
The goal of drug therapy has always been to achieve optimal drug efficacy and safety for every individual. Although pharmacotherapy has come far, this goal remains elusive. From a historical perspective, observations that an individual’s response to a medication was associated with their ethnicity were made, but a genetic link was not pursued until the 1950s and 1960s [Citation1]. Early studies identified disparate drug exposure among individuals’ self-reported membership to different racial/ethnic groups, yet there were other factors that needed elucidation to achieve the goal of personalizing medication therapy. This ignited a multitude of clinical studies linking genetic variation and drug-metabolizing enzyme activity. Thereby the fields of pharmacogenetics and later pharmacogenomics were born. These early studies led to the concept of ‘metabolic phenotype’ in which individual designations such as ‘poor metabolizer’ were assigned. Soon, variation in cytochrome P450 genomes and altered enzyme activity arose as a major factor in drug exposure. However, the need for increased precision in the selection and dosing of medications was still evident.
With the arrival of genomics research, a surge of research activity was undertaken to elucidate the role of genetic variation in drug exposure, disposition, and response. The subtleties of ethnicity still lingered, but most studies continued to be conducted among primarily Caucasian populations. Highly prevalent polymorphisms were identified within the exons of major drug-metabolizing CYP450s such as CYP2D6 and CYP2C19. Intensive research into CYP450-mediated drug metabolism showed that only a handful of CYP450 enzymes were responsible for the majority of drug metabolism. These enzymes are CYP1A2, 2C9, 2C19, 2D6, and 3A4/5 [Citation2]. Of these, CYP2D6 has shown profound variation with hundreds of polymorphisms in addition to copy number variation with new polymorphisms being discovered regularly. Although allele frequencies for CYP2D6 differ among different racial ethnic groups, the large interindividual variability in CYP2D6 activity dwarfs racial/ethnic differences. Not surprisingly, the human CYP2D6 enzyme exhibits approximately 1000-fold activity variation. The profound variability in CYP2D6 corroborated the profound variability in human response to drugs which are primary CYP2D6 substrates. Although significant progress was made, multiple schemes for prediction of CYP2D6 phenotype based on genetics were advanced. Complex examples like CYP2D6 phenotype remain controversial while the low-hanging fruit of pharmacogenomics would seem to be exonic polymorphisms or deletions that have dramatic impacts on function.
Despite controversies in phenotype prediction, the first commercial product to predict metabolic phenotype based on CYP2D6 and CYP2C19 genotype was the Amplichip® [Citation3]. The technology used PCR amplification followed by hybridization to known DNA polymorphisms to identify the subject genotype. Phenotypes of CYP2D6 were then predicted based on genotype and designated as: Poor Metabolizer (PM), Intermediate Metabolizer (IM), Extensive (normal) Metabolizer (EM), and Ultra-rapid Metabolizer (UM). Limitations of such approaches include the lack of accurate results for unknown genotypes and misclassification of phenotype due to errors in genetic predictions [Citation4,Citation5]. Because metabolic phenotype is subject to environmental exposures (concomitant disease(s) and drug therapy, dietary or lifestyle choices), genetic predictions are often extrapolated based on in vitro or other available data. These extrapolations become even more tenuous when complex genotypes are present, such as gene duplications containing alleles coding for enzymes with poorly characterized activity [Citation6]. Today, a multitude of commercial genetic panels are produced for clinical application, accompanying the results are recommendations for avoiding or preferring certain drugs, and/or dose adjustments. Utilization of these tests remains sporadic in modern clinical practice. As with the ethnic group approach, there has been a realization that genetic testing has value, but lacks the precision for optimal medication selection and dose for every individual. There remains a clinical need to achieve even greater precision to maximize drug efficacy and safety.
CYP3A5 variants are exemplary targets of investigation in pharmacogenetic/omic study. Only the CYP3A5*1 allele codes for active enzyme, while all other variants found code for inactive enzyme. If we could assess CYP3A5 activity independently, it would be an ideal pharmacogenetic target for drug–gene interaction studies. However, study of CYP3A5 activity is much more complex than it would first appear. Functional CYP3A5 is rare among Caucasian populations and even when expressed in Caucasians it is not expressed to a large extent. Since most early pharmacogenetic studies were conducted on primarily Caucasians, CYP3A5 activity has been largely neglected until recently [Citation7]. Study of CYP3A5 activity is also complicated by the fact that its closely related homologue CYP3A4 is highly expressed in most individuals and is highly homologous in structure and function. The enzyme pair are extremely important, as they are responsible for metabolizing approximately 50% of drugs. Unlike other CYP450 enzymes, CYP3A4 does not have a clear link between genetics and enzyme function as few polymorphisms have been found. Another perplexing aspect of CYP3A4/5 activity is the lack of consistency in measures of their variability. Studies range from estimates of 10-fold in small populations up to 50-fold or even 200-fold in other studies [Citation8]. Proposed explanations for the large ranges of variability include enzyme induction from environment or other drugs and drug–drug interactions. Some overlooked aspects of the variability are the fact that the two enzymes have similar, but not exact, specificity. Therefore, substrates will be preferentially metabolized by one over the other, such as the way tacrolimus is preferentially metabolized by CYP3A5. When one considers the enhanced variability due to the relative contributions of CYP3A4/5 to metabolism along with the relative expression of CYP3A4/5 in different populations, then enhanced variability among different studies examining different drugs in different populations seems likely.
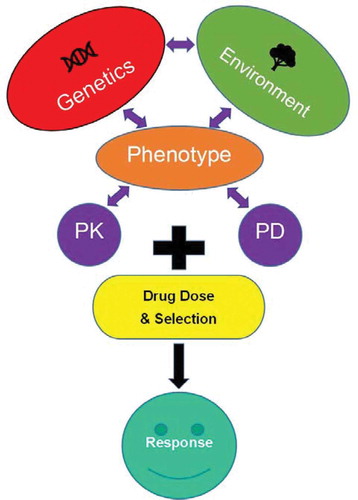
A common assumption made in population genetics is that different spans of DNA share the same chance of mutation. This assumption is less true for genes coding products on two extremes [Citation9]. First, genes with vital physiologic functions are less likely to vary because variants could be lethal. Second, this assumption is often violated when genes code for products with strong environmental interaction. The CYP450 xenobiotic metabolizing families of enzymes are examples of genes with strong environmental interaction. As such, there is a higher rate of mutation and humans from different regions of the world exhibit strong differences in variant allele composition and frequency. However, enzyme function, vis-a-vis metabolic phenotype, is dynamic and a function of epigenetic, transcriptional, translational, and posttranscriptional regulatory mechanisms. In this complex network, enzymes may interact with other proteins subject to a similar constellation of biological forces. Phenotype is the result of the complex interplay between genetics and environment (See ). Regarding drug response, an individual’s respective phenotypes can then be categorized as pharmacokinetic (PK) or pharmacodynamic (PD) in nature. The resulting drug response will then be a function of the PK/PD phenotypic traits interaction with the drug selected and its dose. Only future approaches which contemplate systems biology will be able to predict PK and PD phenotypes accurately at an individual level on a broad scale. To get there, we have quite a long way to go.
Figure 1. Major components determining an individual’s response to pharmacotherapy. The individual’s genome, environmental factors including concomitant disease(s) (e.g. infection, inflammation, hepatitis, tumor), the impact of drugs that induce or inhibit liver enzymes, and dietary/lifestyle choices (e.g. cigarette smoking), along with the pharmacokinetic and pharmacodynamic parameters of a medication and its dose.
With the increasing ease of large-scale genotyping, genome-wide association studies with thousands of participants have been initiated. However, these studies need to encompass global populations to characterize the entire range of human variability. The 1000 Genomes Project is an example of a study with a global focus [Citation10]. In addition, methods to identify polygenic responses need to be developed. Another issue with many genetic studies is that they do not account for environmental influences. The large ‘All of Us’ project entails a large U.S. cohort study which will incorporate monitoring of biomarkers, environment, and genetics to examine relationships with heath and disease [Citation11]. These efforts represent steps in the right direction.
One necessary tool for future systems biology approaches will be readily available phenotyping tools to predict drug response quickly, easily, and accurately. CYP450 enzyme activity/metabolic phenotype is a prime example of an enzyme system which can be accurately determined in vivo. After a subject ingests a substrate probe, metabolic phenotype can be assessed by measuring the parent to product ratio at a specific time post ingestion. Direct assessment of metabolic phenotype addresses the PK component of drug response as outlined in . Modeling programs such as SimCYP, a physiologically based PK model, can perform in vitro to in vivo extrapolation and provide the vital link between in vitro metabolic enzyme activity and metabolic phenotype [Citation12]. Such programs are an excellent example of integrating systems biology and clinical characteristics to predict population level PK and PD responses. Next steps should focus on integrating PK and PD phenotypic characterization in a similar manner to predict an individual’s drug response.
1. Expert opinion
Despite major advances, the goal of individualized drug therapy remains elusive. Pharmacogenetics began with observed racial/ethnic differences in drug response, toxicity, and pharmacokinetics. Scientists discovered genetic polymorphisms in drug-metabolizing enzymes were associated with significant differences in drug clearance and exposure. Soon pharmacogenomic studies expanded the breadth of genes investigated and found new genetic variants responsible for variability in response. Early on, these fields identified that cytochrome P450 drug-metabolizing enzymes played a vital role in drug metabolism and exposure. CYP450 enzymes are the rate-limiting step in the elimination of most drugs. Within this enzyme family, a short list of CYP450 enzymes are responsible for most drug metabolism (CYP1A2, 2C9, 2C19, 2D6, and 3A4/5). A common procedure in pharmacogenetics research was prediction of categorical metabolic phenotypes based on genotypes. Categories such as ‘poor metabolizer’ were conferred when someone carried an allele or alleles(s) associated with lower enzyme activity for a specific CYP450 isoform.
Although genotyping techniques to predict metabolic phenotype account for variability, there are several major problems with this approach. Many variants have been identified which have an unknown or ambiguous impact on phenotype. New polymorphisms with questionable activity are even more problematic when they are found in complex genes such as those exhibiting copy number variants. In the case of CYP2D6, there are so many polymorphisms, including copy number variants, it is quite difficult to link genotype with phenotype. Another major issue is that genotyping does not account for any of the many environmental factors (concomitant disease(s) and drug therapy, dietary or lifestyle choices) which may impact phenotype. While genetics are fixed, phenotype is dynamic and is forged by the interplay between genetics and environment. Challenges in genotypic approaches and a lack of clinical utility outcomes studies have slowed clinical adoption of genotyping approaches. Future approaches that integrate systems biology data, environmental data, and clinical data offer the most promise to accurately characterize PK and PD phenotypes on an individual basis. Once we have characterized an individuals’ phenotype accurately, drug dose and selection can then be optimized.
Declaration of interest
J McGraw is the President and Chief Scientific Officer of Microlitics LLC, a diagnostic company start-up which does not have marketed products as of yet. Armin Gerhardt is Chief Executive officer of Microlitics LLC, a diagnostic company start-up which does not have marketed products as of yet. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer declaration of interest
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Meyer UA. Pharmacogenetics - five decades of therapeutic lessons from genetic diversity. Nat Rev Genet. 2004;5(9):669–676. PubMed PMID: 15372089.
- McGraw J. Chapter 16 - CYP450 and ethnicity A2. In: Padmanabhan, Sandosh, editor. Handbook of pharmacogenomics and stratified medicine. San Diego: Academic Press; 2014. p. 323–340.
- de Leon J. AmpliChip CYP450 test: personalized medicine has arrived in psychiatry. Expert Rev Mol Diagn. 2006;6(3):277–286. PubMed PMID: 16706732.
- Saladores PH, Precht JC, Schroth W, et al. Impact of metabolizing enzymes on drug response of endocrine therapy in breast cancer. Expert Rev Mol Diagn. 2013;13(4):349–365. PubMed PMID: 23638818.
- De Andrés F, Terán S, Hernández F, et al. To genotype or phenotype for personalized medicine? CYP450 drug metabolizing enzyme genotype–phenotype concordance and discordance in the Ecuadorian population. OMICS: J Integr Biol. 2016;20(12):699–710. .
- Gao J, Tian X, Zhou J. et al. From genotype to phenotype: cytochrome P450 2D6-mediated drug clearance in humans. Mol Pharm. 2017; 14(3): 649–657. Epub 2017/ 02/18 PubMed PMID: 28211700.
- Flockhart DA, Rae JM. Cytochrome P450 3A pharmacogenetics: the road that needs traveled. Pharmacogenomics J. 2003;3:3.
- McGraw J, Waller D. Cytochrome P450 variations in different ethnic populations. Expert Opin Drug Metab Toxicol. 2012;8(3):371–382. PubMed PMID: 22288606.
- De S, Lopez-Bigas N, Teichmann SA. Patterns of evolutionary constraints on genes in humans. BMC Evol Biol. 2008;8:275. Epub 2008/10/09. PubMed PMID: 18840274; PubMed Central PMCID: PMCPMC2587479.
- The Genomes Project C. A global reference for human genetic variation. Nature.2015;526:68. https://www.nature.com/articles/nature15393#supplementary-information
- Kuehn BM. Study recruitment to accelerate precision medicine. JAMA. 2018;319(4):332.
- Barter ZE, Tucker GT, Rowland-Yeo K. Response to “Ethnic-specific in vitro-in vivo extrapolation and physiologically based pharmacokinetic approaches to predict cytochrome P450-mediated pharmacokinetics in Chinese population: opportunities and challenges”. Clin Pharmacokinet. 2014;53(2):203. Epub 2013/ 11/21.