
ABSTRACT
Introduction
Cell transplantation is a promising curative therapeutic strategy whereby impaired organ function can be restored without the need for whole-organ transplantation. A key challenge in allotransplantation is the requirement for life-long systemic immunosuppression to prevent rejection, which is associated with serious adverse effects such as increased risk of opportunistic infections and the development of neoplasms. This challenge underscores the urgent need for novel strategies to prevent graft rejection while abrogating toxicity-associated adverse events.
Areas covered
We review recent advances in immunoengineering strategies for localized immunomodulation that aim to support allograft function and provide immune tolerance in a safe and effective manner.
Expert opinion
Immunoengineering strategies are tailored approaches for achieving immunomodulation of the transplant microenvironment. Biomaterials can be adapted for localized and controlled release of immunomodulatory agents, decreasing the effective dose threshold and frequency of administration. The future of transplant rejection management lies in the shift from systemic to local immunomodulation with suppression of effector and activation of regulatory T cells, to promote immune tolerance.
1. Introduction
Advances in cell transplantation promise to restore lost cellular function and reverse diseases that are currently incurable. Advantages of cellular over whole organ transplantation include less tissue mass replacement, minimally invasive procedures, and opportunities for graft immunoengineering. However, with the exception of hematopoietic stem cells (HSCs) [Citation1], cell transplantation strategies have encountered limited success in the clinic.
The major hurdle for transplantation of allogeneic cells and tissues is the requirement of systemic immunosuppression to prevent rejection, which represents a double-edge sword. Specifically, systemic immunosuppression regimens can abrogate acute transplant rejection and delay immune rejection of the allograft [Citation2], albeit at the expense of organ toxicity, increased risk of infections, and neoplasm development [Citation3–5]. Moreover, there is a struggle to balance optimal dosing to avoid under- and over immunosuppression. Thus, a collective goal in the modern era of transplantation is to determine effective anti-rejection approaches that present high safety profiles with minimal immunosuppression-derived toxicity [Citation2].
The standard of care in contemporary immunosuppression regimens typically involves the use of antibody therapy for induction, followed by maintenance with a combination of immunosuppressive agents targeting various rejection pathways [Citation6]. This strategy minimizes agent-associated morbidity while maximizing overall effectiveness. Antibody induction therapy includes T cell-depleting and non-depleting agents. Depleting antibodies are typically used as they provide aggressive immunosuppression in the initial days after transplantation, preventing acute rejection [Citation6,Citation7]. Common T cell depleting antibodies used in solid organ transplantation are polyclonal rabbit antithymocyte globulin (rATG; Thymoglobulin) or alemtuzumab (anti-CD52 monoclonal antibody (mAb)) [Citation8,Citation9]. rATG is mostly used in patients with high risk for delayed graft function (longer cold ischemia time or expanded transplant indication). On the contrary, basilizimab, a non-depleting monoclonal antibody, is used for low-risk patients. Basiliximab is directed against the interleukin-2-receptor (CD25) which is only expressed on activated T cells, inhibiting T cell proliferation and differentiation without causing depletion [Citation10,Citation11]. In some cases, although not as effective, induction is achieved with high doses of agents typically used for maintenance [Citation6]. Common maintenance immunosuppression agents include calcineurin inhibitors (CNI), antiproliferative/antimetabolites, glucocorticoids, mammalian target for rapamycin (mTOR) inhibitors, and T cell costimulation blockers [Citation6,Citation7]. The combinations and doses of maintenance immunosuppressants vary between transplant centers, although a widely adopted regimen for whole organ transplants consists of a combination of tacrolimus, mycophenolate mofetil (MMF), and corticosteroids [Citation2]. Maintenance immunosuppression regimens are established within the first 3 months after transplantation. Reduction in maintenance therapy is not recommended, since it frequently leads to late acute rejection of the allograft. However, some patients may require a modification of the initial maintenance regimen to avoid graft loss. For instance, long-term immunosuppression with CNI may contribute to chronic graft dysfunction due to toxicity-associated adverse events [Citation12,Citation13].
Toxicity-associated complications are the major drawbacks of systemic immunosuppression, ultimately leading to death of patients with functioning grafts, predominantly due to cancer (29%), followed by cardiovascular diseases (23%) and opportunistic infections (12%) [Citation14]. CNI, which are the backbone of immunosuppressive therapy, can cause renal cell toxicity and cellular senescence [Citation15], contributing to premature decline in allograft function in kidney transplant patients and chronic renal failure among patients with nonrenal transplant [Citation16]. Furthermore, although glucocorticoids are effective suppressors of the immune system, they induce metabolic disorders such as hyperglycemia, hyperlipidemia, and hypertension [Citation17]. Therefore, a more tailored approach is needed to mitigate adverse events associated with standard immunosuppression.
In this context, using immunomodulatory therapies to achieve immune tolerance instead of suppression have gained significant interest. Creating an immune tolerogenic microenvironment decreases the risk of acute and chronic graft rejection after solid organ transplantation and can improve transplant survival. Negative costimulatory molecules such as cytotoxic T-lymphocyte associated protein 4 (CTLA-4), programmed cell death 1 (PD-1), or co-inhibitory receptor B and T lymphocyte attenuator (BTLA), and CD160 are investigated for their immunomodulatory effects, as they are capable of immune response inhibition while maintaining peripheral tolerance [Citation18]. Moreover, regulatory T cell (Treg) therapy is investigated in the clinic [Citation19], as Tregs play a major role in inducing and maintaining immune tolerance early after transplantation [Citation20,Citation21].
Further, advances in biomaterials, membranes, and surface functionalization, have led to immunoengineered strategies for immunosuppression-free protection from rejection. Among these, cell encapsulation technologies can prevent rejection by creating a physical barrier between the transplanted graft and the hosts immune cells. However, poor oxygen permeability due to lack of direct transplant vascularization compromises long-term graft viability.
The aforementioned challenges yielded the premise of site-specific immunomodulation for transplantation protection []. This review highlights emerging localized immune modulation approaches to protect allografts, including biomaterial functionalization, local release, and co-delivery of immunomodulatory agents or cells, targeted delivery of nanoparticles, hypoimmunogenic therapeutic cells, and integration of local immunosuppression and cell transplantation within macroencapsulation systems.
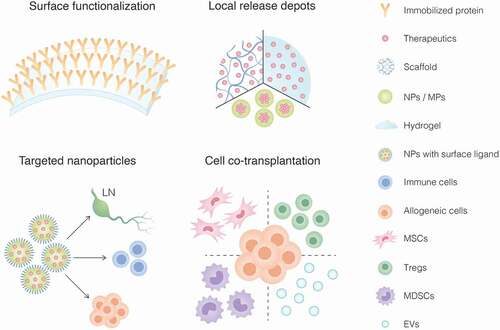
Figure 1. Immunoengineering strategies to achieve local immunomodulation in cell transplantation including 1) surface functionalization of biomaterials with immunomodulatory factors, 2) immunosuppressant-loaded biomaterial platforms (i.e. scaffolds, hydrogels, nanoparticles/microparticles) for in situ deployment and release, 3) nanoparticles with surface ligands for targeted delivery of immunomodulatory agents to lymph nodes (LN), immune cells or allogeneic graft; and 4) co-transplantation of immunomodulatory cells such as mesenchymal stem/stromal cells (MSCs), regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs) or extracellular vesicles (EVs).
2. Cell encapsulation
Cell encapsulation is an extensively researched strategy for immunosuppression-free rejection prophylaxis, specifically for islet transplantation to manage type 1 diabetes [Citation22–26]. Cell encapsulation strategies have shown successful outcomes in clinical trials [Citation27–29], albeit for limited duration. The overarching principle of encapsulation is to provide a physical barrier that isolates the transplanted cells from host immune attack; thus, overcoming the need for immunosuppression. Based on the scale of the encapsulation system, biomaterial-based strategies can be classified as (I) nanoencapsulation, in which cell surfaces are individually coated with a nanoscale immune isolation layer; (II) microencapsulation, where one or a few cells are embedded within a permselective scaffold or microbeads; and (III) macroencapsulation, where a large mass of cells is usually confined within an implantable device [Citation23].
Nanoencapsulation minimizes the coating thickness and improves diffusion for oxygen, nutrients, and insulin. However, nanoscale permselectivity is minimal as it does not provide complete protection from cytokines [Citation30]. Microencapsulation provides the advantage of a high surface-to-volume ratio, allowing efficient diffusion of nutrients and cell waste products. However, they require curative transplant volumes >100 mL, creating a major hurdle in clinical deployment as very few sites in the body can hold such large volumes in an orthotopic setting [Citation22]. In contrast, macroencapsulation devices allow for flexibility in implantation site with potential for minimally invasive subcutaneous (SC) insertion. A potential drawback for these systems is the formation of pericapsular fibrotic overgrowth as a result of the foreign body reaction (FBR) to the device material. Fibrotic tissue formation reduces oxygen and nutrient supply to the grafted cells, which is already limited by lack of direct vascularization, creating a hypoxic environment inhospitable for long-term cell viability. Inadequate oxygen supply is a major limiting factor for clinical translation and implementation of cell encapsulation systems. Thus, enhanced oxygen delivery strategies are being actively developed and tested [Citation31]. Some systems such as β-Air (Beta-O2) have attempted to address encapsulation anoxia via exogenous oxygen supplementation through external ports, which has demonstrated clinical feasibility for daily refilling [Citation32]. However, lack of direct cell apposition to blood vessels impair mass transport critical for metabolic activity, thus affecting graft viability and function [Citation32,Citation33].
Newly developed nanofiber technologies for islet transplantation reduced fibrotic and allogeneic responses to nanofiber-integrated cell encapsulation (NICE) devices implanted in the intraperitoneal (IP) space [Citation34]. The NICE device protected human pluripotent stem cell-derived functional β cells (sc-β cells) from xeno-rejection and reversed diabetes up to 60 days in immunocompetent mice [Citation34]. Compared to SC and epididymal fat pad (EFP) deployment, implantation in the IP space achieved a significantly thinner fibrotic layer and allowed laparoscopic device retrieval. However, IP implantation compromised vasculature development, which limited transplanted cells viability. To improve transplant viability, the same group developed an ‘inverse breathing’ encapsulation device, where CO2 released from respiring cells is recycled into O2 by lithium peroxide (Li2O2) particulates, in a self-regulated manner [Citation35]. The SC implanted device uses silicon-based gas exchangeable materials to improve oxygen delivery to encapsulated islets, thus prolonging graft survival in multiple xenograft models [Citation35]. However, these encapsulation devices require substantially more islets (>2-4 times) to achieve normal glycemia when compared to clinical intraportal pancreatic islet transplantation, which renders translation of these methods challenging.
The high metabolic and oxygen demands of cell transplants prompted a shift in the field, with most efforts focused on direct vascularization macroencapsulation technologies. As an example, devices such as Cell Pouch (Sernova) or Pec-Direct (Viacyte) were engineered to allow for direct vascularization into the device with the goal of improving engraftment and long-term function [Citation29,Citation36]. However, these approaches require systemic immunosuppression, which reverts to the problem of toxicity.
Evidently, achieving adequate graft oxygenation and metabolic sustenance for long-term survival with clinically feasible transplant volumes remains an unresolved challenge to clinical deployment of current encapsulation technologies.
3. Immunoengineering strategies for localized immunomodulation
The need for systemic immunosuppression in direct vascularization approaches motivated efforts to develop novel local immunomodulatory strategies. As key immune rejection events occur at the graft site, localized immunosuppression represents a rational, targeted approach of clinical relevance. The concept of local treatment of allografts to prevent immune rejection has been studied for a few decades [Citation37–40]. Animal studies in the 1990s explored intra-arterial deployment of corticosteroids and CNI in allograft models to achieve localized delivery to cardiac and VCA grafts [Citation37,Citation38]. Two decades later, the feasibility of local immunosuppression via a biohybrid device for islet transplantation and central release of drug was studied for the first time [Citation41]. Localized immunosuppression could attenuate inflammation within the graft environment to protect transplanted cells, sparing the patient from lifelong systemic immunosuppression and its associated complications. Specifically, engineering of biomaterials offers in situ immunomodulation opportunities through immobilization of surface ligands, local release of therapeutic agents through particles/scaffolds/hydrogels, and targeted delivery of nanocarriers [].
Table 1. Local delivery strategies used for therapeutic delivery of immunomodulation.
3.1. Surface functionalization of biomaterials
Surface functionalization entails the addition of a variety of factors to the surface of biomaterials, offering sustained presentation of immunomodulatory factors within the transplant microenvironment. As an example, engineered microgels and scaffolds with addition of streptavidin (SA) coupled with Fas ligand (FasL) allowed for controlled loading, presentation, and retention of SA-FasL protein within the graft microenvironment [Citation42,Citation43]. The Fas pathway is an immune checkpoint which inhibits uncontrolled inflammation [Citation44]. In this context, microgels consisting of poly(ethylene glycol) (PEG) hydrogel beads modified with SA-FasL were co-transplanted with islets [Citation42]. Alternatively, FasL-modified microporous scaffolds were loaded with islets [Citation43]. In diabetic mice, both engineered biomaterials prolonged survival of allogeneic islets with median survival times of 31 or 46 days, compared to 14 days when transplanted with unmodified biomaterials [Citation42,Citation43]. Strikingly, the addition of a short course of rapamycin enhanced islet survival for over 200 days and restored euglycemia. Mice receiving the microgel engineered with SA-FasL had decreased numbers of CD4+ and CD8+ effector T cells (Teff) and increased numbers of Tregs at the graft site [Citation42]. A similar approach using the PEG microgel platform leveraged programmed death ligand 1 (PDL1) [Citation45]. The PDL1/PD1 costimulatory pathway inhibits alloimmune response and induces transplant tolerance. PEG microgels conjugated with SA-PDL1 for controlled delivery of PDL1 within the graft, in combination with a short course of systemic rapamycin, achieved survival of allogeneic islets up to 100 days. PDL1 delivery increased intragraft Tregs [Citation45], consistent with the role of PDL1 as a Treg inductor [Citation46].
An approach to promote in situ Treg differentiation was investigated via functionalization of TGF-β to alginate scaffolds [Citation47]. When these scaffolds were co-transplanted with allogeneic fibroblasts, local generation of Foxp3+ Tregs was superior to constructs lacking TGF-β [Citation47]. While functionalization approaches demonstrate potential efficacy to achieve local immunomodulation, a short course of systemic immunosuppression was still required to abrogate overall immune response and prevent allorejection [Citation42,Citation43,Citation45]. These results indicate that a multi-targeted combinatorial approach affecting multiple pathways is necessary to achieve graft protection and prolonged function. Additional studies in large animal models are also necessary to demonstrate translatability.
3.2. Therapeutics with potential application for local immunomodulation
To date, there is no gold standard for immunosuppression after cell transplantation. Due to the complexity of the immune response toward allogeneic cells, clinical immunosuppression in a local setting is likely to require a multi-targeted, combinatorial drug approach. For pancreatic islet transplantation, current clinical protocols generally include systemic T cell depletion, a tumor necrosis factor α (TNF-α) blocker, tacrolimus, and sirolimus [Citation48,Citation49]. While local immunosuppression could subvert systemic toxicity, the effect of chronic local exposure of some agents on long-term cell viability and function remains detrimental. For example, the known toxicities of tacrolimus and sirolimus on pancreatic islet and renal cell viability, and impaired angiogenesis preclude their use in a localized setting [Citation15,Citation50–54]. Thus, elucidation of the safety profile of potential immunosuppressants is key for their future application in a localized setting. On that account, several other clinical immunosuppressants are appealing for use in a localized setting to prevent allograft rejection, improve graft survival through recruitment of anti-inflammatory cells, minimize toxicity associated adverse events, and improve overall patient quality of life []. We review a few of these promising agents as follows.
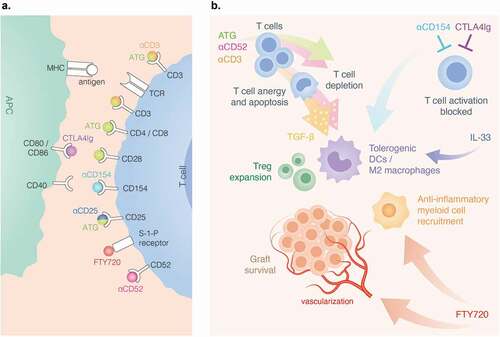
Figure 2. Therapeutics for local immunosuppression and sites of action. A) anti-CD3 binds to CD3 receptor on T cells. rATG binds to CD3. CD152, CD4, CD8, CD25 (IL-2 R) and CD28. CTLA4Ig binds to CD80 and CD86 on APCs, antagonizing CD28 costimulation. Anti-CD154(CD40L) disrupts CD40-CD40L costimulation. FTY720 engagement of S-1-P internalizes the receptors. B) rATG and anti-CD52 induce preferential inhibition of T cells. anti-CD52 and anti-CD3 trigger T cell anergy and apoptosis. APCs that engulf apoptotic vesicles from anergic T cells secrete TGF-β that promotes Treg and tolerogenic DC expansion. Anti-CD154 and CTL4Ig block T cell activation. Anti-CD154 and IL-33 promote a tolerogenic microenvironment. FTY720 induces vascularization and recruits anti-inflammatory myeloid cells.
3.2.1. Anti-inflammatory cytokines
Anti-inflammatory cytokines possess regenerative and immunoregulatory properties during inflammatory states. Namely, TGF-β has an essential role in inducing activated CD4+CD25− cells to express Foxp354. In this context, encapsulation strategies for local TGF-β delivery were tested in models of allotransplantation and rheumatoid arthritis (RA) [Citation55,Citation56]. Intragraft TGF-β-loaded PLGA MPs failed to provide long-term allograft survival in a model of vascularized composite allotransplantation. However, graft survival was prolonged to >300 days when TGF-β was administered alongside IL-2 and rapamycin-loaded MPs [Citation55]. Moreover, release of TGF-β1 from islet-containing PLGA scaffolds transplanted into the EFP, mitigated the immune response against allogeneic islets, decreasing production of inflammatory cytokines and leukocyte infiltration to the graft [Citation57]. Similarly, co-delivery of TGF-β1 MPs and allogeneic islets within the same transplant site, showed minimal T cell infiltration in addition to increased Treg levels in the EFP proximal mesenteric lymph nodes when compared to controls [Citation58]. However, local release of this monotherapy was insufficient to substantially delay graft rejection when compared to untreated controls [Citation58]. Thus, a local multi-drug approach in combination with TGF-β1 is likely needed to induce long-term engraftment.
Localized delivery of IL-33, an M2-polarizing cytokine has been investigated for its immunomodulatory effects in adipose tissue [Citation59]. IL-33 has both pro- and anti- inflammatory properties on alloimmune responses that drive graft-versus-host disease (GVHD). In addition, blocking the pro-inflammatory functions of IL-33 may enhance regulatory properties that contribute to improved transplant outcomes [Citation60]. Loading IL-33 into islet containing PLG scaffolds implanted into the EFP, had a mixed inflammatory effect on graft acceptance in diabetic mice model [Citation59]. Local IL-33 delivery resulted in local upregulation of Tregs, shifting their percentage of total CD4+ T cells from 40% to 75%. In addition, cytotoxic CD8+ T cells were downregulated, which prolonged graft survival from 14 to 33 days, compared to control scaffolds [Citation59]. Even though transplanted islets showed extended survival, IL-33 delayed engraftment for 7 days. The delay in engraftment appears to be intrinsic to IL-33 given that a similar delay was also noted in syngeneic transplants used as controls [Citation59]. IL-33 might induce innate cell populations that inhibit cell engraftment, although its mechanism remains unclear.
3.2.2. T cell costimulation blockers
Blockade of T-cell costimulation pathways allows modulation of adaptive immune responses that can suppress allograft rejection with less off-target effects than other immunosuppressive strategies [Citation61]. CTLA4Ig is a clinically adopted fusion protein that binds to the B7 receptor on antigen presenting cells (APCs), effectively antagonizing CD28 costimulation and T cell activation [Citation62], which can induce tolerance [Citation63]. CTLA4Ig (belatacept) is clinically approved for kidney transplant rejection prophylaxis [Citation64], and has shown success for islet transplantation in clinical trials [Citation65]. CTLA4Ig in a local setting could circumvent toxicity due to systemic inhibition of normal immunoregulatory signaling of endogenous CTLA4 [Citation66] and inhibit early events of alloimmune response [Citation67]. In line with this, local delivery of CTLA4Ig demonstrated long-term allotransplanted islets survival and diabetes reversal in diabetic rodents [Citation68,Citation69]. Local delivery of CTL4Ig from human-derived acellular dermal matrix (ADM) constructs significantly prolonged islet allograft survival when implanted under the kidney capsule (KC). Co-transplantation of allogeneic islets in this matrix resulted in restored euglycemia for >150 days, in addition to Treg expansion and reduction in pro-inflammatory cytokines (IL-6 and IL-17) in serum [Citation69].
Anti-CD154 (CD40L) disrupts the CD40-CD154 (CD40L) costimulatory pathway, halting pro-inflammatory cytokine production, Teff activation, and B-cell class switch, while promoting Treg expansion [Citation70]. Unlike chemical immunosuppressants such as rapamycin and tacrolimus, anti-CD154 does not suppress graft function and prevents formation of alloantibodies and autoantibodies to the therapeutic monoclonal antibody [Citation71]. Importantly, co-treatment with CTLA4Ig and anti-CD154 prolonged islet allograft survival due to the synergistic effect of blocking both costimulatory pathways [Citation72–75]. The use of anti-CD154 is limited due to thromboembolic complications in clinical trials [Citation76]; however, emerging second-generation agents that prevent thromboembolism while maintaining immunomodulatory action have been engineered [Citation77]. We posit that these thromboembolic events and other complications could be avoided with local anti-CD154 delivery.
3.2.3. T cell depleting antibodies
rATG (or Thymoglobulin) is a lymphocyte-depleting, polyclonal antibody preparation, approved by the U.S. Food and Drug Administration (FDA) for induction immunosuppression in kidney transplantion [Citation7,Citation49]. rATG targets a myriad of T cell ligands including CD3, CD152, CD4, CD8, CD25, and CD28[Citation78]. Clinically, rATG is administered transiently, as it induces profound immunosuppression with cytokine storm and increases infection risks [Citation79]. Local dosing of rATG can obviate systemic toxicity associated with T cell depletion as well as prevent Teff cell activation or induce in situ T cell apoptosis [Citation80].
Anti-CD3 is an FDA-approved immunosuppressant for cardiac and renal transplantation [Citation81]. The main effect of anti-CD3 treatment is the induction of T cell anergy and apoptosis, with preferential depletion of conventional T cells while preserving Tregs [Citation82]. Macrophages and APCs that internalize apoptotic vesicles from anergic T cells increase transforming growth factor (TGF-β) production which, in turn, promotes a tolerogenic microenvironment via Treg expansion and APC polarization into immunosuppressive phenotype [Citation83]. The immunomodulatory effects of anti-CD3 on both T cells and APCs, support its use in a localized setting to promote immune tolerance at the graft microenvironment.
3.2.4. Corticosteroids
Anti-inflammatory glucocorticoids, such as dexamethasone (Dex), are often incorporated into biomaterial platforms due to their pleiotropic effects on the implant microenvironment [Citation84–86]. Dex promotes anti-inflammatory macrophage phenotypes which in turn inhibit proinflammatory leukocytes [Citation86,Citation87]; however, it can also globally suppress cellular migration and delay vasculogenesis [Citation88]. In an attempt to overcome the negative impacts of local delivery and assess effect on host cell remodeling, Dex was paired with proangiogenic 17β-estradiol (E2) in macroporous polydimethylsiloxane (PDMS) scaffolds [Citation89]. Implantation of Dex-E2 scaffolds on the EFP of mice, resulted in decreased host cell infiltration and enhanced graft vascularization when compared to monotherapy Dex scaffolds after 14 days of implantation [Citation89]. In another study, co-localization of Dex-loaded PLGA micelles and allogeneic islets under the KC, in addition to systemic CTLA4Ig therapy, led to long-term islet allograft survival and restoration of euglycemia in diabetic mice [Citation90]. While the study is noteworthy, the KC is not a clinically relevant transplantation site. As such, further studies to validate the clinical translatability of results are warranted.
3.2.5. Sphingosine 1-phosphate receptor modulators
FTY720 is the first sphingosine 1-phosphate receptor-1 (S1P) agonist, a new class of FDA-approved immunomodulators for multiple sclerosis [Citation91]. Fingolimod (FTY720) isolates lymphocytes in secondary lymphoid tissues, thus reducing circulation of T and B cells, effectively preventing alloimmune response [Citation92]. Moreover, FTY720 tightens the endothelial barrier, preventing leukocyte transmigration to the graft [Citation93]. Systemic toxicity of FTY720 is associated with the production of reactive oxygen species (ROS) [Citation94], rendering local delivery an attractive approach [Citation95]. Implantation of poly(lactic-co-glycolic acid) (PLGA) films loaded with FTYT20 in the site of injury, reduced proinflammatory cytokine secretion, and increased regenerative cytokines (IL-5, IL-10, and granulocyte colony-stimulating factor [GCSF]), leading to enhanced regeneration of ischemic tissues through recruitment of anti-inflammatory monocytes [Citation96]. However, co-delivery of FTY720 with islets in macroporous PDMS scaffolds negatively impacted the cell engraftment in a dose-dependent associated cytotoxicity [Citation97]. To address this, Bowers et al. [Citation98] developed FTY720-loaded poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) and polycaprolactone (PCL) nanofiber constructs that surround a pocket formed by decellularized porcine dermis. Islets were loaded to the internal pocket of the construct via ultrasound-assisted imaging, 2 weeks after IP implantation. This preconditioning period established a regenerative microenvironment conducive to islet engraftment with clearance of FTY720 by the time of islet transplant, thus avoiding toxic effects.
Beyond inhibition of T cell activation, some drugs used for induction of anergy and management of early immune responses can promote Treg responses. The immunological impact of drugs like rATG and alemtuzumab (anti-CD52 mAb) is preferential inhibition of T and B cell activation over Treg expansion [Citation99]. Specifically, rATG induction therapy reduces the absolute number of Tregs, favorably altering the Treg to T cell ratio [Citation100]. Conversely, alemtuzumab can induce conversion of T cells to Tregs, increase anti-inflammatory cytokines IL-4, IL-10, and TGF-β, and suppress proinflammatory cytokines IFN-γ and IL-17 [Citation99]. The need for preservation of Treg function concomitantly to the inhibition of T cell activation should therefore be considered of great relevance when selecting immunosuppressants in the context of transplantation. In addition, research is being conducted to induce immune tolerogenic microenvironments, as an alternative approach to immunosuppression. Wang et al. recently reviewed localized immunomodulation for transplantation of insulin-producing cells [Citation101]. In summary, the opportunities deriving from the use of immunomodulatory agents in a local setting are largely unexplored, which provides a fertile field for scientific discoveries.
3.3. Targeted nanoparticles
Targeted delivery of drugs to lymph nodes has been explored for its potential to increase the efficacy of immune and cancer therapies while reducing associated toxicities [Citation102,Citation103]. Moreover, the ability of actively targeting encapsulated cargo to a subset of immune cells offers the opportunity to modulate the immune system without compromising systemic immunity [Citation104]. Targeted therapeutic delivery through the use of nanoparticles (NPs) has recently been applied to provide tighter control over the delivery of immunomodulators in the context of allograft acceptance [Citation105]. NPs can be engineered in size and properties that preferentially drain via the lymphatics to the lymph nodes, generating an efficient passive targeting strategy. Additionally, NPs may be actively targeted to immune-relevant sites via surface functionalization with selective ligands [].
Table 2. Targeted nanoparticle strategies for drug delivery and immunomodulation.
Given the pathophysiology of transplant rejection, targeting NPs to lymph nodes and specific subsets of immune cells is an attractive immunomodulatory strategy. In a recent study, the surface of NPs was coated with anti-CD2/CD4 antibodies. These anti-CD2/CD4 PLGA NPs specifically target CD4+ T cells and deliver TGF-β and IL-2104, [Citation106]. After IP administration of CD4 targeted NPs, mice displayed increased Treg population frequencies in the mesenteric lymph nodes and spleen, whereas non-coated NPs at equivalent doses failed to achieve immune inhibition and tolerance [Citation104,Citation106]. In another approach, targeted release of TGF-β and IL-2 can increase local concentration of cytokines available to cells within nanoscale ligand-receptor distances, mitigating high dose-related toxicity while retaining high bioactivity [Citation104]. Alternatively, tacrolimus was incorporated into PLGA NPs to prevent acute rejection after heart transplantation [Citation107]. A single SC injection of tacrolimus-loaded NPs resulted in 50% of allograft survival over 28 days, achieving superior efficacy over intravenous (IV) and intragastric (IG) administration. This demonstrated that SC administration was the most efficient route to deliver PLGA NPs and enhance tacrolimus therapeutic effect. In another attempt to overcome the lack of selectivity and toxicity, targeted delivery of cyclosporin A (CsA) to lymph nodes was achieved by coupling CsA-loaded polylactic (PLA) NPs to allogeneic dendritic cells (DCs), which migrated from the SC injection site to draining lymph nodes [Citation108]. This strategy aims to further improve the efficacy of DCs for immunotherapy by loading them with NPs that can carry different classes of drugs with no systemic release.
In efforts to target immune-cell-rich tissues, anti-CD3-loaded NPs were coated with MECA79 antibody which binds peripheral node addressin (PNAd) molecules, targeting them to peripheral lymph nodes [Citation109]. The use of anti-CD3 in a transplantation setting is of interest, as it inhibits T cell expansion while inducing Tregs [Citation110]. IV administration of MECA79-coated anti-CD3-loaded NPs significantly prolonged heart allograft survival to a median survival of >100 days, when compared to other forms of delivery [Citation109]. This indicates efficient trafficking of MECA79-NPs to lymph nodes with subsequent release of anti-CD3. Furthermore, PEG-b-poly(propylene sulfide) (PEG-b-PPS) polymersome (PS) nanocarriers for SC delivery, significantly altered cellular biodistribution, redirecting rapamycins mechanism of action from immunosuppression to tolerance induction [Citation111]. While standard oral rapamycin administration modulates T cells, rapamycin-loaded PS NPs delivered SC, drained into brachial lymph nodes where they were up-taken by APCs. As a result, APCs developed an anti-inflammatory phenotype with high MHCII for presentation to CD4+ T cell receptors, albeit without costimulatory molecules. Without costimulatory activation, acute rejection causes CD4+ T cells to become anergic or tolerogenic CD8+ Tregs [Citation111]. SC delivery of rapamycin-loaded PS provided a tolerogenic state that allowed long-term survival of allogeneic islets transplanted intraportally in mice [Citation111]. This study highlighted delivery of engineered NPs to repurpose drugs with known mechanisms of action and enhance therapeutic effect while mitigating adverse events.
Most of the studies investigating the use of NPs in transplantation have relied on systemic delivery with increased concentration in targeted sites. However, the treatment of donor organs prior to transplantation is a recently explored paradigm in transplant optimization [Citation105,Citation112]. Ex vivo incubation of mouse tracheal and aortic allografts with rapamycin-loaded PEG micellar NPs prevented transplant fibrosis and vasculopathy for 28 days post-transplantation [Citation112]. Here, rapamycin NPs were coated with cyclic arginine-glycine-aspartate moieties (cRGD) specific for αVβ3 integrins located on the surface of endothelial and epithelial cells, thus, enhancing receptor-mediated endocytosis as compared to untargeted constructs [Citation112]. Similarly, MMF was employed in a system of direct delivery and sustained release with NPs [Citation113]. MMF is commonly used as a maintenance drug after renal transplantation as it inhibits inosine monophosphate dehydrogenase and blocks purine metabolism, preventing lymphocyte proliferation [Citation114]. Perfusion of donor mouse hearts with MMF-loaded PEG-PLGA NPs prior to transplantation significantly reduced IL-6, TNF-α, and IFN-γ, as well as lymphocyte infiltration with intact vasculature in comparison to control and free MMF groups [Citation113]. Altogether, these studies demonstrate that during standard procurement procedures, allografts can be treated with NPs prior to transplantation to reduce immune rejection and promote long-term survival.
Targeted drug delivery using NPs provides significant advantage over free-form administration by minimizing exposure of immunosuppressive agents to non-target tissues. However, despite increasing drug concentration at targeted sites, >95% of systemically administered NPs accumulate elsewhere in the body [Citation115]. Moreover, nanoscale delivery systems are often susceptible to rapid clearance by the reticuloendothelial system (RES), further limiting their therapeutic effect [Citation115]. Additionally, their use for chronic applications inevitably requires repeated administrations, which may pose concerns related to invasiveness of delivery, and poor adherence to treatment. These disadvantages have delayed translation of polymeric NPs into the clinic with only a small number being the FDA approved for cancer, iron-replacement therapies, delivery of imaging agents, fungal treatments, and macular degeneration. Nevertheless, numerous clinical trials are currently testing nanocarriers for cancer and gene therapy vaccines [Citation116].
3.4. Hydrogel depot formulations
Another strategy for molecule-controlled release is using hydrogel systems as depot formulations to modulate the local microenvironment at the delivery site. Hydrogels possess excellent versatility in terms of providing multiple mechanisms for controlled drug release, as reviewed by Li and Mooney [Citation117]. Only a limited number hydrogel drug delivery systems have reached clinical translation, with bone regeneration being the most impactful therapeutic application [Citation117]. In these systems, controlled release of drugs is achieved through either network degradation, swelling, or mechanical deformation, which increase mesh size and promote outward drugs diffusion [Citation117].
Localized immunosuppression with tacrolimus-loaded intragraft triglycerol monostearate (TGMS) hydrogels preserved function of allotransplanted hindlimb in rats for over 280 days without compromising kidney function, a hallmark side effect of tacrolimus therapy [Citation118]. In a similar attempt, the SC co-delivery of tacrolimus-loaded PLGA MPs and pancreatic islets within Matrigel, effectively suppressed immune rejection by inhibiting T cell activation and macrophage proliferation in the local transplant microenvironment, restoring normoglycemia in diabetic mice [Citation119]. In addition, the SC injected Matrigel may also provide an immune-protective barrier to transplanted islets and microspheres [Citation119].
A recent study demonstrated effective IV delivery of immunosuppressive acid labile prodrugs targeted toward a SC injected azido-modified alginate gel depot [Citation120]. In this study, prodrugs of tacrolimus, rapamycin, and MMF were designed to contain a clickable head, a water-soluble segment and a degradable linker responsive to lower pH indicative of inflammation. For the clickable head, dibenzocyclooctin (DBCO) was introduced as it can react with azido groups via Click chemistry. DBCO-modified prodrugs were successfully captured by azido-modified alginate hydrogels and cleared from the bloodstream after 12 h. This approach allowed IV deployment of fresh prodrugs into SC implanted hydrogels. Thus, refilling of hydrogel depots was achieved through noninvasive IV injection, which strategy could be used in other settings for drug supplementation once the payload is exhausted. However, efficacy demonstration of this approach for allotransplantation remains to be elucidated.
Induction of tolerance to islet allografts through local delivery of immunomodulators with hydrogels has been explored [Citation121,Citation122]. Injectable hydrogel Matrigel was used for local delivery of immunomodulatory liposomal clodronate in conjunction with pancreatic islets [Citation121]. Clodronate causes macrophage depletion and decreases expression of adhesion molecules in the synovial lining of patients with longstanding RA [Citation123]. In particular, the protection imparted by clodronate after islet transplantation was key for long-term survival of islets (>60 days) [Citation121]. Local release of clodronate lowered concentration of proinflammatory cytokines in the islet-containing Matrigel [Citation121]. In a similar attempt, Pathak et al. explored local co-delivery of clodronate and tacrolimus with a composite hydrogel using MPs embedded in Matrigel to induce tolerogenic DCs and protect xenogeneic islets from rejection [Citation122]. Local SC co-delivery of islets and immunosuppressants embedded in the Matrigel resulted in decreased expression of CD40, CD80, CD86, and MHCII, characteristic of a tolerogenic DC phenotype, in both the graft and draining lymph nodes. Consequently, after tolerogenic induction and subsequent secretion of TGF-β and IL-10 by tolerogenic DCs, Tregs were increased in the spleen, draining lymph nodes, and graft [Citation122]. Similarly, local delivery of indomethacin and methotrexate was tested using a composite system of polyethyleneimine NPs within a Pluronic hydrogel in a rodent model of RA [Citation124]. After intra-articular injection, controlled release of drugs was prolonged with slower release rates in the composite hydrogel system as compared to NPs alone [Citation124]. These results highlight composite hydrogels as a strategy to develop local and controlled-release systems through addition of secondary barriers to regulate drug diffusion.
4. Co-transplantation with immunomodulatory cells and extracellular vesicles
Tolerance induction protocols employing co-transplantation of allogeneic cells with immunomodulatory cells have the potential to reduce maintenance immunosuppression regimens. We review a few of these graft supportive cells as follows.
4.1. Co-delivery with mesenchymal stem cells (MSCs)
MSCs are multipotent stem cells widely investigated for their pivotal role in vascular regeneration and immunomodulation [Citation125]. The co-delivery of MSCs promotes graft survival and tolerance induction to third-party cardiac [Citation126], kidney [Citation127], endothelial cell induced vascular structure [Citation128], corneal [Citation129], and islet [Citation130] allografts. These graft-beneficial effects can be attributed to the ability of MSCs to induce a pro-tolerogenic Treg phenotype [Citation126,Citation127,Citation129] and reduce the inflammatory alloimmune responses [Citation128]. Although MSCs co-transplantation delays allograft rejection, survival beyond 100 days has yet to be achieved.
To achieve tolerance induction with cell co-delivery strategies, bone marrow-derived MSCs (BM-MSCs) are preclinically and clinically studied for co-transplantation with islets [Citation131,Citation132]. The source of MSC plays a key role in the outcome of graft survival. In fact, co-transplantation of autologous BM-MSCs protected against alloimmune response and prolonged intrahepatic islet engraftment [Citation131]. A randomized controlled trial by Tan et al. [Citation133] demonstrated that treatment with autologous BM-MSCs as induction therapy, IV infused at day 0 and 14 after kidney transplantation, reduced CNI immunosuppression requirement. Additionally, when compared with anti-IL-2 receptor antibody induction therapy, autologous BM-MSCs decreased incidence of acute graft rejection and risk for opportunistic infection, improving estimated renal function at 1-year follow-up[Citation133]. Further, a recent case provided evidence that infusion of autologous BM-MSCs was associated with safe complete discontinuation of maintenance immunosuppression after renal transplantation, allowing a state of immune tolerance [Citation134]. Overall, preclinical models have shown the feasibility of effective MSC therapy in transplantation with early clinical trials supportive of their safety.
4.2. Co-delivery with myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) are a heterogenous population of immunosuppressive cells from the myeloid lineage, characterized by expression of CD11b, CD33, CD34, and vascular endothelial factor (VEGF) receptor 1[Citation135]. MDSCs are upregulated by mediators of inflammation such as IL-2, GCSF, TGF-β, and PGE2[Citation136]. Upon exposure to stimulatory molecules, MDSCs can suppress T cells and promote transplant tolerance via intragraft Treg expansion [Citation137]. Studies have demonstrated that MDSCs secrete IL-10, CTLA4 and IFN-γ to increase the number of Foxp3+ Tregs. Treg functions are upregulated by the interaction of PDL1 on MDSCs with PD1 on Tregs [Citation138]. MDSCs are shown to prolong graft survival in corneal [Citation139], islet [Citation140], cardiac [Citation141], kidney [Citation142], and skin [Citation143] transplantation models and induce transplant tolerance. However, the effector function of MDSCs after transplantation remains poorly understood. Transwell assays showed that MDSCs produce soluble TGF-β to modulate T cells, as well as ROS to regulate Th2 immune responses [Citation138]. Induction of transplant tolerance using clinical immunosuppressants combined with the immunomodulatory effects of MDSCs in a local setting warrants further investigation.
4.3. Co-delivery with regulatory T cells
Tregs are a unique subset of immune cells (CD4+CD25+Foxp3+) that play an important role in maintaining homeostasis and tolerance. Tregs suppress immune responses through DC modulation, inhibitory cytokine secretion, and cytolysis and metabolic disruption of Teffs [Citation19]. Over the last years, some Treg-based cell therapies have reached clinical trials [Citation19]. Treg cell therapies reduced requirement of induction immunosuppression for renal transplantation and maintenance immunosuppression for liver transplantation in phase I/II clinical trials [Citation144,Citation145]. Although still in the early stages, preliminary results from these trials showed significant reduction of infection rates in patients [Citation144]. These results underscore Treg therapy potential to minimize the burden of systemic immunosuppression in transplant patients.
4.4. Cell-free immunomodulation with extracellular vesicles
Extracellular vesicles (EVs) produced by both immune and non-immune cells have an important role in the regulation of immunity. EVs possess both immunostimulatory and immunosuppressive properties, and therefore can be employed as immunomodulatory agents for transplantation [Citation146]. Administration of DC-derived EVs before and after transplantation promoted overall immune tolerance to kidney allografts [Citation147]. Use of immature DC-derived exosomes significantly improved graft survival and reduced inflammatory response by promoting Tregs and limiting CD4+ T cell infiltration [Citation147].
Similar to MSCs, MSC-derived EVs are capable of modulating T cell functions and immune responses. Recent efforts have focused on the therapeutic effects of MSCs through the secretion of paracrine factors, instead of engraftment and differentiation into functional cells [Citation148]. In vivo studies with MSC-derived EVs showcase their potential to be used as alternative to MSC-based therapies [Citation148]. MSC-derived EVs possess immunoregulatory properties that prevent acute graft-versus-host-disease (aGVHD) [Citation149–151]. IV administration of BM-MSC-EVs in previously induced aGVHD mice resulted in a temporary increase of circulating Tregs as well as levels of TNF-α and IFN-γ in plasma, peaking at day 9 after administration and prolonging mice survival until experimental endpoint of 30 days, whereas untreated mice did not survive beyond day 15149. In another study, aGVHD mice that received systemic infusion of EVs showed improvement with suppression of GVHD-associated pathology in the colon, particularly of inflammatory cell infiltration to the lamina propia [Citation150]. Moreover, EVs derived from umbilical cord-derived MSCs (UC-MSCs) reduced circulating CD8+ T cells as well as serum levels of IL-2, TNF-α, and IFN-γ after IV administration to recipient mice receiving allogeneic HSCs for aGVHD induction [Citation151].
In comparison to whole cell-based therapies, MSC-EVs employed as cell-free therapies may offer specific advantages for patient safety such as lower propensity to trigger immune responses and inability to form tumors [Citation152]. Moreover, different methods for preservation of EVs are under investigation to extend their shelf life and reduce storage-related costs compared to cell-based therapies [Citation153]. However, the characterization and application of MSC-derived EVs is limited by low-yield and labor-intensive isolation techniques, as well as lack of standardized isolation protocols [Citation149]. In fact, MSC-derived EVs paracrine factor content greatly varies depending on the techniques used for isolation and the activation status of MSCs [Citation149]. Currently, EVs have yet to be assessed in clinical trials, although a number of studies have examined the effect of vesicles on isolated human organs, demonstrating a potential reparative effect [Citation154].
5. Other emerging strategies for local immunomodulation
Although the transplantation of immunoprotected therapeutic cells may provide a translatable solution, the lack of blood supply to the microenvironment hinders adequate graft oxygenation. In an effort to achieve adequate graft oxygenation, prevascularization was induced using MSCs prior to allogeneic transplantation in cell encapsulation macrodevices [Citation155–157]. In this context, to eliminate the adverse effects of systemic immunosuppression while preserving the advantages of direct graft vascularization, Paez et al. developed a neovascularized implantable cell homing and encapsulation (NICHE) platform [Citation68,Citation158]. The NICHE is a transcutaneously replenishable, dual-reservoir implantable device that integrates direct vascularization and localized delivery of soluble immunosuppressants [] [Citation68,Citation155]. Two independent drug and cell reservoirs are interconnected by a nanoporous membrane that enables uninterrupted and controlled immunosuppressant elution for months. The NICHE deployment strategy consists of a prevascularization phase after SC implantation with MSCs, followed by transcutaneous loading of immunosuppressants and therapeutic cells into the drug and cell reservoirs, respectively [Citation68]. Local release of CTLA4Ig immunosuppressant maintained allogeneic Leydig cell viability in immunocompetent rats, with low lymphocyte infiltration within the NICHE. Importantly, confinement of immunosuppressant to the transplant microenvironment limited systemic exposure while maintaining allogeneic cell survival up to 31 days [Citation68]. Furthermore, NICHE efficacy for allotransplantation of islets and long-term diabetes reversal was demonstrated in an immunocompetent rat model [Citation159]. Transplantation of allogeneic islets within prevascularized NICHE restored normoglycemia in rats for >150 days and local immunosuppressant delivery prevented immune rejection without inducing systemic immunosuppression. Specifically, local anti-lymphocyte serum (ALS) and CTLA4Ig delivery reduced immune cell infiltration and prolonged graft survival similarly to standard systemic delivery, albeit with unchanged circulating T cell and Treg levels. Moreover, for translatability efforts, ease for deployment and short-term allogeneic islet engraftment were demonstrated in a fully MHC-mismatched primate model. Partial immunosuppression was achieved with addition of anti-CD154 to the immunosuppressive cocktail, effectively reducing leukocyte infiltration in fully vascularized devices and localized drug delivery abrogating systemic drug exposure.
Figure 3. Other emerging strategies to overcome systemic immunosuppression in cell transplantation. A) The neovascularized implantable cell homing and encapsulation (NICHE) platform integrates localized delivery of immunosuppressants and direct vascularization. The drug and cell reservoirs are interconnected via nanoporous membranes for controlled release of immunosuppressants into the cell reservoir providing local protection of allogeneic cells against immune cells. Self-sealing silicone ports allow minimally invasive transcutaneous refilling of drug and cell reservoirs. B) Upregulation of CD47 via lentiviral transduction and knock-out (KO) of MHCI and MHCII via CRISPR-Cas9 technology, creates hypoimmunogenic induced pluripotent stem cells (iPSCs) that do not elicit immune responses in allogeneic recipients.
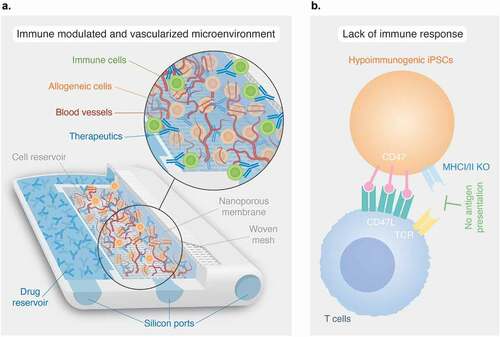
An alternative approach entails engineering hypoimmunogenic pluripotent stem cells as a source of universally compatible cell or tissue grafts. This concept stemmed from drawbacks in pluripotent stem cell reprogramming [Citation160]. Reprogramming technologies allowed the generation of autologous human induced pluripotent stem cells (hiPSCs) for patient-specific treatments. However, hiPSCs production is laborious, costly and is only practical for certain conditions [Citation160]. Instead, the use of allogeneic stem cell therapies can target large patient populations but would require strong immunosuppression. For these reasons, research has focused on generating hypoimmunogenic cell lines that allow transplantation to multiple recipients, while attenuating immune rejection response. Deuse et al. [Citation160] found that syncytiotrophoblast cells, which are intrinsically hypoimmunogenic due to contact between maternal blood and fetal tissue, have downregulation of MHCI and MHCII and upregulation of CD47 molecules. This led to the design of hiPSCs with MHCI and MHCII knockout (KO) using CRISPR-Cas9 technology via the B2M and CIITA gene, and lentiviral transduction for upregulation of CD47 []. These gene-edited hiPSCs did not induce an immune response in HLA mismatched allogeneic humanized mice recipients. Moreover, differentiation of hiPSCs into cardiomyocytes and epithelial cells did not cause immune rejection throughout the 50 days study duration [Citation160].
As an innovative solution to donor organ shortage, gene editing via CRISPR-Cas9 technology has been leveraged to reduce immunogenicity of porcine tissues, making xenotransplantation a potentially feasible solution [Citation161,Citation162]. Yang et al. reported up to 65 simultaneous edits allowing the removal of all copies of the porcine endogenous retrovirus (PERV) pol gene from the pig genome, rendering them more compatible with humans [Citation161,Citation162]. The long-term goal is to produce universal donor cell lines for replacement therapies as a solution to organ shortages, with efforts to advance xenotransplantation into clinical trials [Citation163].
6. Conclusion
Emerging local immunoengineering strategies have the potential to significantly enhance cell transplantation outcomes. However, a great overarching challenge in cell allotransplantation remains overcoming the complexity of the immune response. Addressing knowledge gaps in the crosstalk mechanisms that occur during graft rejection as well as developing complementary and synergistic approaches that target multiple components of the alloimmune response is crucial to achieve long-term protection of transplanted cells without the risk of systemic adverse events.
7. Expert opinion
Cell transplantation is undergoing an important transition from the traditional approach that targets systemic immune responses to more effective and less toxic methods of immune modulation at the local graft microenvironment. While cell encapsulations that provide a physical barrier against immune rejection have been extensively explored to avoid systemic immunosuppression, lack of direct graft vascularization precluded their successful development. Subsequent efforts were made to achieve graft oxygenation reviving the idea of using cell replacement therapy in conjunction with a method aimed to deter immune cells from attacking the grafted area.
Advances in bio- and immunoengineering have spurred tailored approaches to address these challenges. Specifically, biomaterials and surface functionalization provide versatile tools to locally modulate the immune response, while stimulating the growth of a functional vasculature network. It is at the graft microenvironment that nanocarriers, scaffolds, and hydrogels can be adapted for local and personalized release of immunomodulatory agents directed against acute and chronic transplant rejection.
In this context, local delivery of immunomodulatory agents presents a unique opportunity to address the limitations of systemic delivery avoiding toxicity, rapid clearance, and off-target effects. For this, while biomaterials functionalization can provide transient therapeutic efficacy that is relevant in the short term against chronic rejection, platforms that allow for sustained, and potentially life-long immunomodulation are needed for extending life of the transplant. Here, simple mechanisms for drug refilling or supplementation may be fundamental for long-term clinical deployment. Nonetheless, it is still unclear if local therapeutic release is sufficient to regulate the alloimmune response. In addition, local confinement of immunosuppressants impose potential toxicity to the graft. Thus, safety profiles for local drug release are required as with systemically deployed agents.
Additionally, local immunomodulation opens the door to new developments in immunotherapeutic agents. Designed to achieve antigen-specific tolerance and maintenance of Tregs as opposed to immunosuppression, these may shorten the course of anti-rejection therapy while achieving long-term graft viability. This could be achieved in concert with providing secondary immune modulation through cells such as MSCs, which provide the distinct advantage of physiological and continuous production of cytokines to modulate the immune microenvironment. Alternatively, sub-cellular components such as extracellular vesicles could be used aiming at reducing activation of T and B cells. The benefits could be multiple, including improving adherence to treatment, limiting treatment fatigue, and visits to the healthcare centers, and improvement of patient quality of life.
It must be noted, however, that the solution to long-term successful cell transplantation may require a multipronged approach including biomaterials, cells, and therapeutics, which is likely to render the clinical translation complex. As an example, the process of producing therapeutic cells to meet clinical standards is cumbersome, expensive, and complex in itself. To this end, the prospected use of engineered hypoimmunogenic cells is attractive as it does not require third-party biomaterials or supporting cells; however, the genetic modifications to achieve cell immortalization could compromise the long-term cell physiology. Engineering universal cells that can evade immune rejection hold great potential in overcoming donor shortage; however, despite the advances in genomic editing technologies, a risk of developing genomic instability over time still exists. Moreover, the idea of having cells highly capable of evading the immune system impose safety concerns for clinical applications.
Novel immunoengineering strategies have set a new standard in the field of transplantation, and we believe that future immunosuppression protocols should shift from systemic to local immune modulating strategies. Shifting to local delivery systems that allow multi-targeted strategies to modulate the immune response in the transplant microenvironment will address the limitations of current encapsulation technologies for clinical cell transplantation. Promoting these new methodologies will ultimately lead to clinically significant increases in treatment quality resulting in overall improved patient care.
Article highlights
The requirement of systemic immunosuppression to prevent transplant rejection is a major hurdle in allotransplantation.
Novel immunoengineering strategies offer a unique opportunity to prevent transplant rejection without the adverse events of systemic immunosuppression regimens.
Localized immunosuppression represents a targeted approach to tackle immune rejection specifically at the graft site.
Local immunomodulation is designed to achieve antigen-specific tolerance and maintenance of Tregs as opposed to immunosuppression.
The future of transplant rejection lies in the shift from systemic to local immune modulating strategies to promote transplant tolerance in a safe an effective manner.
Declaration of interest
J Paez-Mayorga, C Ying Xuan Chua, and A Grattoni are inventors of intellectual properties licensed by NanoGland, LLC. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med 2006 Apr 27;354(17):1813–1826.
- Pilch NA, Bowman LJ, Taber DJ. Immunosuppression trends in solid organ transplantation: the future of individualization, monitoring, and management. Pharmacother J Human Pharmacol Drug Ther 2021;41(1):119–131.
- Fishman JA. Infection in Organ Transplantation. Am J Transplant 2017 Apr;17(4):856–879.
- Nellore A, Fishman JA. The microbiome, systemic immune function, and allotransplantation. Clin Microbiol Rev 2016 Jan;29(1):191–199.
- Santangelo ML, Criscitiello C, Renda A, et al. Immunosuppression and multiple primary malignancies in kidney-transplanted patients: a single-institute study. Biomed Res Int 2015;2015:183523.
- Enderby C, Keller CA, and . An overview of immunosuppression in solid organ transplantation. Am J Manag Care. 2015;21(1 Suppl):12–23.
- Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med 2004 Dec 23; 351(26):2715–2729.
- Thymoglobulin [prescribing information]. Cambridge: Genzyme Corporation; 2020.
- Campath (alemtuzumab) [prescribing information]. Cambridge: Genzyme Corporation; 2020 October.
- Simulect (basiliximab) [prescribing information]. East Hanover: Novartis Pharmaceuticals Corporation; 2020 August.
- Knight RJ, Kerman RH, Schoenberg L, et al. The selective use of basiliximab versus thymoglobulin in combination with sirolimus for cadaveric renal transplant recipients at low risk versus high risk for delayed graft function. Transplantation 2004 Sep 27 78(6):904–910.
- Brakemeier S, Kannenkeril D, Durr M, et al. Experience with belatacept rescue therapy in kidney transplant recipients. Transpl Int 2016 Nov;29(11):1184–1195.
- Martinez JM, Pulido LB, Bellido CB, et al. Rescue immunosuppression with mammalian target of rapamycin inhibitor drugs in liver transplantation. Transplant Proc 2010 Mar;42(2):641–643.
- Hariharan S, Israni AK, Danovitch G. Long-term survival after kidney transplantation. N Engl J Med 2021 Aug 19; 385(8):729–743.
- Koppelstaetter C, Kern G, Leierer G, et al. Effect of cyclosporine, tacrolimus and sirolimus on cellular senescence in renal epithelial cells. Toxicol In Vitro 2018 Apr;48:86–92.
- Ojo AO. Renal disease in recipients of nonrenal solid organ transplantation. Semin Nephrol 2007 Jul;27(4):498–507.
- Fardet L, Flahault A, Kettaneh A, et al. Corticosteroid-induced clinical adverse events: frequency, risk factors and patient’s opinion. Br J Dermatol 2007 Jul;157(1):142–148.
- Li XC, Rothstein DM, Sayegh MH. Costimulatory pathways in transplantation: challenges and new developments. Immunol Rev 2009 May;229(1):271–293.
- Kawai K, Uchiyama M, Hester J, et al. Regulatory T cells for tolerance. Hum Immunol 2018 May;79(5):294–303.
- Massart A, Ghisdal L, Abramowicz M, et al. Operational tolerance in kidney transplantation and associated biomarkers. Clin Exp Immunol 2017 Aug;189(2):138–157.
- Romano M, Fanelli G, Albany CJ, et al. Present, and future of regulatory t cell therapy in transplantation and autoimmunity. Front Immunol 2019;10:43.
- Paez-Mayorga J, Lukin I, Emerich D, et al. Emerging strategies for beta cell transplantation to treat diabetes. Trends Pharmacol Sci 2021 Dec 6;43(3):221–233.
- Wu S, Wang L, Fang Y, et al. Advances in encapsulation and delivery strategies for islet transplantation. Adv Healthc Mater 2021 Oct;10(20):e2100965.
- Farina M, Alexander JF, Thekkedath U, et al. Cell encapsulation: overcoming barriers in cell transplantation in diabetes and beyond. Adv Drug Deliv Rev. 2019 Jan 15;139:92–115. DOI:10.1016/j.addr.2018.04.018.
- Dimitrioglou N, Kanelli M, Papageorgiou E, et al. Paving the way for successful islet encapsulation. Drug Discov Today 2019 Mar;24(3):737–748.
- Desai T, Shea LD. Advances in islet encapsulation technologies. Nat Rev Drug Discov 2017 May;16(5):338–350.
- Sneddon JB, Tang Q, Stock P, et al. Stem cell therapies for treating diabetes: progress and remaining challenges. Cell Stem Cell 2018 Jun 1; 22(6):810–823.
- Rickels MR. Stem cell-derived islets: next steps for histologic and functional assessment during development as a cellular therapy for the treatment of diabetes. Diabetes 2019 May;68(5):901–903.
- Shapiro AMJ, Thompson D, Donner TW, et al. Insulin expression and C-peptide in type 1 diabetes subjects implanted with stem cell-derived pancreatic endoderm cells in an encapsulation device. Cell Reports Medicine 2021 ;2(12):100466.
- O’Sullivan ES, Vegas A, Anderson DG, et al. Islets transplanted in immunoisolation devices: a review of the progress and the challenges that remain. Endocr Rev 2011 Dec;32(6):827–844.
- Papas KK, De Leon H, Suszynski TM, et al. Oxygenation strategies for encapsulated islet and beta cell transplants. Adv Drug Deliv Rev 2019 Jan 15;139:139–156. DOI:10.1016/j.addr.2019.05.002.
- Carlsson PO, Espes D, Sedigh A, et al. Transplantation of macroencapsulated human islets within the bioartificial pancreas betaAir to patients with type 1 diabetes mellitus. Am J Transplant 2018 Jul;18(7):1735–1744.
- Ludwig B, Reichel A, Steffen A, et al. Transplantation of human islets without immunosuppression. Proc Natl Acad Sci U S A 2013 Nov 19; 110(47):19054–19058.
- Wang X, Maxwell KG, Wang K, et al. A nanofibrous encapsulation device for safe delivery of insulin-producing cells to treat type 1 diabetes. Sci Transl Med 2021 Jun 2;13(596).
- Wang LH, Ernst AU, Flanders JA, et al. An inverse-breathing encapsulation system for cell delivery. Sci Adv 2021 May;7(20). doi:10.1126/sciadv.abd5835.
- Gamble A, Pepper AR, Bruni A, et al. The journey of islet cell transplantation and future development. Islets 2018 Mar 4; 10(2):80–94.
- Ruers TJ, Daemen MJ, Thijssen HH, et al. Sensitivity of graft rejection in rats to local immunosuppressive therapy. Transplantation. 1988 Dec;46(6):820–825.
- Shirbacheh MV, Jones JW, Harralson TA, et al. Pharmacokinetics of intra-arterial delivery of tacrolimus to vascularly isolated rabbit forelimb. J Pharmacol Exp Ther 1999 Jun;289(3):1196–1201.
- Freise CE, Clemmings S, Clemens LE, et al. Demonstration of local immunosuppression with methylprednisolone in the sponge matrix allograft model. Transplantation 1991 Aug;52(2):318–325.
- Gruber SA. Locoregional immunosuppression of organ transplants. Immunol Rev 1992 Oct;129(1):5–30.
- Buchwald P, Bocca N, Marzorati S, et al. Feasibility of localized immunosuppression: 1. Exploratory studies with glucocorticoids in a biohybrid device designed for cell transplantation. Pharmazie. 2010 Jun;65(6):421–428.
- Headen DM, Woodward KB, Coronel MM, et al. Local immunomodulation Fas ligand-engineered biomaterials achieves allogeneic islet graft acceptance. Nat Mater. 2018 Aug;17(8):732–739.
- Skoumal M, Woodward KB, Zhao H, et al. Localized immune tolerance from FasL-functionalized PLG scaffolds. Biomaterials. 2019 Feb;192:271–281.
- Waring P, Mullbacher A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol. 1999 Aug;77(4):312–317.
- Coronel MM, Martin KE, Hunckler MD, et al. Immunotherapy via PD-L1-presenting biomaterials leads to long-term islet graft survival. Sci Adv. 2020 Aug;6(35):eaba5573.
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010 Jul;236(1):219–242.
- Orr S, Strominger I, Eremenko E, et al. TGF-β affinity-bound to a macroporous alginate scaffold generates local and peripheral immunotolerant responses and improves allocell transplantation. Acta Biomater. 2016 ;45:196–209.
- Hering BJ, Clarke WR, Bridges ND, et al. Phase 3 Trial of Transplantation of Human Islets in Type 1 Diabetes Complicated by Severe Hypoglycemia. Diabetes Care 2016 Jul;39(7):1230–1240.
- Shapiro AM, Pokrywczynska M, Ricordi C. Clinical pancreatic islet transplantation. Nat Rev Endocrinol. 2017 May;13(5):268–277.
- Niclauss N, Bosco D, Morel P, et al. Rapamycin impairs proliferation of transplanted islet beta cells. Transplantation. 2011 Apr 15; 91(7):714–722.
- Nishimura R, Nishioka S, Fujisawa I, et al. Tacrolimus inhibits the revascularization of isolated pancreatic islets. PLoS One. 2013;8(4):e56799.
- Berman DM, Ruiz P Jr., Blandino-Rosano M, et al. Steroid-free immune suppression impairs glycemic control in a healthy cynomolgus monkey. Cell Transplant. 2019 Mar;28(3):262–268.
- Xu C, Niu YJ, Liu XJ, et al. Tacrolimus reversibly reduces insulin secretion, induces insulin resistance, and causes islet cell damage in rats. Int J Clin Pharmacol Ther. 2014 Jul;52(7):620–627.
- Shevach EM, Davidson TS, Huter EN, et al. Role of TGF-Beta in the induction of Foxp3 expression and T regulatory cell function. J Clin Immunol. 2008 Nov;28(6):640–646.
- Fisher JD, Balmert SC, Zhang W, et al. Treg-inducing microparticles promote donor-specific tolerance in experimental vascularized composite allotransplantation. Proceedings of the National Academy of Sciences 2019;116( 51):25784–25789.
- Bassin EJ, Buckley AR, Piganelli JD, et al. TRI microparticles prevent inflammatory arthritis in a collagen-induced arthritis model. PLOS ONE. 2020;15(9):e0239396.
- Liu JMH, Zhang J, Zhang X, et al. Transforming growth factor-beta 1 delivery from microporous scaffolds decreases inflammation post-implant and enhances function of transplanted islets. Biomaterials. 2016 Feb;80:11–19.
- Li Y, Frei AW, Labrada IM, et al. Immunosuppressive PLGA TGF-beta1 microparticles induce polyclonal and antigen-specific regulatory t cells for local immunomodulation of allogeneic islet transplants. Front Immunol. 2021;12:653088.
- Liu JMH, Zhang X, Joe S, et al. Evaluation of biomaterial scaffold delivery of IL-33 as a localized immunomodulatory agent to support cell transplantation in adipose tissue. J Immunol Regen Med. 2018 Mar;1:1–12.
- Liew FY, Girard J-P, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016 ;16(11):676–689.
- Schroder PM, Fitch ZW, Schmitz R, et al. The past, present, and future of costimulation blockade in organ transplantation. Curr Opin Organ Transplant. 2019 Aug;24(4):391–401.
- Bluestone JA, St Clair EW, Turka LA. CTLA4Ig: bridging the basic immunology with clinical application. Immunity. 2006 Mar;24(3):233–238.
- Judge TA, Tang A, Spain LM, et al. The in vivo mechanism of action of CTLA4Ig. J Immunol. 1996 Mar 15;156(6):2294–2299.
- Hardinger KL, Sunderland D, Wiederrich JA. Belatacept for the prophylaxis of organ rejection in kidney transplant patients: an evidence-based review of its place in therapy. Int J Nephrol Renovasc Dis. 2016;9:139–150.
- Posselt AM, Szot GL, Frassetto LA, et al. Islet transplantation in type 1 diabetic patients using calcineurin inhibitor-free immunosuppressive protocols based on T-cell adhesion or costimulation blockade. Transplantation. 2010 Dec 27; 90(12):1595–1601.
- McDonald-Hyman C, Turka LA, Blazar BR. Advances and challenges in immunotherapy for solid organ and hematopoietic stem cell transplantation. Sci Transl Med. 2015 Mar 25; 7(280):280rv2.
- Tikkanen JM, Lemstrom KB, Koskinen PK. Blockade of CD28/B7-2 costimulation inhibits experimental obliterative bronchiolitis in rat tracheal allografts: suppression of helper T cell type1-dominated immune response. Am J Respir Crit Care Med. 2002 Mar 1; 165(5):724–729.
- Paez-Mayorga J, Capuani S, Hernandez N, et al. Neovascularized implantable cell homing encapsulation platform with tunable local immunosuppressant delivery for allogeneic cell transplantation. Biomaterials. 2020 Oct;257:120232.
- Zhang W, Gorantla VS, Campbell PG, et al. Biopatterned CTLA4/Fc matrices facilitate local immunomodulation, engraftment, and glucose homeostasis after pancreatic islet transplantation. Diabetes. 2016;65(12):3660–3666.
- Pinelli DF, Ford ML. Novel insights into anti-CD40/CD154 immunotherapy in transplant tolerance. Immunotherapy. 2015;7(4):399–410.
- Kenyon NS, Chatzipetrou M, Masetti M, et al. Long-term survival and function of intrahepatic islet allografts in rhesus monkeys treated with humanized anti-CD154. Proc Natl Acad Sci U S A. 1999 Jul 6; 96(14):8132–8137.
- Larsen CP, Elwood ET, Alexander DZ, et al. Long-term acceptance of skin and cardiac allografts after blocking CD40 and CD28 pathways. Nature. 1996 May 30; 381(6581):434–438.
- Habiro K, Kotani M, Omoto K, et al. Mechanism of allorecognition and skin graft rejection in CD28 and CD40 ligand double-deficient mice. Transplantation. 2003 Sep 15; 76(5):854–858.
- Kim SC, Wakwe W, Higginbotham LB, et al. Fc-silent anti-CD154 domain antibody effectively prevents nonhuman primate renal allograft rejection. Am J Transplant. 2017 May;17(5):1182–1192.
- Elwood ET, Larsen CP, Cho HR, et al. Prolonged acceptance of concordant and discordant xenografts with combined CD40 and CD28 pathway blockade. Transplantation. 1998 Jun 15; 65(11):1422–1428.
- Boumpas DT, Furie R, Manzi S, et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003 Mar;48(3):719–727.
- Ramanujam M, Steffgen J, Visvanathan S, et al. Phoenix from the flames: rediscovering the role of the CD40-CD40L pathway in systemic lupus erythematosus and lupus nephritis. Autoimmun Rev. 2020 Nov;19(11):102668.
- Mohty M. Mechanisms of action of antithymocyte globulin: t-cell depletion and beyond. Leukemia. 2007 Jul;21(7):1387–1394.
- Nanmoku K, Shinzato T, Kubo T, et al. Effect of rabbit antithymocyte globulin on acute and chronic active antibody-mediated rejection after kidney transplantation. Transplant Proc. 2019 Oct;51(8):2602–2605.
- Woodside KJ, Hu M, Gugliuzza KK, et al. T-lymphocyte apoptosis is increased by non-interleukin-2-dependent induction in human mixed lymphocyte cultures. Transplant Proc. 2005 May;37(4):1949–1952.
- Mullard A. FDA approves 100th monoclonal antibody product. Nat Rev Drug Discov. 2021 Jul;20(7):491–495.
- Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol. 2011 Aug 15; 187(4):2015–2022.
- Kuhn C, Weiner HL. Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside. Immunotherapy. 2016 Jul;8(8):889–906.
- Bartneck M, Peters FM, Warzecha KT, et al. Liposomal encapsulation of dexamethasone modulates cytotoxicity, inflammatory cytokine response, and migratory properties of primary human macrophages. Nanomedicine. 2014 Aug;10(6):1209–1220.
- Yang C, Mariner PD, Nahreini JN, et al. Cell-mediated delivery of glucocorticoids from thiol-ene hydrogels. J Control Release. 2012 Sep 28; 162(3):612–618.
- Jiang K, Weaver JD, Li Y, et al. Local release of dexamethasone from macroporous scaffolds accelerates islet transplant engraftment by promotion of anti-inflammatory M2 macrophages. Biomaterials. 2017 Jan;114:71–81.
- Martinez FO, Sica A, Mantovani A, et al. Macrophage activation and polarization. Front Biosci. 2008 Jan 1; 13(13):453–461.
- Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond). 1998 Jun;94(6):557–572.
- Liang JP, Accolla RP, Jiang K, et al. Controlled release of anti-inflammatory and proangiogenic factors from macroporous scaffolds. Tissue Eng Part A. 2021 Oct;27(19–20):1275–1289.
- Kuppan P, Kelly S, Polishevska K, et al. Co-localized immune protection using dexamethasone-eluting micelles in a murine islet allograft model. Am J Transplant. 2020 Mar;20(3):714–725.
- Chun J, Hartung H-P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101.
- Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010 Nov;9(11):883–897.
- Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004 Jul;4(7):1019–1025.
- Takasaki T, Hagihara K, Satoh R, et al. More than just an immunosuppressant: the emerging role of FTY720 as a novel inducer of ROS and Apoptosis. Oxid Med Cell Longev. 2018 ;2018:4397159.
- Lu B, Gao Q, Liu R, et al. Effect of a new drug releasing system on microencapsulated islet transplantation. Int J Clin Exp Pathol. 2015;8(10):12390–12399.
- Awojoodu AO, Ogle ME, Sefcik LS, et al. Sphingosine 1-phosphate receptor 3 regulates recruitment of anti-inflammatory monocytes to microvessels during implant arteriogenesis. Proc Natl Acad Sci U S A. 2013 Aug 20; 110(34):13785–13790.
- Frei AW, Li Y, Jiang K, et al. Local delivery of fingolimod from three-dimensional scaffolds impacts islet graft efficacy and microenvironment in a murine diabetic model. J Tissue Eng Regen Med. 2018 Feb;12(2):393–404.
- Bowers DT, Olingy CE, Chhabra P, et al. An engineered macroencapsulation membrane releasing FTY720 to precondition pancreatic islet transplantation. J Biomed Mater Res B Appl Biomater. 2018 Feb;106(2):555–568.
- Furukawa A, Wisel SA, Tang Q. Impact of immune-modulatory drugs on regulatory T cell. Transplantation. 2016 Nov;100(11):2288–2300.
- Tang Q, Leung J, Melli K, et al. Altered balance between effector T cells and FOXP3+ HELIOS+ regulatory T cells after thymoglobulin induction in kidney transplant recipients. Transpl Int. 2012 Dec;25(12):1257–1267.
- Wang X, Brown NK, Wang B, et al. Local immunomodulatory strategies to prevent allo-rejection in transplantation of insulin-producing cells. Adv Sci (Weinh). 2021 Sep;8(17):e2003708.
- Azzi J, Yin Q, Uehara M, et al. Targeted delivery of immunomodulators to lymph nodes. Cell Rep. 2016 May 10; 15(6):1202–1213.
- Hunter Z, McCarthy DP, Yap WT, et al. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano. 2014 Mar 25; 8(3):2148–2160.
- Horwitz DA, Bickerton S, Koss M, et al. Suppression of murine lupus by CD4+ and CD8+ Treg cells induced by t cell-targeted nanoparticles loaded with interleukin-2 and transforming growth factor β. Arthritis Rheumatol. 2019 Apr;71(4):632–640.
- Hussain B, Kasinath V, Madsen JC, et al. Intra-organ delivery of nanotherapeutics for organ transplantation. ACS Nano. 2021 Oct 29; 15(11):17124–17136.
- McHugh MD, Park J, Uhrich R, et al. Paracrine co-delivery of TGF-beta and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials. 2015 Aug;59:172–181.
- Deng C, Jin Q, Wu Y, et al. Immunosuppressive effect of PLGA-FK506-NPs in treatment of acute cardiac rejection via topical subcutaneous injection. Drug Deliv. 2021 Dec;28(1):1759–1768.
- Azzi J, Tang L, Moore R, et al. Polylactide-cyclosporin A nanoparticles for targeted immunosuppression. FASEB J. 2010 Oct;24(10):3927–3938.
- Bahmani B, Uehara M, Jiang L, et al. Targeted delivery of immune therapeutics to lymph nodes prolongs cardiac allograft survival. J Clin Invest. 2018 Nov 1; 128(11):4770–4786.
- Bisikirska B, Colgan J, Luban J, et al. TCR stimulation with modified anti-CD3 mAb expands CD8+ T cell population and induces CD8+CD25+ Tregs. J Clin Invest. 2005 Oct;115(10):2904–2913.
- Burke JA, Zhang X, Bobbala S, et al., Subcutaneous nanotherapy repurposes the immunosuppressive mechanism of rapamycin to enhance allogeneic islet graft viability. Nat Nanotechnol. 2022 17(3): 319–330.
- Zhu P, Atkinson C, Dixit S, et al. Organ preservation with targeted rapamycin nanoparticles: a pre-treatment strategy preventing chronic rejection in vivo. RSC Adv. 2018 Jul 23 8(46):25909–25919.
- Uehara M, Bahmani B, Jiang L, et al. Nanodelivery of mycophenolate mofetil to the organ improves transplant vasculopathy. ACS Nano. 2019Nov 26; 13(11):12393–12407.
- Heemann U, Kliem V, Budde K, et al. Mycophenolate mofetil maintenance therapy in renal transplant patients: long-term results of the TranCept STAY study. Clin Transplant. 2012 Nov-Dec;26(6):919–926.
- Park K. Facing the truth about nanotechnology in drug delivery. ACS Nano. 2013 Sep 24; 7(9):7442–7447.
- Anselmo AC, Mitragotri S. Nanoparticles in the clinic: an update. Bioeng Transl Med. 2019 Sep;4(3):e10143.
- Li J, Mooney DJ. Designing hydrogels for controlled drug delivery. Nat Rev Mater. 2016 Dec;1(12). DOI:10.1038/natrevmats.2016.71.
- Dzhonova DV, Olariu R, Leckenby J, et al. local injections of tacrolimus-loaded hydrogel reduce systemic immunosuppression-related toxicity in vascularized composite allotransplantation. Transplantation. 2018 Oct;102(10):1684–1694.
- Pathak S, Regmi S, Gupta B, et al. Single synchronous delivery of FK506-loaded polymeric microspheres with pancreatic islets for the successful treatment of streptozocin-induced diabetes in mice. Drug Deliv. 2017 Nov;24(1):1350–1359.
- Wang H, Sobral MC, Snyder T, et al. Clickable, acid labile immunosuppressive prodrugs for in vivo targeting. Biomater Sci. 2020 Jan 1; 8(1):266–277.
- Haque MR, Lee DY, Ahn CH, et al. Local co-delivery of pancreatic islets and liposomal clodronate using injectable hydrogel to prevent acute immune reactions in a type 1 diabetes. Pharm Res. 2014 Sep;31(9):2453–2462.
- Pathak S, Acharya S, Regmi S, et al. Particulate-based single-dose local immunosuppressive regimen for inducing tolerogenic dendritic cells in xenogeneic islet transplantation. Adv Healthc Mater 2021 Jan;10(2):e2001157.
- Barrera P, Blom A, van Lent PL, et al. Synovial macrophage depletion with clodronate-containing liposomes in rheumatoid arthritis. Arthritis Rheum. 2000 Sep;43(9):1951–1959.
- Yin N, Guo X, Sun R, et al. Intra-articular injection of indomethacin–methotrexate in situ hydrogel for the synergistic treatment of rheumatoid arthritis. J Mat Chem B. 2020;8(5):993–1007.
- Wang Y, Chen X, Cao W, et al. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat Immunol. 2014 Nov;15(11):1009–1016.
- Obermajer N, Popp FC, Soeder Y, et al. Conversion of Th17 into IL-17A(neg) regulatory T cells: a novel mechanism in prolonged allograft survival promoted by mesenchymal stem cell-supported minimized immunosuppressive therapy. J Immunol. 2014 Nov 15; 193(10):4988–4999.
- Casiraghi F, Azzollini N, Todeschini M, et al. Localization of mesenchymal stromal cells dictates their immune or proinflammatory effects in kidney transplantation. Am J Transplant. 2012 Sep;12(9):2373–2383.
- Souidi N, Stolk M, Rudeck J, et al. Stromal cells act as guardians for endothelial progenitors by reducing their immunogenicity after co-transplantation. Stem Cells. 2017 May;35(5):1233–1245.
- Lohan P, Murphy N, Treacy O, et al. Third-party allogeneic mesenchymal stromal cells prevent rejection in a pre-sensitized high-risk model of corneal transplantation. Front Immunol. 2018;9:2666.
- Arzouni AA, Vargas-Seymour A, Nardi N, et al. Using mesenchymal stromal cells in islet transplantation. Stem Cells Transl Med. 2018 Aug;7(8):559–563.
- Kenyon NS, Willman MA, Han D, et al. Extended survival versus accelerated rejection of nonhuman primate islet allografts: effect of mesenchymal stem cell source and timing. Am J Transplant. 2021 Nov;21(11):3524–3537.
- Wang H, Strange C, Nietert PJ, et al. Autologous mesenchymal stem cell and islet cotransplantation: safety and efficacy. Stem Cells Transl Med. 2018 Jan;7(1):11–19.
- Tan J, Wu W, Xu X, et al. Induction therapy with autologous mesenchymal stem cells in living-related kidney transplants: a randomized controlled trial. Jama. 2012 Mar 21; 307(11):1169–1177.
- Casiraghi F, Perico N, Gotti E, et al. Kidney transplant tolerance associated with remote autologous mesenchymal stromal cell administration. Stem Cells Transl Med. 2020 Apr;9(4):427–432.
- Hutchinson JA, Geissler EK. Now or never? The case for cell-based immunosuppression in kidney transplantation. Kidney Int. 2015 Jun;87(6):1116–1124.
- Scalea JR, Lee YS, Davila E, et al. Myeloid-derived suppressor cells and their potential application in transplantation. Transplantation. 2018 Mar;102(3):359–367.
- Qin J, Arakawa Y, Morita M, et al. C-C chemokine receptor type 2-dependent migration of myeloid-derived suppressor cells in protection of islet transplants. Transplantation. 2017 Aug;101(8):1793–1800.
- Cao P, Sun Z, Feng C, et al. Myeloid-derived suppressor cells in transplantation tolerance induction. Int Immunopharmacol. 2020 Jun;83:106421.
- He Y, Wang B, Jia B, et al. Effects of adoptive transferring different sources of myeloid-derived suppressor cells in mice corneal transplant survival. Transplantation. 2015 Oct;99(10):2102–2108.
- Marigo I, Bosio E, Solito S, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010 Jun 25; 32(6):790–802.
- Nakamura T, Nakao T, Yoshimura N, et al. Rapamycin prolongs cardiac allograft survival in a mouse model by inducing myeloid-derived suppressor cells. Am J Transplant. 2015 Sep;15(9):2364–2377.
- Dugast AS, Haudebourg T, Coulon F, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. 2008 Jun 15; 180(12):7898–7906.
- Carretero-Iglesia L, Bouchet-Delbos L, Louvet C, et al. Comparative study of the immunoregulatory capacity of in vitro generated tolerogenic dendritic cells, suppressor macrophages, and myeloid-derived suppressor cells. Transplantation 2016 Oct;100(10):2079–2089.
- Sawitzki B, Harden PN, Reinke P, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet. 2020 May 23; 395(10237):1627–1639.
- Todo S, Yamashita K, Goto R, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016 Aug;64(2):632–643.
- Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014 Mar;14(3):195–208.
- Pang XL, Wang ZG, Liu L, et al. Immature dendritic cells derived exosomes promotes immune tolerance by regulating T cell differentiation in renal transplantation. Aging (Albany NY). 2019 Oct 26 11(20):8911–8924.
- Keshtkar S, Azarpira N, Ghahremani MH. Mesenchymal stem cell-derived extracellular vesicles: novel frontiers in regenerative medicine. Stem Cell Res Ther. 2018 Mar 9; 9(1):63.
- Dal Collo G, Adamo A, Gatti A, et al. Functional dosing of mesenchymal stromal cell-derived extracellular vesicles for the prevention of acute graft-versus-host-disease. Stem Cells. 2020;38(5):698–711.
- Fujii S, Miura Y, Fujishiro A, et al. Graft-versus-host disease amelioration by human bone marrow mesenchymal stromal/stem cell-derived extracellular vesicles is associated with peripheral preservation of naive t cell populations. Stem Cells. 2018 Mar;36(3):434–445.
- Wang L, Gu Z, Zhao X, et al. Extracellular vesicles released from human umbilical cord-derived mesenchymal stromal cells prevent life-threatening acute graft-versus-host disease in a mouse model of allogeneic hematopoietic stem cell transplantation. Stem Cells Dev. 2016 Dec 15; 25(24):1874–1883.
- Rani S, Ryan AE, Griffin MD, et al. Mesenchymal stem cell-derived extracellular vesicles: toward cell-free therapeutic applications. Mol Ther. 2015 May;23(5):812–823.
- Wu JY, Li YJ, Hu XB, et al. Preservation of small extracellular vesicles for functional analysis and therapeutic applications: a comparative evaluation of storage conditions. Drug Deliv. 2021 Dec;28(1):162–170.
- Grange C, Bellucci L, Bussolati B, et al. Potential applications of extracellular vesicles in solid organ transplantation. Cells. 2020;9(2):369.
- Farina M, Chua CYX, Ballerini A, et al. Transcutaneously refillable, 3D-printed biopolymeric encapsulation system for the transplantation of endocrine cells. Biomaterials. 2018 Sep;177:125–138.
- Farina M, Ballerini A, Fraga DW, et al. 3D printed vascularized device for subcutaneous transplantation of human islets. Biotechnol J. 2017 Sep;12(9):1700169.
- Paez-Mayorga J, Capuani S, Farina M, et al. Enhanced in vivo vascularization of 3d-printed cell encapsulation device using platelet-rich plasma and mesenchymal stem cells. Adv Healthc Mater 2020 Oct;9(19):e2000670.
- Grattoni A, Paez-Mayorga J, Capuani S. Transcutaneously refillable cell confinement platform with local trophic factor delivery. U.S. Provisional Pat. Ser. No. 63/043,439. United States. 2020 June 24, 2020.
- Paez-Mayorga J, Campa-Carranza J, Capuani S, et al. Implantable niche with local immunosuppression for islet allotransplantation achieves type 1 diabetes reversal. Nature Portfolio [Preprint]. 2022. https://doi.org/10.21203/rs.3.rs-1285406/v1
- Deuse T, Hu X, Gravina A, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol. 2019 Mar;37(3):252–258.
- Niu D, Wei HJ, Lin L, et al. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science. 2017 Sep 22; 357(6357):1303–1307.
- Yang L, Güell M, Niu D, et al. Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science. 2015 Nov 27; 350(6264):1101–1104.
- Porrett PM, Orandi BJ, Kumar V, et al. First clinical-grade porcine kidney xenotransplant using a human decedent model. American Journal of Transplantation. 2022;22(4): 1037–1053. doi:10.1111/ajt.16930.