation23–27]. The same is true for the pairs of bonded atoms (C, H, etc.); this opens good possibilities of transferring scale factors for the atoms in similar environment.
Note that it is also possible to apply the procedure for each symmetry block individually, yielding somewhat better frequency fitting, similar to what is often done for the standard scaling approach. Small values of certain scale factors indicate the possibility of stipulating them as fixed zero elements in minimization procedure. We also hope that new calculations for other molecular systems will clarify some questions of modelling Cartesian scale factors and their transferability in a series of related molecules.
New numerical algorithm for the calculation of scale factors for the molecular force fields expressed in Cartesian coordinates is developed and implemented as a part of the software package SPECTRUM [Citation33]. This algorithm allows to reduce the difficulties related to the variable choice of different sets of internal coordinates. Though we have demonstrated numerical implementation of the proposed approach on the examples of rather simple molecules, the method does not have limitations on the molecular size. Moreover it is possible to use different models and special constraints for obtaining Cartesian scale factors which reveal transferability or in-pair equality properties similar to the properties of the scale factors in internal coordinates. These properties are very important for the calculations of vibrational spectra for related compounds, especially among large biological molecules, associates, polymers and nanostructures.
Acknowledgement
Authors are grateful to the Russian Foundation on Basic Research for the financial support (Project No 18-03-00412a).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Bright Wilson Jr. E, Decius JC, Cross PC. Molecular vibrations, the theory of infrared and Raman vibrational spectra. New York: Dover Publications; 1980.
- Volkenstein MV, Gribov LA, El'yashevich MA, et al. Vibrations of molecules. Moscow: Nauka; 1972.
- Tikhonov AN, Goncharskii AV, Stepanov VV, et al. Numerical methods for the solution of ill-posed problems. Dordrecht: Kluwer; 1995.
- Tikhonov AN, Leonov AS, Yagola AG. Nonlinear ill-posed problems. London: Chapman & Hall; 1997.
- Yagola AG, Kochikov IV, Kuramshina GM, et al. Inverse problems of vibrational spectroscopy. (Inverse and III-posed problems). Zeist: VSP; 1999; p. 297.
- Kuramshina GM, Mogi T, Takahashi H. Structures and vibrational spectra of 5H-dibenz[b,f]azepine and 5H-dibenzo[a,d]cycloheptene-5-ol on the basis of quantum mechanical calculations. J Mol Struct. 2003;661-662:121–139.
- Hadamard HJ. Le probleme de Cauchy et les equations aux derivees partielles lineaires hyperbolique. Hermann, Paris; 1932.
- Kochikov IV, Kuramshina GM, Pentin IA, et al. Regularizing algorithm of the inverse vibrational problem solution. Dokl Akad Nauk SSSR. 1981;261(5):1104–1106.
- Kochikov IV, Kuramshina GM, Pentin YA, et al. A stable method for the calculation of the force fields of polyatomic molecules in dependent coordinates. Theor Exp Chem. 1984;20(1):66–72.
- Kochikov IV, Yagola AG, Kuramshina GM, et al. Force-fields and mean amplitudes of vibration of chromium, molybdenum and tungsten oxotetrafluorides. J Mol Struct Theochem. 1984;106(3–4):355–360.
- Kochikov IV, Kuramshina GM, Pentin YA, et al. Calculation of force fields of polyatomic molecules by the Tikhonov regularization method. Dokl Akad Nauk SSSR. 1985;283(4):850–854.
- Kochikov IV, Kuramshina GM, Yagola AG. Stable numerical methods of solving certain inverse problems of vibrational spectroscopy. USSR Comput Math Math Phys. 1987;27(11-12):33–40.
- Kochikov IV, Kuramshina GM, Pentin YA, et al. Regularizing algorithms for molecular force field calculations. J Mol Struct. 1992;272:13–33.
- Kochikov IV, Kuramshina GM, Pentin YA. Application of Tikhonov’s regularization method to the calculation of force fields of polyatomic molecules. Russ J Phys Chem. 1987;61(4):451–457.
- Kochikov IV, Kuramshina GM. A complex of programs for the force field calculations of polyatomic molecules by the Tikhonov regularization method. Vestnik Moskovskogo Universiteta, Seria 2, Khimia. 1985;26(4):354–358.
- Kuramshina GM, Weinhold F, Kochikov IV, et al. Joint treatment of ab initio and experimental data in molecular force field calculations with Tikhonov’s method of regularization. J Chem Phys. 1994;100(2):1414–1424.
- Kochikov IV, Tarasov YI, Spiridonov VP, et al. Extension of a regularizing algorithm for the determination of equilibrium geometry and force field of free molecules from joint use of electron diffraction, molecular spectroscopy and ab initio data on systems with large-amplitude oscillatory motion. J Mol Struct. 1999;485-486:421–443.
- Kochikov IV, Tarasov YI, Spiridonov VP, et al. The use of ab initio anharmonic force fields in experimental studies of equilibrium molecular geometry. J Mol Struct. 2000;550-551:429–438.
- Kochikov IV, Tarasov YI, Spiridonov VP, et al. The equilibrium structure of thiophene by the combined use of electron diffraction, vibrational spectroscopy and microwave spectroscopy guided by theoretical calculations. J Mol Struct. 2001;567-568:29–40.
- Kuramshina GM, Yagola AG. Regularizing algorithms for solving nonlinear ill-posed problems of vibrational spectroscopy. Eurasian J Math Comput Appl. 2016;4(2):14–36.
- Kuramshina GM, Yagola AG. Applications of regularizing algorithms in structural chemistry. Eurasian J Math Comput Appl. 2017;5(3):53–72.
- Pulay P, Fogarasi G, Pongor G, et al. Combination of theoretical ab initio and experimental information to obtain reliable harmonic force constants. scaled quantum mechanical (QM) force fields for glyoxal, acrolein, butadiene, formaldehyde, and ethylene. J Am Chem Soc. 1983;105:7037–7047.
- Stepanova AV, Kochikov IV, Kuramshina GM. New approach for the correction of Ab initio molecular force fields In cartesian coordinates. Int J Quantum Chem. 2009;109(1):28–33.
- Kochikov IV, Kuramshina GM, Stepanova AV. Scaled ab initio molecular force fields in Cartesian coordinates: application to benzene, pyrazine and isopropylamine molecules. Asian Chem Lett. 2009;13(3):143–154.
- Kochikov IV, Stepanova AV, Kuramshina GM. Ab initio molecular force fields fitted in Cartesian coordinates to experimental frequencies of isotopic species using symmetry constraints: application to indole and pyrrole molecules. Struct Chem. 2019;30(2):605–614.
- Kuramshina GM, Takahashi H. Ab initio and DFT theoretical studies of pyridoxale-5′-phosphate methylamine Shiff base isomers. J Mol Struct. 2005;735-736:39–51.
- Kuramshina GM, Vakula OA, Vakula NI, et al. Vibrational spectra and force field of Methyl -2-(2′-pyridyl)benzimidazole: experimental data and density functional theory investigation in comparison with 2-(2′-pyridyl)benzimidazole. Struct Chem. 2016;27(1):209–219.
- Pradeep K, Sengupta K, Krimm S. Vibrational analysis of peptides, polypeptides, and proteins. XXXII. α-poly(L-glutamic acid). Biopolymers. 1985;24:1479–1491.
- Frisch MJ, Trucks GW, Schlegel HB, et al. //Gaussian 09, Revision D.01. Wallingford (CT): Gaussian, Inc.; 2013.
- Jensen F. Introduction to computational chemistry. Chichester: John Wiley and Sons; 1999.
- http://www.chemcraftprog.com. Version 1.8 (build 486).
- Kochikov IV, Kuramshina GM, Stepanova AV, et al. Regularized scaling factor method for calculating molecule force fields. Moscow Univer Phys Bull c/c of Vestnik – Moskovskii Universitet Fizika I Astronimiia. 1997;52:28–33. ALLERTON PRESS, INC.
- Yagola AG, Kochikov IV, Kuramshina GM, et al. Inverse problems of vibrational spectroscopy. Moscow: KURS; 2017.
- Zeroka D, Jensen JO, Samuels AC. Infrared spectra of some isotopomers of isopropylamine: A theoretical study. Int J Quantum Chem. 1999;72(2):109–126.
- Baker J, Jarzecki AA, Pulay P. Direct scaling of primitive valence force constants: an alternative approach to scaled quantum mechanical force fields. J Phys Chem A. 1998;102:1412–1424.