
ABSTRACT
Introduction
Drug delivery to solid tumors remains a significant therapeutic challenge. Mesenchymal stem/stromal cells (MSCs) home to tumor tissues and can be employed as tumor targeted drug/gene delivery vehicles. Reportedly, therapeutic gene- or anti-cancer drug-loaded MSCs have shown remarkable anti-tumor effects in preclinical studies, and some clinical trials for assessing therapeutic MSCs in patients with cancer have been registered.
Areas covered
In the present review, we first discuss the source and interdonor heterogeneity of MSCs, their tumor-homing mechanism, and the route of MSC administration in MSC-based cancer therapy. We then summarize the therapeutic applications of MSCs as a drug delivery vehicle for therapeutic genes or anti-cancer drugs and the drug delivery mechanism from drug-loaded MSCs to cancer cells.
Expert opinion
Although numerous preclinical studies have revealed significant anti-tumor effects, several clinical trials assessing MSC-based cancer gene therapy have failed to demonstrate corroborative results, documenting limited therapeutic effects. Notably, a successful clinical outcome with MSC-based cancer therapy would require the interdonor heterogeneity of administered MSCs to be resolved, along with improved tumor-homing efficiency and optimized drug delivery efficiency from MSCs to cancer cells.
1. Introduction
Ongoing medical research is persistently striving to overcome various types of cancers. However, cancer remains one of the leading causes of death worldwide, with an estimated 19.3 million new cases and 10.0 million cancer-related deaths reported in 2020 [Citation1]. Chemotherapy and immunotherapy play a vital role in cancer therapy, along with surgical intervention and radiation therapy. Various types of anti-cancer agents, including chemotherapeutics, hormones and molecular targeted agents, have been approved for clinical use [Citation2]. However, most of these anti-cancer agents are systemically administered and distribute not only to tumor tissues but also to normal tissues, where they may induce side effects. In addition, some types of cancers do not exhibit any response to anti-cancer agents because the delivery efficiency of the agents is very low [Citation3,Citation4]. Therefore, tumor-targeted drug delivery is extremely important to improve the safety and efficacy of anti-cancer agents. Several cancer-targeting methods have been developed, including passively targeted nanocarriers (e.g. liposomes and micelles) and actively targeted therapeutics (e.g. antibody-drug conjugates [ADCs]). Although the tumor accumulation of nanocarriers and ADCs is basically based on the enhanced permeability and retention (EPR) effect [Citation5], effective cancer therapy has been achieved only in some cancer types. Several unsolved issues limit the therapeutic effects of nanocarriers and ADCs. For example, the high interstitial fluid pressure in tumor tissues significantly limited the accumulation of nanocarriers in inner tumor tissue regions [Citation3]. In addition, the abundant stroma in some tumor types potently suppressed the uptake of nanocarriers and the binding of ADCs to surface receptors of cancer cells [Citation6]. Therefore, cancer-targeting methods that do not depend on the EPR effect are currently in demand.
Cells with tumor-homing ability have recently been recognized as a drug delivery vehicle for cancer therapy. Some types of immune cells, stem cells, and progenitor cells are known to possess tumor-homing abilities [Citation7,Citation8]. Endogenous monocytes, neutrophils, and lymphocytes are recruited from the bone marrow (BM) or lymphoid tissues to tumor tissues, where they contribute to immune reactions. In addition, exogenously transplanted leukocytes and monocytes migrate to tumor tissues [Citation7–9]. In 2000, Aboody et al. reported that neural stem cells injected into the frontoparietal lobe of the brain migrated to brain tumor tissues in a mouse model of glioma [Citation10]. Furthermore, systemically administrated hematopoietic progenitor cells and fibroblast-derived neural stem cells were detected in the tumors of tumor-bearing mice [Citation11,Citation12]. These cells are guided by chemokines secreted from solid tumor tissues and spontaneously invade tumors. Therefore, some cell types with tumor-homing ability can be developed as drug delivery vehicles independent of the EPR effect.
Among the various types of tumor-homing cells, mesenchymal stem/stromal cells (MSCs) are the most attractive candidates for drug delivery vehicles for cancer therapy, as they demonstrate tumor tropism, a self-renewing/proliferative capacity, low immunogenicity, and low risk of tumorigenesis (). According to the minimal criteria proposed by the International Society for Cell & Gene Therapy (ISCT), MSCs are plastic adherent cells that express CD73, CD90, and CD105, but do not express CD34, CD45, CD14, CD11b, CD79a, CD19, and HLA-DR, and at least differentiate into adipocytes, osteoblasts, and chondrocytes [Citation13,Citation14]. MSCs can be isolated from various tissues, including BM, adipose tissue (AD), and umbilical cord (UC; UC blood and Wharton’s jelly) [Citation15] or induced from pluripotent stem cells [Citation16], and rapidly expanded on a clinical scale using a standard monolayer culture method [Citation15]. The risk of tumor formation with MSCs is considered extremely low owing to their genetic stability [Citation17], and several in vivo tumorigenicity tests [Citation18] and long-term follow-up of patients with MSC transplantation [Citation19] revealed no tumor formation. Moreover, low immunogenicity is another unique characteristic of MSCs, and allogeneic MSC transplantation rarely causes rejection because MSCs express low levels of major histocompatibility complex (MHC) class I and exceptionally low levels of MHC class II and costimulatory molecules (CD40, CD80, and CD86) [Citation20,Citation21]. Furthermore, in animal models, MSCs homed to various primary tumors and metastatic foci, including Kaposi’s sarcoma, Ewing sarcoma, fibrosarcoma, glioma, melanoma, lung cancer, hepatocellular cancer, stomach cancer, colon cancer, pancreatic cancer, renal cell carcinoma, breast cancer, and ovarian cancer [Citation8,Citation14,Citation15,Citation22–24]. Accordingly, the use of MSCs would be useful for improving the safety and efficacy of anti-cancer agents through the enhanced delivery efficiency of anti-cancer agents to tumor tissues. Many researchers have evaluated the application of MSCs as a drug delivery vehicle for anti-cancer drugs and therapeutic genes ().
Figure 1. The properties and therapeutic applications of MSCs as a vehicle for anti-cancer agents. (a) MSCs can be isolated from various tissues or induced from pluripotent stem cells. The International Society for Cell & Gene Therapy (ISCT) has defined minimum criteria for MSCs: adhesion to plastic plates, expression of CD73, CD90, CD105, no expression of CD34, CD45, CD14, CD11b, CD79a, CD19, and HLA-DR, and differentiation into adipocyte, osteoblast, and chondrocyte. MSCs are useful as a source of tumor-targeted drug delivery vehicles owing to their high self-renewing or proliferation potency, low immunogenicity, low risk of tumorigenesis, and tumor tropism. (b) Various engineered MSCs. MSCs are employed as a cancer-targeting vehicle for therapeutic genes, oncolytic virus, anti-cancer drugs, and drug-encapsulated nanoparticles. MSCs, mesenchymal stem/stromal cells
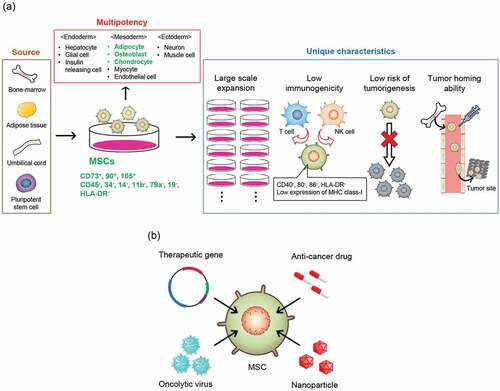
Based on their self-renewal and proliferative capacity, low immunogenicity, low risk of tumorigenesis, and tumor tropism, MSCs are considered a next-generation drug delivery vehicle for cancer therapeutics. In the present review, we describe the properties and therapeutic applications of MSCs as drug delivery vehicles for anti-cancer drugs during cancer therapy.
2. Properties of MSCs
Understanding the properties of MSCs is valuable for the application of MSCs as a drug delivery vehicle. In this chapter, we first classify the literature according to the MSC source, tumor homing mechanisms, and MSC administration route. Next, we summarize a few recent studies on the influence of exogenously administered MSCs on the tumor microenvironment.
2.1. MSC source
2.1.1. Tissue sources of MSCs
MSCs can be isolated from various tissues, with BM, AD, and UC considered major sources. In regenerative medicine, MSCs have been used to treat autoimmune diseases, cardiovascular diseases, neurological disorders, and severe inflammatory diseases [Citation15,Citation25]. More than a thousand clinical trials have been registered. BM-, AD-, or UC-derived MSCs have been employed in over 90% of clinical trials [Citation25]. Furthermore, BM- and AD-derived MSCs have been approved for clinical use to treat acute graft versus host disease (GVHD), osteoarthritis clone disease, and spinal cord injury in the European Union and Japan (Alofisel®, Stemirac®, Temcell®). In research assessing MSC-based drug delivery, BM-, AD-, and UC-derived MSCs have been frequently utilized.
BM is the most common source of MSCs, and BM-derived MSCs have been used for over 20 years in the field of regenerative medicine [Citation25]. However, BM-derived MSCs present some unfavorable aspects and disadvantages when compared with AD and UC as sources of MSCs. For example, BM aspiration is highly invasive, and the volume of collectible BM is extremely low (15 mL/kg from a healthy donor). Moreover, BM contains a relatively low number of MSCs (0.001‒0.01% of total BM cells) [Citation25–27]. In comparison, the collection of AD tissue is much less invasive, and the yield of MSCs can be 100‒500-fold greater than that of BM [Citation27–29]. UC can be obtained as perinatal medical waste with a high yield (3 × 105 cells/cm of fresh Wharton’s jelly) [Citation30]. All MSCs prepared from these tissues share typical characteristics, such as tumor-homing, tissue repair, and immune regulation, and AD- and UC-derived MSCs have been increasingly used in recent years [Citation25]. Conversely, some comparative studies have reported that BM-, AD-, and UC-derived MSCs present unique characteristics in terms of the proliferation rate, the expression level of immunosuppressive cytokines, and tumor tropism [Citation27–31]. However, it is challenging to systematically characterize MSCs depending only on their source, as MSCs from any source are highly heterogeneous. In addition, the method employed for isolating MSCs can affect the collectible cell population, and standard isolation protocols are yet to be established [Citation30,Citation32]. Furthermore, the donors’ age and health condition [Citation21], cell culture conditions, and cell passage numbers [Citation21,Citation33] greatly impact the MSC characteristics. Therefore, selecting the tissue source of MSCs may be insufficient to govern their use as a drug delivery vehicle for cancer. Instead, it is important to select MSCs with high tumor-homing ability or to optimize the cell culture protocol to improve or maintain the tumor-homing ability for the therapeutic application of MSCs in cancer therapy.
2.1.2. Preparation of homogenous MSCs
As described above, the interdonor heterogeneity of MSCs could reduce the drug delivery efficiency of MSC-based drug delivery and could hinder clinical applications. Zyrafete et al. succeeded in reducing interdonor heterogeneity and established a cell bank of clinical-grade MSCs by mixing and pooling cells isolated from the BM of eight healthy donors [Citation34]. This cell bank can supply 500 doses of MSCs for the treatment of GVHD. Pluripotent stem cells can also be a source of homogenous MSCs on a larger scale. Several protocols to induce MSCs from embryonic stem (ES) cells and induced pluripotent stem (iPS) cells have been developed [Citation16], and MSCs differentiated from these pluripotent cells reportedly present characteristics similar to those of primary MSCs [Citation35]. Adrian et al. succeeded in establishing an MSC supply system using iPS cells, which could generate 29 million clinical doses (2 × 106 cells/kg) of good manufacturing practice (GMP)-grade MSCs, CYP-001. In a phase I clinical trial, the safety of CYP-001 was demonstrated in patients with GVHD [Citation35]. Therefore, pluripotent stem cell is an attractive source to prepare homogenous MSC-products from a perspective of scalability, and pluripotent stem cell-derived MSCs will be becoming more widely used in the near future.
2.1.3. Autologous vs. allogeneic transplantation of MSCs
Both autologous and allogeneic MSCs have been assessed in clinical trials for MSC-based cancer gene therapy (). Autologous transplantation carries no risk of immune rejection. Several studies have reported that the survival time of syngeneic MSCs transplanted into mice was longer than that of allogeneic MSCs [Citation20]. Zangi et al. reported that transplantation of allogeneic MSCs induced a memory T cell response [Citation36]. However, some drawbacks associated with the use of autologous MSCs for transplantation tend to persist. First, the processes of an expanded culture or engineering of MSCs and validation of the final product require a prolonged period of several weeks and high cost. It is difficult to obtain sufficient BM or AD tissues in patients with low body weight and high or low age. Furthermore, MSCs isolated from patients with diabetes, rheumatoid arthritis, or systemic lupus erythematosus have shown impaired function [Citation20]. Conversely, allogeneic MSCs are available as ‘off-the-shelf’ cell products prepared on a large scale. In addition, they can reduce the interdonor heterogeneity, as it is possible to prepare clinical doses of MSCs from one tissue source and avoid off-specification products. Recently, several cell banks of allogeneic MSCs have been established, and GMP-grade MSCs have been easily obtained [Citation30,Citation34,Citation35].
Table 1. Clinical trials of MSC-related cancer gene therapy registered in Clinical Trials.gov. The data were searched in April 2021
2.2. Tumor-homing mechanism of MSCs
2.2.1. Tumor-homing mechanism of systemically administered MSCs
The homing of MSCs to tumor tissues demonstrates some similarities to leukocyte migration in inflamed tissues. Systemically administered MSCs are guided by chemokines secreted from solid tumors and activated following interaction with the endothelium [Citation14,Citation15,Citation38,Citation39]. Some chemokines in the blood flow and cell adhesion molecules on the endothelial surface of tumor blood vessels induce the rolling of MSCs, as well as their adherence to the endothelium [Citation15,Citation38,Citation39]. After adhesion, MSCs migrate into tumor tissues through the vessel wall [Citation15,Citation39,Citation40]. Reportedly, several types of chemokine receptors exist on MSCs, such as C-C chemokine receptor (CCR)-1, CCR-2, CCR-4, CCR-7, CCR-9, and C-X-C chemokine receptor (CXCR)-1, CXCR-4, CXCR-6, and CXCR-7 [Citation15,Citation38,Citation39]. CXCR-4, the stromal cell-derived factor-1 (SDF-1) receptor, has been well investigated and is considered to play a major role in MSC homing [Citation15,Citation39]. However, SDF-1 is constantly secreted from the BM and various other tissues. Lourenco et al. reported that the macrophage migration inhibitory factor (MIF)-CXCR-4 axis plays a crucial role in the tumor-homing of MSCs [Citation41]. MIF is overexpressed in several tumor types, and the authors demonstrated that the MIF-CXCR4 axis induced MSC homing to tumor tissues by activating the mitogen-activated protein kinase (MAPK) pathway. Both galectin-1 and very late antigen-4 (VLA-4, integrin α4β1) are known to play key roles during rolling and adhesion [Citation39]. Galectin-1 and VLA-4 on MSCs bind to P-selectin and vascular cell adhesion molecule-1 expressed on the surface of vascular endothelial cells, respectively, and mediate MSC rolling and endothelial adhesion. After adhesion to the endothelium, MSCs express matrix metalloproteinase (MMP)-2 and transmigrate across the vessel wall [Citation39,Citation40]. Typically, the transmigration of leukocytes requires 10‒20 min, whereas that of MSCs takes 60‒120 min [Citation39].
2.2.2. Improvement of MSC tumor-homing
In addition to understanding the tumor-homing mechanism of MSCs, attempts have been made to improve the tumor-homing property of MSCs by employing gene and cell engineering techniques. Several studies have reported that the overexpression of chemokine receptors, including CCR-1, CXCR-4, and CXCR-7, enhanced the homing ability of MSCs to target sites [Citation38,Citation40]. Modification of the cell surface with SLeX, a P-selectin ligand, E-selectin-binding peptide, and antibodies against P-selectin and VCAM1 facilitated the rolling and adhesion of MSCs to the vascular endothelium at the target site, resulting in an improved homing rate [Citation38,Citation40]. Priming methods can upregulate the expression of migration-related genes without gene transfer. Feng et al. revealed that hypoxia priming (1.5% O2) improved the tumor-homing of MSCs in a glioblastoma mouse model [Citation42]. Under hypoxic conditions, the disassembly of hypoxia-inducible factor-1α was inhibited, and the expression of target genes relevant to cell proliferation, migration, and invasion was increased. Smith et al. reported that pre-exposure of MSCs to a glioma-conditioned medium containing fibronectin and laminin enhanced their tumor-homing in a glioblastoma mouse model [Citation43]. Loading iron oxide NPs to MSCs is another method to improve the homing efficacy of MSCs to the target site via magnetic guidance. Several studies demonstrated that the number of MSCs homing to the target site was increased by the loading of iron oxide NPs to MSCs and by applying the external magnetic field to the target site [Citation35,Citation44–47]. In addition, several studies demonstrated that the expression of migration-related genes of MSCs were upregulated by only iron oxide NPs loading [Citation44,Citation48] or by the combination of the loading and application of the external magnetic field [Citation49].
2.2.3. Tumor-homing of non-systemically administered MSCs
Several studies have reported that non-systemically administered MSCs migrate to tumor tissues. Intraperitoneally injected MSCs reportedly migrate to tumors in orthotopic ovarian tumor model mice [Citation50], peritoneal dissemination model mice [Citation24,Citation51], and TH-MYCN transgenic mice that spontaneously develop glioblastoma in the superior mesenteric ganglion [Citation52]. Furthermore, Rincón et al. reported that intraperitoneally administered MSCs homed to the subcutaneous tumor in a non-small-cell lung carcinoma cell line (CMT64) xenograft mouse model [Citation53]. However, compared with the tumor-homing of systemically administered MSCs, little is known regarding the cascade and molecular mechanism underlying tumor homing of non-systemically administered MSCs.
2.3. Administration methods and fate of MSCs after administration
2.3.1. Transplantation forms of MSCs
MSC transplantation can be classified into three categories: cell suspensions, multicellular spheroids, and cell sheets. A cell suspension is the most basic form of drug administration. Single or glued cells, constructed from two or multiple cells suspended in saline or ringer’s solutions, are administered. Cell spheroids or multicellular spheroids are three-dimensionally cultured cell aggregates, ranging between 100‒500 μm in size. Multicellular spheroids can present innate cellular functions by resembling in vivo conditions, including high cell-cell interactions and the deposition of extracellular matrices in the three-dimensional structure [Citation54]. Smruthi et al. demonstrated that spheroid formation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-expressing MSCs improved TRAIL expression and the anti-tumor effect in a glioblastoma xenograft mouse model [Citation55]. In addition, cell sheets can improve the innate cellular function and survival rate of transplanted MSCs. However, to our knowledge, few studies have been reported on MSC-based cancer therapy using multicellular spheroids or cell sheets. Therefore, we focus on suspended MSCs and discuss the administration route and biodistribution after administration in the following section.
2.3.2. Administration routes and biodistributions of MSCs
The MSC administration routes are broadly classified into systemic (intravascular) and local administration. Systemic (intravascular) administration route includes intravenous, intra-arterial, and intraportal administration. Most intravenously administered MSCs are temporarily entrapped in the lung vasculature and gradually transferred to the systemic circulation [Citation24,Citation39]. The ex vivo culturing of MSCs increases the cell size and expression of cell adhesion molecules [Citation56], and these morphological and biological changes are considered major causes for the lung entrapment of intravenously administered MSCs [Citation39,Citation56]. Accordingly, the intravenous injection of MSCs can efficiently target lung tumors. Conversely, several studies have demonstrated that reducing the lung entrapment of MSCs improved the homing rate to the targets outside the lungs. They include intravenous administration of MSCs by cell surface coating with heparin [Citation57], avoidance of cell hypertrophy by employing a modified culture method [Citation38,Citation56], and blocking of cell adhesion proteins with antibodies [Citation56]. Intra-arterially injected MSCs are predominantly distributed to first-pass organs, and intraportally administered MSCs are mainly distributed in the liver. Therefore, local intra-arterial and intraportal administrations are effective in targeting tumors in specific organs or tissues. However, both intra-arterial and intraportal administration require invasive surgical intervention, associated with a high risk of bleeding complications. Intravenous administration is the least invasive and useful for repeated administrations. In clinical trials of regenerative medicine, the intravenous administration route is the most frequently employed [Citation25], and many clinical trials have shown the safety of intravenous injection of MSCs (1‒2 × 106 cells/kg) [Citation58].
In a preclinical study of MSC-based drug delivery for cancer therapy, local (non-intravascular) administration routes, including intracranial, nasal, peritoneal, peritumoral, thecal, tumoral, and subcutaneous administration routes, have been reported. Intra-tumoral and peritumoral administration routes are conceptually the simplest and can efficiently deliver MSCs to target tumors. These routes are suitable for easily detectable tumors, such as skin tumors. Intracranial administration is attractive for targeting brain tumors such as glioblastoma, as it can bypass the blood-brain barrier (BBB). Among primary brain tumors, glioblastoma is the most malignant tumor and is not curable with surgery alone, as glioblastoma cells invade surrounding brain tissues. Temozolomide is the only standard anti-cancer drug for glioblastoma, but the five-year survival rate with standard therapy composed of radiotherapy plus temozolomide is only 9.8% [Citation59]. Thus, MSC-based drug delivery is expected to be a novel strategy for improving the efficacy of glioblastoma therapy. Although several studies have reported that systemically administered MSCs cross the BBB and are homed to the brain tumor [Citation15,Citation60], a comparative study by Nakamizo et al. revealed that the intracranial administration of interferon (IFN)-β-expressing MSCs significantly prolonged the survival of glioblastoma model mice when compared with intravenous administration [Citation61]. Intranasal administration of MSCs, which has been reported to bypass the BBB [Citation62], may also present an alternative route for targeting brain tumors.
2.3.3. Disappearance of MSCs from the body
It has been reported that most MSCs disappear from the body by undergoing necrotic cell death within one week of administration [Citation63,Citation64], in which shear stress, lack of nutrients, hypoxia, and detachment-induced apoptosis have been observed [Citation63,Citation65]. Although MSCs can generally escape T cell and natural killer (NK) cell-mediated destruction, MSCs may be phagocytosed by monocytes or macrophages, because some studies demonstrated that MSCs were phagocytosed by monocytes/macrophages under in vitro culture conditions [Citation66,Citation67].This rapid disappearance could avoid unexpected adverse effects attributed to MSCs and may be beneficial to establish the safety of MSCs as a drug delivery vehicle. However, regulating the survival duration of administered MSCs is necessary to deliver therapeutic MSCs to tumor sites efficiently.
2.4. The influence of administered MSC on tumor tissues
2.4.1. Differentiation capacity of MSCs to cancer-associated fibroblasts
Although MSCs are expected to be developed as drug delivery vehicles, several studies have reported that MSCs support the construction of the tumor microenvironment by differentiation into cancer (carcinoma-, tumor-)-associated fibroblasts (CAFs) [Citation23,Citation68], as well as accelerate tumor growth and metastasis by stimulating tumor cells via direct cell-cell interactions and tumor cell-derived extracellular vesicles (EVs) [Citation68–71]. CAFs closely resemble myofibroblasts and express α-smooth muscle actin (α-SMA) and fibroblast activation protein (FAP) [Citation72]. CAFs reportedly promote tumor growth and suppress tumor immunity through matrix remodeling and immune crosstalk [Citation72]. Several studies have demonstrated that CAFs originate from MSCs. Quante et al. and Raz et al. revealed that MSCs recruited from the BM differentiated into CAFs [Citation23,Citation68]. In the tumor tissue, transforming growth factor (TGF)-β1 and SDF-1 induce the differentiation of MSCs into CAFs. These reports suggest that endogenous MSCs naturally possess tumor tropism and pro-tumor activity. In addition, several studies have reported that other tissue-derived MSCs, such as AD-derived MSCs, possess the ability to differentiate into CAF-like cells under in vitro culture conditions [Citation20]. However, it remains unclear whether MSCs can be recruited from tissues other than the BM for differentiation into CAFs.
Although endogenous MSCs may act as pro-tumorigenic cells, MSC administration might minimally impact tumor growth. While induction from MSCs to CAFs required 4‒30 days [Citation73–75], most MSCs disappeared from the body within a week [Citation63,Citation64]. In addition, the differentiation of MSCs increased the expression of MHC molecules [Citation21], which could result in immune rejection. Therefore, the risk that exogenously administered MSCs can be a primary source of CAFs could be low, especially when allogeneic MSCs are employed.
2.4.2. Influence of MSC-derived EVs on tumor progression
EVs, including exosomes, microvesicles, and microparticles, are cell-released and lipid bilayer-delimited nanoparticles [Citation76]. EVs contain various proteins, lipids, and nucleic acids and can mediate cell-cell interactions. Several studies have suggested that EVs secreted from MSCs in tumor tissues promote tumor growth and metastasis. It has been reported that microRNA-15a, 21, 34a, 191, 125a, 140, and 222/223 in MSC-derived EVs improved the resistance to anti-cancer drugs, proliferation, migration of cancer cells, and angiogenesis by tumor stromal cells [Citation69]. However, studies have also reported that MSC-derived EVs possess anti-tumor effects [Citation69]. Therefore, it remains unclear whether MSC-derived EVs exhibit pro- or anti-tumorigenic effects.
Although endogenous MSC-derived EVs may act as pro-tumorigenic vesicles, MSC administration might have little effect on tumor growth. The safe dose of MSCs ranges between 1‒2 × 106 cells/kg [Citation58], whereas MSC-derived EVs were administered at doses equivalent to EVs that can be collected from 4‒7 × 107 cells/kg MSCs [Citation70,Citation71]. Based on these numbers, it can be stated that EVs secreted from administered MSCs have little impact on tumor progression. Therefore, at safe doses of MSCs, pro-tumorigenicity of MSCs is considered extremely low. However, undesired or unexpected adverse effects cannot be completely ruled out, and the effect of MSC transplantation prior to its clinical application should be carefully tested.
2.4.3. Suppression of anti-tumor immunity by MSC administration
The influence of MSCs on tumor immunity needs to be cautiously monitored. MSCs potently suppress the activity of lymphocytes, promote the polarization of monocytes/macrophages toward an anti-inflammatory phenotype, suppress the migration and maturation of dendritic cells, and induce regulatory T cells. The immunomodulatory properties of MSCs are beneficial for treating inflammatory and autoimmune diseases, but may be unbeneficial for cancer treatment. Chen et al. reported that intravenously administered MSCs suppressed T cell immunity via the programmed cell death-1 (PD-1)/PD-L1 pathway and reduced the survival of a multiple myeloma cell line (5TGM1) multiple myeloma model mice [Citation77]. Furthermore, another group reported that MSCs suppressed cell proliferation and IL-6 expression by NK cells in a human hepatocellular carcinoma cell line (BEL7404) and LM3 xenograft model mice [Citation78]. These reports indicate that MSC administration may inhibit the cancer immunity cycle [Citation79] and the cancer-NK cell immunity cycle [Citation80]. Several studies have suggested that MSCs promote the polarization of monocytes/macrophages toward a different subpopulation called tumor-associated macrophages (TAMs), which promote angiogenesis, tumor cell growth and invasion, and suppress cytolytic T cell responses [Citation20,Citation21]. In contrast, the undesirable effect of MSC administration on tumor immunity can be suppressed by gene engineering of MSCs. Chen et al. reported that administration of MSCs suppressed T cell responses and promoted tumor growth; this effect was prevented by knocking down PD-L1 using shRNA [Citation77]. Yu et al. reported that co-transplantation of MSCs and mouse breast cancer cell line 4T1 cells promoted tumor growth, although administration of MSCs overexpressing Sirt1, the closest mammalian homolog of the yeast enzyme Sir2, suppressed tumor growth by recruiting NK cells [Citation81]. Li et al. demonstrated that FAPα is a critical molecule for the recruitment of monocytes/macrophages and polarization into TAM by MSCs [Citation82]. The elucidation of molecular mechanisms underlying interactions between MSCs and immune cells will provide strategies for preventing tumor immune suppression mediated by MSCs.
3. MSC-based drug delivery for cancer therapy
Several studies on MSC-based drug delivery for cancer therapy have been reported. In the following subsections, we describe strategies and issues involved in the application of MSCs as a vehicle for therapeutic genes and anti-cancer drugs.
3.1. Cancer gene therapy using gene-engineered MSCs
3.1.1. Preclinical study of MSC-based cancer gene therapy
Cancer gene therapy involves the administration of vectors carrying a therapeutic gene encoding an anti-tumor protein or the administration of genetically modified cells expressing an anti-tumor protein. As efficient gene delivery to the tumor tissue is a key step in cancer gene therapy, tumor-homing MSCs have been considered a vehicle for the therapeutic gene (, ). In this subsection, we describe some therapeutic genes, as well as their therapeutic mechanisms in MSC-based cancer gene therapy. In 2002, Studeny et al. demonstrated that the administration of IFN-β-expressing MSCs suppressed tumor growth [Citation83]. Elevated tumor concentrations of IFN-β can suppress tumor cell growth by inducing apoptosis, cell cycle arrest, and activation of the immune response. In general, the systemic administration of IFN-β has shown limited therapeutic effects owing to its short half-life in blood and low delivery efficiency to the tumor site. The authors demonstrated that subcutaneous co-injection of IFN-β-expressing MSCs markedly reduced the volume of the human melanoma cell line A375SM tumor in mice. This result suggests that MSCs can sustainably release therapeutic proteins at the tumor site. Furthermore, intravenously injected IFN-β-expressing MSCs were detected inside the tumor tissue in a lung metastasis mouse model, and intravenous cell injections prolonged the survival of lung metastasis model mice. These results demonstrate the potential of cancer gene therapy using MSCs. TRAIL is a cytokine of the tumor necrosis factor (TNF) family that initiates apoptosis by binding to its death receptors and selectively induces apoptosis of cancer cells. Previous studies have demonstrated that TRAIL-expressing MSCs show considerable anti-tumor effects in tumor-bearing mice [Citation15,Citation20]. Reportedly, herpes simplex virus-thymidine kinase (HSV-tk), a prodrug-activating suicide gene, phosphorylates ganciclovir, a prodrug antiviral agent, in HSV-tk-expressing cells and induces cellular apoptosis. This suicide system is widely applied to eliminate transplanted cells from the host to enhance the safety of cell-based therapy and regulate the function of transplanted cells [Citation8,Citation84]. Phosphorylated ganciclovir in HSV-tk-expressing cells is transferred to neighboring cells via gap junctions and induces apoptosis of neighboring cells [Citation8]. Phosphorylated ganciclovir reportedly competes with deoxyguanosine triphosphate and thus can selectively induce apoptosis in actively dividing cells such as tumor cells without significantly impacting normal tissues/cells. The regulation of gene expression by specific promoters enables avoiding the ‘off-target effect’ and improving the safety of the MSC-based cancer gene therapy. Tie-2, CCL5 (RANTES), and alpha-fetoprotein promoter have been employed in preclinical and clinical studies [Citation85–87]. Oncolytic viruses are defined as viruses that selectively replicate in and kill cancer cells and are harmless to normal tissues/cells. Once oncolytic virus infection occurs, an anti-tumor immune response is activated, and immunogenic cell death of the infected cells and neighboring tumor cells is induced. Thus, oncolytic virus therapy can be expected to afford a vaccination effect [Citation88]. MSCs can be developed as a suitable vehicle for oncolytic viruses, as several oncolytic viruses are immunogenic and lack tumor tropism [Citation88]. However, many genetically engineered oncolytic viruses are designed to be activated in cancer cells. Some oncolytic viruses have been modified to efficiently infect and replicate MSCs [Citation89].
Figure 2. Manufacture of engineered MSCs for anti-cancer therapy. Autologous MSCs are isolated from the patient’s tissue, including BM and AD. The number of MSCs that can be collected differ between source tissues. Several weeks are needed for MSC expansion and gene engineering. After drug loading and quality check, therapeutic gene-modified or drug-loaded MSCs are administered to the patient. Allogeneic MSCs are induced from allogeneic iPS cells or isolated from healthy donor tissue such as BM, AD, and UC. Although allogeneic iPS cell-derived MSCs were previously employed, a project for establishing cell banks for autologous iPS cells is ongoing [Citation98,Citation139]. iPS cells can supply greater doses of MSCs from a single cell bank. Allogeneic tissue-derived- and iPS cell-derived MSCs are processed to the final product in the factory. The stored allogeneic MSC-product can be supplied on demand. AD, adipose tissue; BM, bone marrow; iPS, induced pluripotent stem; MSCs, mesenchymal stem/stromal cells; UC, umbilical cord
![Figure 2. Manufacture of engineered MSCs for anti-cancer therapy. Autologous MSCs are isolated from the patient’s tissue, including BM and AD. The number of MSCs that can be collected differ between source tissues. Several weeks are needed for MSC expansion and gene engineering. After drug loading and quality check, therapeutic gene-modified or drug-loaded MSCs are administered to the patient. Allogeneic MSCs are induced from allogeneic iPS cells or isolated from healthy donor tissue such as BM, AD, and UC. Although allogeneic iPS cell-derived MSCs were previously employed, a project for establishing cell banks for autologous iPS cells is ongoing [Citation98,Citation139]. iPS cells can supply greater doses of MSCs from a single cell bank. Allogeneic tissue-derived- and iPS cell-derived MSCs are processed to the final product in the factory. The stored allogeneic MSC-product can be supplied on demand. AD, adipose tissue; BM, bone marrow; iPS, induced pluripotent stem; MSCs, mesenchymal stem/stromal cells; UC, umbilical cord](/cms/asset/a29ee665-a710-4d69-9757-f928157fd341/iedd_a_1960309_f0002_oc.jpg)
Table 2. Gene types and their therapeutic mechanisms in MSC-based cancer gene therapy
3.1.2. Clinical trials of MSC-based cancer gene therapy
The study of MSC-based cancer gene therapy has entered the clinical stage. According to Clinical Trials.gov, 11 clinical trials on MSC-based cancer gene therapy have been registered (, the data were searched in April 2021.). All studies in phases I and II trials evaluated the safety and tolerance of the administered gene-modified MSCs. A phase I/II clinical trial by von Einem et al. demonstrated the safety of intravenous administered HSV-tk-expressing autologous MSCs and ganciclovir (NCT02008539). Another phase I trial revealed the safety of oncolytic virus-loaded autologous MSCs (NCT01844661). However, no therapeutic effects were observed in these studies. Although several preclinical studies have demonstrated significant anti-tumor effects, these effects have not been corroborated in most clinical trials. This can be attributed to numerous possible reasons, one of which is the MSC dose employed. Niess’s group administered HSV-tk expressing MSCs at a dose of 5 × 105 cells/mouse to hepatocellular carcinoma xenograft model mice, which is equivalent to 2.5 × 107 cells/kg [Citation85]; however, the mean dose in clinical trials was 2.65 × 106 cells/kg [Citation85]. The low homing efficiency and interdonor heterogeneity of MSCs could affect their therapeutic potential. Schweizer et al. reported that intravenously administered MSCs were not detected at the tumor site in patients with prostate cancer [Citation99]. This result indicates the necessity for improving the homing ability of MSCs. Melen et al. reported that clinical outcomes were influenced by the expression of chemokine receptors related to migration. Oncolytic virus-loaded autologous MSCs were intravenously administered to 13 pediatric patients. Clinical responses were observed in 5 patients, with no response recorded in 8 patients. The authors compared the gene and protein expression profiles of cells between responders and non-responders and reported that the expression levels of CCR1, CXCR1, and CXCR4 were higher in the response group than that in non-responders [Citation100].
3.2. Application of MSCs as a vehicle for anti-cancer drugs and functionalized nanoparticles
3.2.1. MSC loading of anti-cancer drugs
MSCs have been employed not only as gene vehicles but also as vehicles for anti-cancer drugs such as doxorubicin (DOX), paclitaxel (PTX), and gemcitabine (). In most studies, free anti-cancer drugs or drug-encapsulated nanoparticles (NPs) were loaded into the cytoplasm of MSCs through passive diffusion or endocytosis (). Free anti-cancer drugs can be easily loaded into MSCs by simple incubation with anti-cancer drugs for several hours [Citation101]. Then, loaded drugs are released from MSCs via passive diffusion, drug efflux transporters such as p-glycoprotein (P-gp) [Citation14,Citation102], EVs [Citation101], or tunneling nanotubes [Citation103] (). The expression of P-gp reportedly varies (), suggesting substantial interdonor heterogeneity in expression. Some studies have revealed that anti-cancer drugs were released from MSCs via P-gp; however, P-gp expression was scarcely examined in most of these studies. Free anti-cancer drug-loaded MSCs suppressed tumor cell growth following co-culture with tumor cells and local administration to tumor-bearing mice [Citation101,Citation102,Citation104–107]. However, few studies have succeeded in suppressing tumor growth following systemic administration, and it may be challenging to efficiently deliver drugs using free drug-loaded MSCs owing to the short retention time of the loaded drug. Although drugs are rapidly released from drug-loaded MSCs, systemically administered MSCs reach the tumor tissue approximately 4‒6 h after administration [Citation24,Citation108]. The use of drug-encapsulated NPs that sustainably release the drug can improve the retention time of drug loading in the cell. Zhang et al. reported that the loading of polyamidoamine dendrimer-DOX (DOX-PAMAM) could delay DOX release from MSCs by 12‒48 h when compared with free DOX [Citation109]. Some NPs, such as PAMAM dendrimers, silica NPs, and liposomes, may be suitable for improving drug retention in MSCs due to their low initial burst release. NPs loaded to MSCs are released extracellularly in an intact form via exocytosis and EVs [Citation48,Citation110–112] or may only release the drug after disassembling the NPs in the cellular endosome/lysosome [Citation112,Citation113] (). However, the drug delivery efficiency of NP-loaded MSCs to tumor cells was not high. The in vitro anti-tumor effect of NP-loaded MSCs was lower than that of NPs, possibly due to poor drug release from MSCs. For example, the IC50 values of PTX-PLGA-NPs and PTX-PLGA-NP-loaded MSCs against the human lung adenocarcinoma cell line A549 were 2.1 nM and 22 nM, respectively [Citation114]. The stimulus-triggered drug release system is useful for improving drug release from NP-loaded MSCs. Huang et al. reported that PTX release from SPION-PTX-NP-loaded MSCs was improved by high-frequency magnetic field treatment [Citation60]. This drug release has been attributed to magnetothermally induced structural disruption of SPION-PTX-NP and necrosis of NP-loaded MSCs. Apart from the low in vitro anti-tumor effects, several studies have successfully suppressed tumor growth in tumor-bearing mice by systemic administration of NP-loaded MSCs; the observed anti-tumor effect was superior to that of NPs owing to the improved delivery of anti-cancer drugs to the tumor tissue.
Figure 3. Drug-loading methods of MSCs and the drug delivery route from drug-loaded MSCs to cancer cells. (a) Drug-loading methods for the application of MSCs as a drug delivery vehicle. Free anti-cancer drugs and drug-encapsulated NPs can be loaded on MSCs through passive diffusion, specific transporters, and endocytosis. Drug-encapsulated NPs can be modified to the surface of MCSs using cell surface modification methods. (b) The intercellular drug delivery mechanism. NPs modified on the surface of MSCs are taken up by neighboring cancer cells through endocytosis. Drugs loaded within MSCs are directly delivered to neighboring cancer cells through gap-junction and tunneling nanotubes (direct delivery). MSCs release loaded drugs to the outside of cells through passive diffusion, specific transporters, exocytosis, and EVs, and released drugs are then taken up by cancer cells (indirect delivery). EVs, extracellular vesicles; MSCs, mesenchymal stem/stromal cells; NPs, nanoparticles
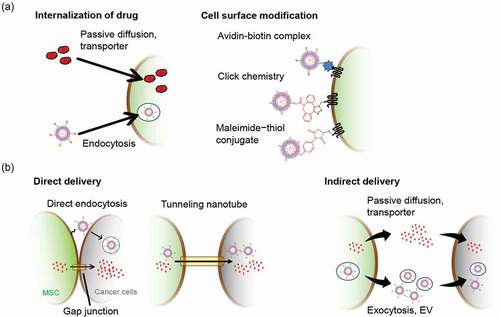
Table 3. Loading methods and therapeutic applications of drug-loaded MSCs
Table 4. Heterogeneity of P-gp expression in MSCs
3.2.2. Surface modification of MSCs with anti-cancer drugs
Internalization of free anti-cancer drugs and anti-cancer drug-encapsulated NPs is cytotoxic to MSCs and affects the viability and tumor tropism of MSCs [Citation60,Citation102,Citation105,Citation107,Citation109,Citation110]. Some studies have successfully modified the surface of MSCs with anti-cancer drug-encapsulated NPs and reduced the cytotoxicity of loaded drugs toward MSCs (, ). Layek et al. successfully modified the cell surface of MSCs with PTX-PLGA NPs without inducing cytotoxicity by employing a strain-promoted [3 + 2] azide-alkyne cycloaddition (SPAAC) reaction [Citation133], a biorthogonal reaction [Citation134]. Cell surface modification with PTX-PLGA NPs marginally affected the migratory ability of the cells to the tumor cell-conditioned medium. Furthermore, intraperitoneal administration of PTX-NP-loaded MSCs demonstrated a higher therapeutic effect than that of NP administration in peritoneal dissemination model mice, established by intraperitoneal injection of a human ovarian cancer cell line, MA148 cells. Takayama et al. demonstrated that cell surface modification using the avidin-biotin complex method could quickly modify the cell surface with DOX liposomes without significantly influencing the viability, migration, and tumor-homing ability of MSCs [Citation135]. Cell surface modification using the avidin-biotin complex method could stably modify cells with avidinated or biotinylated compounds; over 80% of modified DOX liposomes remained loaded in MSCs at 24 h. Furthermore, DOX liposomes on the surface of MSCs were directly taken up by neighboring murine colon adenocarcinoma colon26 cells via endocytosis (), efficiently suppressing the growth of colon26 cells in subcutaneous xenograft and lung metastasis mouse models. Although few studies have focused on cell surface modification-based drug loading when compared with loading inside cells, these studies indicate that cell surface modification with anti-cancer drugs can be valuable for efficient cancer-targeted drug delivery.
4. Conclusion
MSCs are a useful drug delivery vehicle for cancer therapy owing to their innate properties, including tumor tropism, self-renewal or proliferation potency, low immunogenicity, and lack of tumorigenicity. Several studies have demonstrated that MSCs can deliver therapeutic genes to tumor sites and present excellent anti-tumor effects in preclinical trials [Citation14,Citation15,Citation83]. In addition, recent studies have demonstrated that anti-cancer drug-loaded MSCs possess remarkable anti-tumor effects in various in vivo experiments. Although some issues need to be addressed, MSC-based drug delivery will afford an effective cancer-targeting therapy in the future.
5. Expert opinion
MSCs are expected to be developed as a useful drug delivery vehicle for cancer-targeting therapy, especially for treating cancers with a poor prognosis, such as glioblastoma, multi-organ metastases, peritoneal dissemination, and relapsed/refractory cancers. To obtain reliable therapeutic outcomes with MSCs, improving some aspects of current MSC-based therapy remains crucial.
First, the heterogeneity of MSCs needs to be addressed. Donor-dependent variations in tumor tropism and self-renewal or proliferation potency of MSCs can markedly affect the therapeutic effect of MSC-based therapy. This is because obtaining MSCs with high tumor tropism on a clinical scale from every patient is challenging. Therefore, enhancing the migration ability and improving the self-renewal or proliferation potency of MSCs by employing gene or cell engineering techniques should be considered. Furthermore, the use of allogeneic tissue-derived MSCs can reduce interdonor heterogeneity and allow the preparation of adequate clinical doses of MSCs from a single tissue source after confirming their properties. iPS cells are also an attractive cell source for MSCs, and the therapeutic applications of iPS-derived homogenous MSCs prepared on a large scale will be expanded. In addition, as several previous studies have reported [Citation136,Citation137], the quality control of ex vivo expansion process is one of the keys in the manufacturing process of homogenous MSC products.
Second, the tumor delivery efficiency of MSCs should be improved by regulating their biodistribution. Although the safety of intravenous MSC administration has been demonstrated in some clinical trials, the homing rate of intravenously administered MSCs to the tumor site is as low as 1% or less, except for lung tumors, mainly due to their entrapment in the lung vasculature. Accordingly, some completed clinical trials assessing MSC-based cancer gene therapy failed to demonstrate significant therapeutic effects. In several preclinical studies, intravenously administered gene-modified and drug-loaded MSCs showed significant anti-tumor effects at high doses (0.5‒2 × 106 cells/mouse, equal to 1.5‒6 × 107 cells/kg [Citation138]); however, the safe dose in humans is 1‒2 × 106 cells/kg. Therefore, it may be difficult to considerably increase the MSC dose to avoid the risk of developing adverse effects, such as thrombosis and pulmonary embolism. In order to achieve therapeutic efficacy within the safe dose range of MSCs, it is crucial to regulate the biodistribution and improve the tumor-homing efficiency of administered MSCs. Several studies have succeeded in avoiding lung entrapment and improving the tumor tropism of MSCs by using gene or cell engineering techniques or optimizing MSC culture conditions. The development of these techniques will be one of the keys to achieving successful clinical outcomes of therapeutic gene-modified and drug-loaded MSCs for targeted cancer therapy.
Third, the MSC-based drug delivery should be optimized. Over the last ten years, several methods for loading anti-cancer drugs to MSCs have been developed, and some studies have achieved high loading efficiency of anti-cancer drugs without affecting the MSC characteristics, along with efficient delivery of anti-cancer drugs to the tumor site. However, the efficiency of anti-cancer drug delivery from MSCs to tumor cells in the tumor tissue is extremely low and needs to be improved. Recently, some studies, including our previous report, have elucidated the drug delivery mechanism from drug-loaded MSCs to cancer cells [Citation101,Citation111,Citation135]. Understanding the drug delivery mechanisms and optimizing drug delivery will significantly improve the clinical translation of MSC-based drug delivery.
Article highlights
Cells with tumor-homing ability are expected to act as a tumor-targeted drug delivery vehicle.
Mesenchymal stem/stromal cells (MSCs) are considered the most attractive candidate drug delivery vehicles for cancer therapy owing to their tumor tropism.
Many preclinical studies have demonstrated that gene- or anti-cancer drug-loaded MSCs suppress tumor growth.
Current clinical trials assessing MSCs have reported limited therapeutic effects.
The key to achieving a reliable outcome with MSC-based cancer therapy is to adjust the heterogeneity of MSCs and optimize the drug delivery efficiency from MSCs to cancer cells with the regulation of their biodistribution.
Abbreviations
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Sun J, Wei Q, Zhou Y, et al. A systematic analysis of FDA-approved anticancer drugs. BMC Syst Biol. 2017;11(S5):87.
- Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583–592.
- Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146.
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392.
- Matsumura Y. Cancer stromal targeting (CAST) therapy. Adv Drug Deliv Rev. 2012;64(8):710–719.
- Dong H, Xu X, Wang L, et al. Advances in living cell-based anticancer therapeutics. Biomater Sci. 2020;8(9):2344–2365.
- Kimbrel EA, Lanza R. Next-generation stem cells - ushering in a new era of cell-based therapies. Nat Rev Drug Discov. 2020;19(7):463–479.
- Guo L, Zhang Y, Yang Z, et al. Tunneling nanotubular expressways for ultrafast and accurate M1 macrophage delivery of anticancer drugs to metastatic ovarian carcinoma. ACS Nano. 2019;13:1078–1096.
- Aboody KS, Brown A, Rainov NG, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A. 2000;97(23):12846–12851.
- Tabatabai G, Bähr O, Möhle R, et al. Lessons from the bone marrow: how malignant glioma cells attract adult haematopoietic progenitor cells. Brain. 2005;28(9):2200–2211.
- Bagó JR, Okolie O, Dumitru R, et al. Tumor-homing cytotoxic human induced neural stem cells for cancer therapy. Sci Transl Med. 2017;9(375):eaah6510.
- Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317.
- Cheng S, Nethi SK, Rathi S, et al. Engineered mesenchymal stem cells for targeting solid tumors: therapeutic potential beyond regenerative therapy. J Pharmacol Exp Ther. 2019;370(2):231–241.
- Wu HH, Zhou Y, Tabata Y, et al. Mesenchymal stem cell-based drug delivery strategy: from cells to biomimetic. J Control Release. 2019;294:102–113.
- Vodyanik MA, Yu J, Zhang X, et al. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell. 2010;7(6):718–729.
- Neri S. Genetic stability of mesenchymal stromal cells for regenerative medicine applications: a fundamental biosafety aspect. Int J Mol Sci. 2019;20(10):2406.
- Barkholt L, Flory E, Jekerle V, et al. Risk of tumorigenicity in mesenchymal stromal cell-based therapies‒bridging scientific observations and regulatory viewpoints. Cytotherapy. 2013;15(7):753–759.
- Döring M, Cabanillas Stanchi KM, Lenglinger K, et al. Long-term follow-up after the application of mesenchymal stromal cells in children and adolescents with steroid-refractory graft-versus-host disease. Stem Cells Dev. 2021;30(5):234–246.
- Christodoulou I, Goulielmaki M, Devetzi M, et al. Mesenchymal stem cells in preclinical cancer cytotherapy: a systematic review. Stem Cell Res Ther. 2018;9:336.
- Zhang J, Huang X, Wang H, et al. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res Ther. 2015;6(1):234.
- Bexell D, Scheding S, Bengzon J, et al. Toward brain tumor gene therapy using multipotent mesenchymal stromal cell vectors. Mol Ther. 2010;18(6):1067–1075.
- Quante M, Tu SP, Tomita H, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–272.
- Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27(10):2614–2623.
- Kabat M, Bobkov I, Kumar S, et al. Trends in mesenchymal stem cell clinical trials 2004–2018: Is efficacy optimal in a narrow dose range? Stem Cells Transl Med. 2020;9(1):17–27.
- Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147.
- Vangsness CT Jr, Sternberg H, Harris L. Umbilical cord tissue offers the greatest number of harvestable mesenchymal stem cells for research and clinical application: a literature review of different harvest sites. Arthroscopy. 2015;31(9):1836–1843.
- Fraser JK, Wulur I, Alfonso Z, et al. Fat tissue: an underappreciated source of stem cells for biotechnology. Trends Biotechnol. 2006;24(4):150–154.
- Yoshimura K, Shigeura T, Matsumoto D, et al. Characterization of freshly isolated and cultured cells derived from the fatty and fluid portions of liposuction aspirates. J Cell Physiol. 2006;208(1):64–76.
- Chatzistamatiou TK, Papassavas AC, Michalopoulos E, et al. Optimizing isolation culture and freezing methods to preserve Wharton’s jelly’s mesenchymal stem cell (MSC) properties: an MSC banking protocol validation for the Hellenic Cord Blood Bank. Transfusion. 2014;54(12):3108–3120.
- Mohamed-Ahmed S, Fristad I, Lie SA, et al. Adipose-derived and bone marrow mesenchymal stem cells: a donor-matched comparison. Stem Cell Res Ther. 2018;9(1):168.
- Hua J, Gong J, Meng H, et al. Comparison of different methods for the isolation of mesenchymal stem cells from umbilical cord matrix: proliferation and multilineage differentiation as compared to mesenchymal stem cells from umbilical cord blood and bone marrow. Cell Biol Int. 2013;38(2):198–210.
- Neuhuber B, Swanger SA, Howard L, et al. Effects of plating density and culture time on bone marrow stromal cell characteristics. Exp Hematol. 2008;36(9):1176–1185.
- Kuçi Z, Bönig H, Kreyenberg H, et al. Mesenchymal stromal cells from pooled mononuclear cells of multiple bone marrow donors as rescue therapy in pediatric severe steroid-refractory graft-versus-host disease: a multicenter survey. Haematologica. 2016;101(8):985–994.
- Bloor AJC, Patel A, Griffin JE, et al. Production, safety and efficacy of iPSC-derived mesenchymal stromal cells in acute steroid-resistant graft versus host disease: a phase I, multicenter, open-label, dose-escalation study. Nat Med. 2020;26(11):1720–1725.
- Zangi L, Margalit R, Reich-Zeliger S, et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells. 2009;27(11):2865–2874.
- Ruano D, López-Martín JA, Moreno L, et al. First-in-human, first-in-child trial of autologous MSCs carrying the oncolytic virus Icovir-5 in patients with advanced tumors. Mol Ther. 2020;28(4):1033–1042.
- Krueger TEG, Thorek DLJ, Denmeade SR, et al. Concise review: mesenchymal stem cell-based drug delivery: the good, the bad, the ugly, and the promise. Stem Cells Transl Med. 2018;7(9):651–663.
- Nitzsche F, Müller C, Lukomska B, et al. Concise review: MSC adhesion cascade-insights into homing and transendothelial migration. Stem Cells. 2017;35(6):1446–1460.
- Ullah M, Liu DD, Thakor AS. Mesenchymal stromal cell homing: mechanisms and strategies for improvement. iScience. 2019;15:421–438.
- Lourenco S, Teixeira VH, Kalber T, et al. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J Immunol. 2015;194(7):3463–3474.
- Feng Y, Zhu M, Dangelmajer S, et al. Hypoxia-cultured human adipose-derived mesenchymal stem cells are non-oncogenic and have enhanced viability, motility, and tropism to brain cancer. Cell Death Dis. 2014;5(12):e1567.
- Smith CL, Chaichana KL, Lee YM, et al. Pre-exposure of human adipose mesenchymal stem cells to soluble factors enhances their homing to brain cancer. Stem Cells Transl Med. 2015;4(3):239–251.
- Zhang T, Xu Q, Huang T, et al. New insights into biocompatible iron oxide nanoparticles: a potential booster of gene delivery to stem cells. Small. 2020;16(37):e2001588.
- Arbab AS, Jordan EK, Wilson LB, et al. In Vivo Trafficking and Targeted Delivery of Magnetically Labeled Stem Cells. Hum Gene Ther. 2004;15(4):351–360.
- Kono Y, Takegaki J, Ohba T, et al. Magnetization of mesenchymal stem cells using magnetic liposomes enhances their retention and immunomodulatory efficacy in mouse inflamed skeletal muscle. Int J Pharm. 2021;596:120298.
- Ahn YJ, Kong TH, Choi JS, et al. Strategies to enhance efficacy of SPION-labeled stem cell homing by magnetic attraction: a systemic review with meta-analysis. Int J Nanomedicine. 2019;14:4849–4866.
- Xu C, Feng Q, Yang H, et al. A light-triggered mesenchymal stem cell delivery system for photoacoustic imaging and chemo-photothermal therapy of triple negative breast cancer. Adv Sci (Weinh). 2018;5(10):1800382.
- Silva LHA, Silva MC, Vieira JB, et al. Magnetic targeting increases mesenchymal stromal cell retention in lungs and enhances beneficial effects on pulmonary damage in experimental silicosis. Stem Cells Transl Med. 2020;9(10):1244–1256.
- Mader EK, Butler G, Dowdy SC, et al. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J Transl Med. 2013;11(1):20.
- Malekshah OM, Sarkar S, Nomani A, et al. Bioengineered adipose-derived stem cells for targeted enzyme-prodrug therapy of ovarian cancer intraperitoneal metastasis. J Control Release. 2019;311–312:273–287.
- Kimura K, Kishida T, Wakao J, et al. Tumor-homing effect of human mesenchymal stem cells in a TH-MYCN mouse model of neuroblastoma. J Pediatr Surg. 2016;51(12):2068–2073.
- Rincón E, Cejalvo T, Kanojia D, et al. Mesenchymal stem cell carriers enhance antitumor efficacy of oncolytic adenoviruses in an immunocompetent mouse model. Oncotarget. 2017;8(28):45415–45431.
- Kusamori K, Nishikawa M, Mizuno N, et al. Transplantation of insulin-secreting multicellular spheroids for the treatment of type 1 diabetes in mice. J Control Release. 2014;173:119–124.
- Suryaprakash S, Lao YH, Cho HY, et al. Engineered mesenchymal stem cell/nanomedicine spheroid as an active drug delivery platform for combinational glioblastoma therapy. Nano Lett. 2019;19(3):1701–1705.
- Wang S, Guo L, Ge J, et al. Excess integrins cause lung entrapment of mesenchymal stem cells. Stem Cells. 2015;33(11):3315–3326.
- Kim JC, Tae G. The modulation of biodistribution of stem cells by anchoring lipid-conjugated heparin on the cell surface. J Control Release. 2015;217:128–137.
- Moll G, Ankrum JA, Kamhieh-Milz J, et al. Intravascular mesenchymal stromal/stem cell therapy product diversification: time for new clinical guidelines. Trends Mol Med. 2019;25(2):149–163.
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466.
- Huang WC, Lu IL, Chiang WH, et al. Tumortropic adipose-derived stem cells carrying smart nanotherapeutics for targeted delivery and dual-modality therapy of orthotopic glioblastoma. J Control Release. 2017;254:119–130.
- Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318.
- Li G, Bonamici N, Dey M, et al. Intranasal delivery of stem cell-based therapies for the treatment of brain malignancies. Expert Opin Drug Deliv. 2018;15(2):163–172.
- Takayama Y, Kusamori K, Hayashi M, et al. Long-term drug modification to the surface of mesenchymal stem cells by the avidin-biotin complex method. Sci Rep. 2017;7(1):16953.
- Dhada KS, Hernandez DS, Suggs LJ. In Vivo photoacoustic tracking of mesenchymal stem cell viability. ACS Nano. 2019;13(7):7791–7799.
- Leibacher J, Henschler R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res Ther. 2016;7(1):7.
- de Witte SFH, Luk F, Sierra Parraga JM, et al. Immunomodulation by therapeutic mesenchymal stromal cells (MSC) is triggered through phagocytosis of MSC by monocytic cells. Stem Cells. 2018;36(4):602–615.
- Preda MB, Neculachi CA, Fenyo IM, et al. Short lifespan of syngeneic transplanted MSC is a consequence of in vivo apoptosis and immune cell recruitment in mice. Cell Death Dis. 2021;12:566.
- Raz Y, Cohen N, Shani O, et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J Exp Med. 2018;215(12):3075–3093.
- Zhao R, Chen X, Song H, et al. Dual role of MSC-derived exosomes in tumor development. Stem Cells Int. 2020;2020:8844730.
- Roccaro AM, Sacco A, Maiso P, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123(4):1542–1555.
- Biswas S, Mandal G, Roy Chowdhury S, et al. Exosomes produced by mesenchymal stem cells drive differentiation of myeloid cells into immunosuppressive M2-polarized macrophages in breast bancer. J Immunol. 2019;203(12):3447–3460.
- Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–186.
- Jotzu C, Alt E, Welte G, et al. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal Cell Pathol (Amst). 2010;33(2):61–79.
- Strong AL, Pei DT, Hurst CG, et al. Obesity enhances the conversion of adipose-derived stromal/stem cells into carcinoma-associated fibroblast leading to cancer cell proliferation and progression to an invasive phenotype. Stem Cells Int. 2017;2017:9216502.
- Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. 2017;16(1):31.
- Théry C, Witwer KW, Aikawa E. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guideline. J Extracell Vesicles. 2018;7:1535750.
- Chen D, Tang P, Liu L, et al. Bone marrow-derived mesenchymal stem cells promote cell proliferation of multiple myeloma through inhibiting T cell immune responses via PD-1/PD-L1 pathway. Cell Cycle. 2018;17(7):858–867.
- Chen J, Ji T, Wu D, et al. Author correction: human mesenchymal stem cells promote tumor growth via MAPK pathway and metastasis by epithelial mesenchymal transition and integrin α5 in hepatocellular carcinoma. Cell Death Dis. 2019;10(7):488.
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
- Huntington ND, Cursons J, Rautela J. The cancer-natural killer cell immunity cycle. Nat Rev Cancer. 2020;20(8):437–454.
- Yu Y, Liu Y, Zong C, et al. Mesenchymal stem cells with Sirt1 overexpression suppress breast tumor growth via chemokine-dependent natural killer cells recruitment. Sci Rep. 2016;6(1):35998.
- Li X, Chen M, Lu W, et al. Targeting FAPα-expressing tumor-associated mesenchymal stromal cells inhibits triple-negative breast cancer pulmonary metastasis. Cancer Lett. 2021;503:32–42.
- Studeny M, Marini FC, Champlin RE, et al. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002;62:3603–3608.
- Tsujimura M, Kusamori K, Oda C, et al. Regulation of proliferation and functioning of transplanted cells by using herpes simplex virus thymidine kinase gene in mice. J Control Release. 2018;275:78–84.
- Niess H, Bao Q, Conrad C, et al. Selective targeting of genetically engineered mesenchymal stem cells to tumor stroma microenvironments using tissue-specific suicide gene expression suppresses growth of hepatocellular carcinoma. Ann Surg. 2011;254(5):767–774.
- Yan C, Yang M, Li Z, et al. Suppression of orthotopically implanted hepatocarcinoma in mice by umbilical cord-derived mesenchymal stem cells with sTRAIL gene expression driven by AFP promoter. Biomaterials. 2014;35:3035–3043.
- von Einem JC, Guenther C, Volk HD, et al. Treatment of advanced gastrointestinal cancer with genetically modified autologous mesenchymal stem cells: results from the phase 1/2 TREAT-ME-1 trial. Int J Cancer. 2019;145(6):1538–1546.
- Moreno R. Mesenchymal stem cells and oncolytic viruses: joining forces against cancer. J Immunother Cancer. 2021;9(2):e001684.
- Hammer K, Kazcorowski A, Liu L, et al. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int J Cancer. 2015;137(4):978–990.
- Tsujimura M, Kusamori K, Katsumi H, et al. Cell-based interferon gene therapy using proliferation-controllable, interferon-releasing mesenchymal stem cells. Sci Rep. 2019;9(1):18869.
- Mohme M, Maire CL, Geumann U, et al. Local intracerebral immunomodulation using interleukin-expressing mesenchymal stem cells in glioblastoma. Clin Cancer Res. 2020;26(11):2626–2639.
- Xu G, Guo Y, Seng Z, et al. Bone marrow-derived mesenchymal stem cells co-expressing interleukin-18 and interferon-β exhibit potent antitumor effect against intracranial glioma in rats. Oncol Rep. 2015;34(4):1915–1922.
- Wu Z, Liu W, Wang Z, et al. Mesenchymal stem cells derived from iPSCs expressing interleukin-24 inhibit the growth of melanoma in the tumor-bearing mouse model. Cancer Cell Int. 2020;20(1):33.
- Iida Y, Yoshikawa R, Murata A, et al. Local injection of CCL19-expressing mesenchymal stem cells augments the therapeutic efficacy of anti-PD-L1 antibody by promoting infiltration of immune cells. J Immunother Cancer. 2020;8(2):e000582.
- Zhang J, Hou L, Wu X, et al. Inhibitory effect of genetically engineered mesenchymal stem cells with apoptin on hepatoma cells in vitro and in vivo. Mol Cell Biochem. 2016;416(1–2):193–203.
- Mangraviti A, Tzeng SY, Gullotti D, et al. Non-virally engineered human adipose mesenchymal stem cells produce BMP4, target brain tumors, and extend survival. Biomaterials. 2016;100:53–66.
- Lee HK, Finniss S, Cazacu S, et al. Mesenchymal stem cells deliver synthetic microRNA mimics to glioma cells and glioma stem cells and inhibit their cell migration and self-renewal. Oncotarget. 2013;4(2):346–361.
- Hanatani T, Takasu N. CiRA iPSC seed stocks (CiRA’s iPSC Stock Project). Stem Cell Res. 2020;50:102033.
- Schweizer MT, Wang H, Bivalacqua TJ, et al. A phase I study to assess the safety and cancer-homing ability of allogeneic bone marrow-derived mesenchymal stem bells in men with localized prostate cancer. Stem Cells Transl Med. 2019;8(5):441–449.
- Melen GJ, Franco-Luzón L, Ruano D, et al. Influence of carrier cells on the clinical outcome of children with neuroblastoma treated with high dose of oncolytic adenovirus delivered in mesenchymal stem cells. Cancer Lett. 2016;371:161–170.
- Pascucci L, Coccè V, Bonomi A, et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014;192:262–270.
- Pessina A, Bonomi A, Coccè V, et al. Mesenchymal stromal cells primed with paclitaxel provide a new approach for cancer therapy. PLoS One. 2011;6(12):e28321.
- Kolba MD, Dudka W, Zaręba-Kozioł M, et al. Tunneling nanotube-mediated intercellular vesicle and protein transfer in the stroma-provided imatinib resistance in chronic myeloid leukemia cells. Cell Death Dis. 2019;10(11):817.
- Bonomi A, Sordi V, Dugnani E, et al. Gemcitabine-releasing mesenchymal stromal cells inhibit in vitro proliferation of human pancreatic carcinoma cells. Cytotherapy. 2015;17(12):1687–1695.
- Bonomi A, Silini A, Vertua E, et al. Human amniotic mesenchymal stromal cells (hAMSCs) as potential vehicles for drug delivery in cancer therapy: an in vitro study. Stem Cell Res Ther. 2015;6(1):155.
- Coccè V, Farronato D, Brini AT, et al. Drug loaded gingival mesenchymal stromal cells (GinPa-MSCs) inhibit in vitro proliferation of oral squamous cell carcinoma. Sci Rep. 2017;7(1):9376.
- Clavreul A, Pourbaghi-Masouleh M, Roger E, et al. Human mesenchymal stromal cells as cellular drug-delivery vectors for glioblastoma therapy: a good deal? J Exp Clin Cancer Res. 2017;36(1):135.
- Komarova S, Roth J, Alvarez R, et al. Targeting of mesenchymal stem cells to ovarian tumors via an artificial receptor. J Ovarian Res. 2010;3(1):12.
- Zhang X, Yao S, Liu C, et al. Tumor tropic delivery of doxorubicin-polymer conjugates using mesenchymal stem cells for glioma therapy. Biomaterials. 2015;39:269–281.
- Zhao Y, Tang S, Guo J, et al. Targeted delivery of doxorubicin by nano-loaded mesenchymal stem cells for lung melanoma metastases therapy. Sci Rep. 2017;7(1):44758.
- Wang X, Chen H, Zeng X, et al. Efficient lung cancer-targeted drug delivery via a nanoparticle/MSC system. Acta Pharm Sin B. 2019;9(1):167–176.
- Cao S, Guo J, He Y, et al. Nano-loaded human umbilical cord mesenchymal stem cells as targeted carriers of doxorubicin for breast cancer therapy. Artif Cells Nanomed Biotechnol. 2018;46(sup1):642–652.
- Sadhukha T, O’Brien TD, Prabha S. Nano-engineered mesenchymal stem cells as targeted therapeutic carriers. J Control Release. 2014;196:243–251.
- Moku G, Layek B, Trautman L, et al. Improving payload capacity and anti-tumor efficacy of mesenchymal stem cells using TAT peptide functionalized polymeric nanoparticles. Cancers (Basel). 2019;11(4):491.
- Roger M, Clavreul A, Venier-Julienne MC, et al. Mesenchymal stem cells as cellular vehicles for delivery of nanoparticles to brain tumors. Biomaterials. 2010;31(32):8393–8401.
- Dai T, Yang E, Sun Y, et al. Preparation and drug release mechanism of CTS-TAX-NP-MSCs drug delivery system. Int J Pharm. 2013;456(1):186–194.
- Cao B, Yang M, Zhu Y, et al. Stem cells loaded with nanoparticles as a drug carrier for in vivo breast cancer therapy. Adv Mater. 2014;26(27):4627–4631.
- Kang S, Bhang SH, Hwang S, et al. Mesenchymal stem cells aggregate and deliver gold nanoparticles to tumors for photothermal therapy. ACS Nano. 2015;9(10):9678–9690.
- Levy O, Brennen WN, Han E, et al. A prodrug-doped cellular trojan horse for the potential treatment of prostate cancer. Biomaterials. 2016;91:140–150.
- Wu J, Liu Y, Tang Y, et al. Synergistic chemo-photothermal therapy of breast cancer by mesenchymal stem cell-encapsulated Yolk-Shell GNR@HPMO-PTX nanospheres. ACS Appl Mater Interfaces. 2016;8(28):17927–17935.
- Layek B, Sadhukha T, Panyam J, et al. Nano-engineered mesenchymal stem cells Increase therapeutic efficacy of anticancer drug through true active tumor targeting. Mol Cancer Ther. 2018;17(6):1196–1206.
- Timin AS, Peltek OO, Zyuzin MV, et al. Safe and effective delivery of antitumor drug using mesenchymal stem cells impregnated with submicron carriers. ACS Appl Mater Interfaces. 2019;11(14):13091–13104.
- Aoki M, Kakimoto K, Goto M, et al. Novel therapeutic approach using drug-loaded adipose-derived stem cells for pancreatic cancer. Sci Rep. 2019;9(1):17971.
- Huang L, Xu C, Xu P, et al. Intelligent photosensitive mesenchymal stem cells and cell-derived microvesicles for photothermal therapy of prostate cancer. Nanotheranostics. 2018;3(1):41–53.
- Lenna S, Bellotti C, Duchi S, et al. Mesenchymal stromal cells mediated delivery of photoactive nanoparticles inhibits osteosarcoma growth in vitro and in a murine in vivo ectopic model. J Exp Clin Cancer Res. 2020;39(1):40.
- Li L, Guan Y, Liu H, et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano. 2011;5(9):7462–7470.
- Xu M, Asghar S, Dai S, et al. Mesenchymal stem cells-curcumin loaded chitosan nanoparticles hybrid vectors for tumor-tropic therapy. Int J Biol Macromol. 2019;134:1002–1012.
- Yao S, Li X, Liu J, et al. Maximized nanodrug-loaded mesenchymal stem cells by a dual drug-loaded mode for the systemic treatment of metastatic lung cancer. Drug Deliv. 2017;24(1):1372–1383.
- Pessina A, Sisto F, Coccè V, et al. A mesenchymal stromal cell line resistant to paclitaxel that spontaneously differentiates into osteoblast-like cells. Cell Biol Toxicol. 2011;27(3):169–180.
- Han N, Li Z, Cai Z, et al. P-glycoprotein overexpression in bone marrow-derived multipotent stromal cells decreases the risk of steroid-induced osteonecrosis in the femoral head. J Cell Mol Med. 2016;20(11):2173–2182.
- Kim SG, Jeon CH, Suh HS, et al. P-glycoprotein expression in extracellular matrix formation of chondrogenic differentiation of human adult stem cells. Cell Biol Int. 2007;31(9):1042–1048.
- Bosco DB, Kenworthy R, Zorio DA, et al. Human mesenchymal stem cells are resistant to Paclitaxel by adopting a non-proliferative fibroblastic state. PLoS One. 2015;10(6):e0128511.
- Layek B, Sehgal D, Argenta PA, et al. Nanoengineering of mesenchymal stem cells via surface modification for efficient cancer therapy. Adv Ther. 2019;2(9):1900043.
- Takayama Y, Kusamori K, Nishikawa M. Click chemistry as a tool for cell engineering and drug delivery. Molecules. 2019;24(1):172.
- Takayama Y, Kusamori K, Tsukimori C, et al. Anticancer drug-loaded mesenchymal stem cells for targeted cancer therapy. J Control Release. 2021;329:1090–1101.
- Robb KP, Fitzgerald JC, Barry F, et al. Mesenchymal stromal cell therapy: progress in manufacturing and assessments of potency. Cytotherapy. 2019;21(3):289–306.
- Sensebé L, Gadelorge M, Fleury-Cappellesso S. Production of mesenchymal stromal/stem cells according to good manufacturing practices: a review. Stem Cell Res Ther. 2013;4(3):66.
- Galipeau J, Sensébé L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. 2018;22(6):824–833.
- Galipeau J. The mesenchymal stromal cells dilemma‒does a negative phase III trial of random donor mesenchymal stromal cells in steroid-resistant graft-versus-host disease represent a death knell or a bump in the road? Cytotherapy. 2013;15(1):2–8.