

ABSTRACT
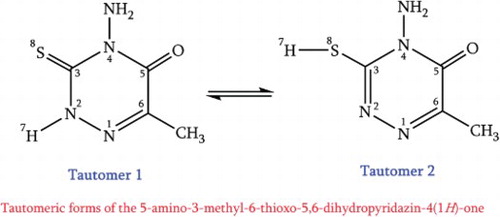
The tautomerization reactions of the 4-amino-6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one studied by means of M06-2x and CBS-QB3 theoretical methods. The measured energy profiles are complemented with kinetic rate coefficients calculations using transition state theory (TST). In line with the optimized tautomers geometries using the CBS-QB3 method, the natural bond orbital (NBO) analysis reveals that the stabilization energies of non-bonding LP(e)S8 to the σ*N2–C3 antibonding orbitals increase from tautomers 1 to 2. Furthermore, the delocalization energies of LP(e)S8→σ*N2–C3 could explain the increase of LP(e)S8 non-bonding orbitals occupancies in the tautomers 1 and 2 (2 > 1). The increase of LP(e)S8→σ*N2–C3 delocalizations could fairly explicate the kinetics of tautomeric pathways 1 and 2 (k2 > k1). Moreover, the HOMO–LUMO energy gap is increased parallel with the decreasing of activation energy barriers. NBO results also show that the kinetics of these processes controlled using LP→σ* resonance energies. Furthermore, nucleus-independent chemical shift (NICS) indices show the calculated reaction and energy barriers are involved by changes in aromaticity characters as well as electron transfer from LP(e)S8 to σ*N2–C3 orbitals, thus these reactions are controlled from both thermodynamic and kinetic viewpoints by the changes aromaticity characters.
GRAPHICAL ABSTRACT
Disclosure statement
No potential conflict of interest was reported by the authors.