
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Objectives: Riociguat is a soluble guanylate cyclase stimulator licensed for the treatment of pulmonary arterial hypertension (PAH), a potentially fatal complication of human immunodeficiency virus infection. This study investigated the inhibitory potency of selected antiretroviral regimens on the metabolic clearance of riociguat.
Methods: The inhibitory potential of the components of six antiretroviral combinations (ATRIPLA® (efavirenz/emtricitabine/tenofovir disoproxil), COMPLERA® (rilpivirine/emtricitabine/tenofovir disoproxil), STRIBILD® (elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil), TRIUMEQ® (abacavir/dolutegravir/lamivudine), and two ritonavir-boosted regimens) on riociguat metabolism were evaluated in recombinant human CYP1A1 and CYP3A4 as well as in human hepatocytes exhibiting both CYP1A1 and CYP3A4 activity. In vitro-in vivo correlation was performed between calculated and observed increases in riociguat exposure in vivo.
Results: Using both in vitro systems, the predicted increase in exposure of riociguat was highest with components of TRIUMEQ® followed by COMPLERA®, ATRIPLA®, STRIBILD®, and the ritonavir-boosted regimens. Further experiments in human hepatocytes confirmed CYP1A1 to be the predominant enzyme in the metabolic clearance of riociguat.
Conclusion: Antiretroviral treatment containing the potent CYP1A1 inhibitor abacavir had the greatest impact on riociguat metabolic clearance. The impact of comedications containing only strong CYP3A4 inhibitors e.g. ritonavir was less pronounced, suggesting a benefit of riociguat over PAH-targeting medications with contraindications for use with strong CYP3A4 inhibitors.
1. Introduction
Pulmonary arterial hypertension (PAH) is a potentially fatal complication of human immunodeficiency virus (HIV) infection [Citation1,Citation2]. The pathology of PAH associated with HIV is assumed to be similar to idiopathic PAH and hence PAH treatment guidelines recommend that PAH-targeted medical therapies should be used [Citation3,Citation4], considering co-morbidities and drug–drug interactions (DDIs) with antiretroviral treatment. Antiretroviral treatment of adult HIV infection is typically initiated as a triple regimen combining two nucleoside reverse transcriptase inhibitors and a third single or boosted drug with a different mode of action, given either as individual agents or in a fixed-dose combination [Citation5]. Pharmacokinetic (PK) DDIs between antiretroviral agents and concomitant medications are common and complex, and several antiretrovirals are potent inducers and/or inhibitors of cytochrome P450 (CYP) enzymes, particularly CYP3A4 () [Citation6–Citation19].
Table 1. CYP enzymes known to interact with antiretroviral drugs. Adapted from [Citation16].
PK interactions have been reported between several PAH-targeted agents and ritonavir or other strong CYP3A4 inhibitors, leading to increases in drug exposure. Agents affected include the phosphodiesterase type 5 inhibitors sildenafil [Citation20,Citation21] and tadalafil [Citation22,Citation23], and the endothelin receptor antagonist (ERA) bosentan [Citation24,Citation25]. An increase in exposure to macitentan, another ERA, is also expected [Citation26,Citation27]. The clinical implications of these interactions are beyond the scope of this study, but these agents have warnings or contraindications for their use with ritonavir or other strong CYP3A4 inhibitors.
Riociguat is a soluble guanylate cyclase stimulator licensed for the treatment of PAH and inoperable or recurrent/persistent chronic thromboembolic pulmonary hypertension [Citation28]. Following oral dosing, riociguat absorption is rapid, with high oral bioavailability, and dose-proportional pharmacokinetics [Citation29]. Riociguat is cleared by renal excretion and oxidative biotransformation. Studies with radiolabeled riociguat have shown that approximately 27–72% of a [14C]riociguat dose is eliminated by CYP-mediated oxidative metabolism. The main biotransformation pathway for riociguat is N-demethylation to form the major pharmacologically active metabolite M1 (Figure S1), catalyzed predominantly by CYP1A1 and CYP3A4 [Citation29,Citation30]. The observed variability in the mass balance study with respect to CYP-mediated metabolism and the observed reduced exposure of riociguat in smokers [Citation31–Citation33] strongly suggested a predominant role of CYP1A1 in metabolism of riociguat as smoking is known to induce CYP1A1 [Citation34,Citation35]. Riociguat is also a substrate of the transport proteins P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). However, as 4–19% of a riociguat dose is eliminated unchanged in urine by glomerular filtration and the fraction unbound in plasma is low [Citation29], active renal secretion of riociguat is unlikely.
The potential for PK interactions between fixed-dose combination antiretrovirals and a single 0.5 mg dose of riociguat was investigated in a clinical study in 41 HIV-infected adults without PAH [Citation36]. Riociguat exposures ranged from no apparent change when co-administered with efavirenz/emtricitabine/tenofovir disoproxil (ATRIPLA®) to an approximately 3-fold increase with abacavir/dolutegravir/lamivudine (TRIUMEQ®) compared with historical healthy volunteers receiving riociguat alone. The effect of TRIUMEQ® on riociguat exposure was unexpected based on information in the literature and labels at the time. While there is broad understanding of the inhibitory potency of antiretroviral agents toward CYP3A4 and efflux transporters, there is a lack of published data on potential interactions between antiretroviral agents and CYP1A1. Two in vitro studies were therefore conducted to understand the mechanisms underlying the influence of antiretroviral agents on the metabolic clearance of riociguat: 1) to evaluate the inhibitory potential of individual components of the antiretroviral regimens on the formation of M1 of riociguat in recombinant human CYP1A1 and CYP3A4 (IC50 determination); and 2) to evaluate the inhibitory potency of antiretroviral combinations on the metabolic in vitro clearance of riociguat in human hepatocytes exhibiting both CYP1A1 and CYP3A4 activity.
2. Materials and method
2.1. Chemicals and reagents
Riociguat, its metabolite M1, and [2H3]M1 were synthesized at Bayer AG (Wuppertal, Germany); 7-hydroxyflavone, clarithromycin, darunavir, efavirenz, elvitegravir, ketoconazole, and lamivudine were purchased from Sigma Aldrich (St. Louis, MO, USA); atazanavir from Toronto Research Chemicals (North York, ON, Canada); rilpivirine from Key Organics Ltd (Camelford, UK); dolutegravir and emtricitabine from Hycultec GmbH (Beutelsbach, Germany); abacavir and tenofovir disoproxil fumarate from Fluorochem Ltd (Hadfield, UK); cobicistat from Haoyuan Chemexpress (Shanghai, P.R. China); ritonavir from Abbott Laboratories (Abbott Park, IL, USA); granisetron from AvaChem Scientific (San Antonio, TX, USA); and midazolam from Roche Pharma AG (Grenzach-Wyhlen, Germany). Nicotinamide adenine dinucleotide phosphate (NADP) disodium salt, glucose-6-phosphate, and glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides were purchased from Roche Diagnostics GmbH (Mannheim, Germany). Recombinant human CYP isoforms CYP1A1 (human CYP1A1+OR supersomes) and CYP3A4 (human CYP3A4+OR+b5 supersomes) were obtained from Corning Inc. (Woburn, MA, USA). Cryopreserved human hepatocytes were obtained from KaLy-Cell (Plobsheim, France). All other reagents and solvents used were from commercial suppliers and were of analytical or high-performance liquid chromatography grade.
2.2. Recombinant human CYP1A1 and CYP3A4 assays
2.2.1. Antiretroviral agents investigated
The individual components of four fixed-dose combinations – efavirenz/emtricitabine/tenofovir disoproxil (ATRIPLA®, Gilead Sciences, Inc., Foster City, CA, USA), rilpivirine/emtricitabine/tenofovir disoproxil (COMPLERA®, Gilead Sciences, Inc.), elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil (STRIBILD®, Gilead Sciences, Inc.), abacavir/dolutegravir/lamivudine (TRIUMEQ®, ViiV Healthcare, Brentford, UK) – and two PI-based combinations (atazanavir/ritonavir/emtricitabine/tenofovir disoproxil and darunavir/ritonavir/emtricitabine/tenofovir disoproxil) were evaluated for their inhibitory potency in a concentration-dependent manner (0.03–1.0 µM and 1.6–50 µM) using riociguat as a substrate. The known CYP1A1 and CYP3A4 inhibitors 7-hydroxyflavone and ketoconazole, respectively, served as positive controls.
2.2.2. Incubation conditions and sample analysis
For IC50 value determination, riociguat (1 µM) was incubated with recombinant human CYP1A1 or CYP3A4 isoforms (2 pmol/mL and 20 pmol/mL, respectively) in the presence of twelve concentrations of test compounds (0.03–1.0 µM and 1.6–50 µM). The assay was performed in a 96-well format on a Genesis Workstation 200 (Tecan, Crailsheim, Germany) as single experiments with two dilution series (up to 1 and up to 50 µM). The mixture of riociguat, antiretroviral agent, and CYP isoforms in phosphate buffer (50 mM, pH 7.4) containing 1 mM EDTA was pre-warmed at 37°C for 5 minutes. Reactions were initiated by the addition of an NADPH-generating system containing NADP (1 mM), glucose 6-phosphate, and glucose 6-phosphate dehydrogenase (5 mM and 0.5 U/mL, respectively). At defined time points (20 minutes for CYP1A1 and 60 minutes for CYP3A4), reactions were quenched with acetonitrile containing the internal standard ([2H3]M1). Samples were then centrifuged, and the supernatants were quantified for M1 formation by liquid chromatography with tandem mass spectrometry using an AB SCIEX API 4000 mass spectrometer (Applied Biosystems, MDS Sciex, Concord, ON, Canada).
To investigate time-dependent inhibition (TDI), the experiments were repeated as described above, but with the tested inhibitors pre-incubated with the recombinant CYP isoforms and NADPH-generating system in the absence of riociguat for 30 minutes at 37°C.
2.2.3. Data analysis
Analytical data were processed by Analyst software (version 1.6.2; Applied Biosystems MDS Sciex). The amount of M1 formed in the presence of the test drugs was expressed as a percentage of the corresponding control values without inhibitor. Linear interpolation was used to determine IC50 values. If <50% inhibition was observed, the data were not extrapolated.
The predicted increase in riociguat exposure in vivo based on these in vitro data was calculated as an area under concentration–time curve ratio (AUCR) using the following equation [Citation37]:
where fm,CYPi is the fraction metabolized (term i indicates multiple involved enzymes, here CYP1A1 or CYP3A4), [I]j is the estimated inhibitor concentration (term j indicates multiple inhibitors of single enzyme, here a single component of the antiretroviral regimens), Ki,j is the in vitro inhibition constant for a single enzyme–inhibitor pair (here for the single component of the antiretroviral regimens against CYP1A1 or CYP3A4, calculated using the equation Ki = IC50/(S/Km+1) assuming competitive inhibition). Ki values were not corrected with fu,inc in the incubation media because calculated fu,inc values based on estimated logP value [Citation38] were for all inhibitors nearly or equal to 1.
2.3. Human hepatocyte study
2.3.1. Antiretroviral agents investigated
Six different antiretroviral combination regimens, as well as clarithromycin and ketoconazole, were investigated ().
Table 2. Fixed-dose antiretroviral regimens (and their individual components) and concentrations evaluated in the human hepatocyte studies.
2.3.2. Incubation conditions and sample analysis
Freshly thawed human hepatocytes with CYP1A1 and CYP3A4 activity were placed in suspension culture (1 × 106 viable cells/mL, Williams E medium fortified with L-glutamine [0.292 g/L] and carbogen [95/5 O2/CO2, v/v]) then pre-incubated for 60 minutes at 37°C with the tested combinations of antiretroviral agents in the absence of the respective substrates. A mixture of probe substrates (0.1 μM riociguat, 0.5 μM granisetron cleared almost solely by CYP1A1 [Citation39] and 0.5 μM midazolam cleared by CYP3A4 [Citation40]) was then added, and the reactions were allowed to proceed. Aliquots of the incubation mixture were removed at 0, 20, 40, 60, 90, 120, and 180 minutes after pre-incubation to obtain time-dependent profiles. Reactions were terminated with acetonitrile and analyzed directly or stored at –20°C until analysis. Experiments were performed in duplicate in two donors.
Samples were centrifuged, and the supernatants were analyzed by liquid chromatography/high-resolution mass spectrometry using a high-resolution mass spectrometer Q ExactiveTM Orbitrap MS (Thermo Fisher Scientific, Bremen, Germany). The parent substrates (riociguat, midazolam, and granisetron) and the metabolite M1 were quantified.
2.3.3. Data analysis
Analytical data were processed by Chromeleon software (version 7.2 SR4; Thermo Scientific, Dreieich, Germany). The depletion kinetics of riociguat, as well as granisetron and midazolam for correlation analysis, were plotted semi-logarithmically as a percentage of the remaining drug vs. incubation time. The obtained slope k (i.e. in vitro rate constant of substrate loss per minute) was used to determine the intrinsic clearance scaled for the whole liver (CLint) [Citation41]:
where k is the rate constant of substrate loss, which is a negative slope of the linear regression from log percentage substrate remaining vs. incubation time [Citation42]. A value of 110 × 106 was used for hepatocyte cell number/g liver [Citation43], 1 × 106 hepatocyte cells/mL of incubation were used, HLW is the average human liver weight per kilogram of body weight, for which a value of 21 g/kg was used [Citation44], and 0.06 is a factor for conversion of CLint units from mL/min/kg to L/h/kg.
Calculated CLint values were scaled to hepatic blood clearance (CLh) disregarding protein binding parameters as follows:
where Qh is hepatic blood flow per kg body weight, for which a value of 1.32 L/h/kg was used [Citation45].
The predicted increase in riociguat exposure in vivo arising from co-administration of the antiretroviral combinations compared with riociguat alone was calculated as AUCR using the following equation based on Rowland and Matin [Citation46], refined by Ito et al. [Citation47]:
where fm,total is the total fraction metabolized, CLh,inh is hepatic blood clearance after incubation with inhibitor, and CLh,w/o inh is hepatic blood clearance after incubation without inhibitor.
2.4. Estimation of fraction metabolized
2.4.1. Estimation of total fraction metabolized (fm,total)
Total fraction metabolized was estimated from the human mass balance with radiolabeled [14C]riociguat after oral administration to four healthy volunteers [Citation29]. Unknown metabolites (1.7–4.2% of dose) were added to the identified metabolites (27.0–72.5% of dose) eliminated by CYP-mediated oxidative metabolism, and this sum was corrected by the total excreted radioactivity of 90.3–94.7%. These calculations resulted in an fm,total of 0.32–0.85.
2.4.2. Estimation of fraction metabolized (fm) for CYP1A1 and CYP3A4
The fractions metabolized via CYP1A1 and CYP3A4 for riociguat were estimated by revisiting data from clinical DDI studies [Citation48] with clarithromycin (a strong CYP3A4 and P-gp inhibitor) [Citation49,Citation50] and with ketoconazole (a strong CYP3A4, strong CYP1A1, and P-gp inhibitor) [Citation51,Citation52]. Concomitant administration of ketoconazole 400 mg led to a 46% increase in Cmax of riociguat and a 150% increase in AUC. Co-administration of clarithromycin led to a marginal increase in riociguat exposure, with no significant change in Cmax and a 42% increase in AUC. The possible inhibitory potency toward the efflux transporters P-gp and BCRP, which might be involved in the unchanged clearance of riociguat, was not incorporated in calculations because of the presumably low impact on transporter-mediated clearance since riociguat has high oral bioavailability and no hints for active renal secretion. An approach described by Ohno et al. [Citation53] and refined by Loue and Tod [Citation54] was applied, using the contribution ratio of the respective CYP enzyme to oral clearance of the substrate (CRi; equal to fraction metabolized [fm,CYPi], referred to as fm later in this paper) and the inhibition ratio (IR) of the inhibitor on the respective enzyme (IRi):
As clarithromycin has no inhibitory potency on CYP1A1, the change in AUC of riociguat from the clinical DDI study with clarithromycin (AUCR 1.41 [90% confidence interval (CI): 1.23–1.63]) [Citation48] was used to calculate the fm value of riociguat via CYP3A4. An IR value of 0.94 for CYP3A4 [Citation54] and 0.0 for CYP1A1 was used. Applying the upper equation, the calculated fm value on CYP3A4 for riociguat was approximately 0.3 (in the range 0.2–0.4, depending on the applied AUCR values within 90% CI).
The observed AUCR value of 2.50 (90% CI: 2.14–2.92) [Citation48] in the clinical drug–drug interaction study with ketoconazole (strong CYP3A4 and CYP1A1 inhibitor) was used to calculate the fm value of riociguat via CYP1A1. An IR value of 0.98 for CYP3A4 [Citation54] and a mean fm,CYP3A4 value of 0.3 (see above) were used. As no clinical data on PK DDI with CYP1A1 substrates with a defined clearance ratio are available, the IR value for ketoconazole and CYP1A1 was calculated using the following equation: IR = [I]/([I]+Ki). Using different input parameters for [I] (total plasma concentration, unbound plasma concentration, and unbound liver inlet concentration) and the calculated Ki value from this in vitro study (53 nM; Ki = IC50/(S/Km+1), Km of riociguat for CYP1A1 ~ 0.2 µM), the IR value for ketoconazole on CYP1A1 ranged from 0.38 to 0.98. Based on these input parameters, the estimated fm value on CYP1A1 (fm,CYP1A1) for riociguat was in the range of 0.23–0.95. However, the overall fraction metabolized for riociguat based on the estimated fm value of CYP1A1 (0.23–0.95) and of CYP3A4 (approximately 0.3) would result in a maximum fm,total of >1.0 (approximately 0.5–1.2). Therefore, based on the results from the human mass balance study and the clinical DDI study with clarithromycin and ketoconazole, several scenarios for fm,CYP1A1 and fm,CYP3A4 were applied in the calculations. To cover the whole range of total fraction metabolized of 0.32–0.85 with different CYP1A1/CYP3A4 contributions the fm,CYP3A4 values of 0.20, 0.30, and 0.40 were complemented with a broad range of fm,CYP1A1 (0.0–0.65) (Tables S3–S5). This is in line with investigations of highly variable CYP1A1 content in human liver microsomes [Citation30,Citation39].
3. Results
3.1. Recombinant CYP1A1 and CYP3A4 assays
IC50 values for individual antiretroviral components for the formation of metabolite M1 in recombinant human CYP1A1 and CYP3A4 in vitro are listed in . Potent in vitro inhibitors of the CYP1A1-mediated M1 formation were ketoconazole and rilpivirine (IC50 < 1 μM). Inhibitory potency in the range of 1–50 µM was also observed for abacavir, ritonavir, efavirenz, cobicistat, and elvitegravir, while atazanavir, darunavir, dolutegravir, emtricitabine, lamivudine, tenofovir disoproxil, and clarithromycin did not demonstrate any inhibitory potency (IC50 > 50 μM). Pre-incubation increased the inhibitory potency of abacavir, rilpivirine, elvitegravir, and efavirenz, indicating a time-dependent effect.
Table 3. Inhibitory potency of antiretroviral drugs on the formation of riociguat metabolite M1 in recombinant human CYP1A1 and CYP3A4.
Potent in vitro inhibitors of the CYP3A4-mediated M1 formation were ritonavir, ketoconazole, cobicistat, darunavir, atazanavir, and rilpivirine (IC50 < 1 μM). Inhibitory potency in the range of 1–50 µM was also observed for clarithromycin, tenofovir disoproxil, elvitegravir, efavirenz, dolutegravir, and abacavir, while emtricitabine and lamivudine did not demonstrate any inhibitory potency for M1 formation (IC50 > 50 μM). Pre-incubation increased the inhibitory potency of clarithromycin, rilpivirine, abacavir, and dolutegravir, indicating a time-dependent effect.
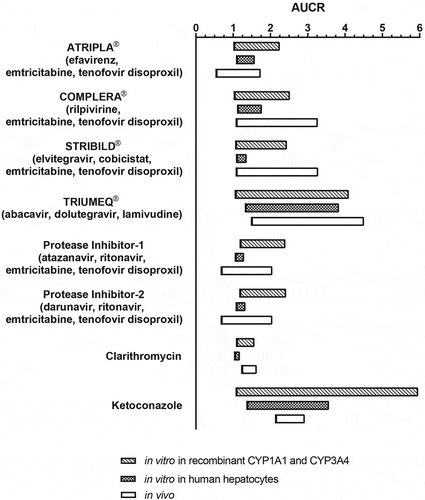
Based on these individually determined IC50 values for single components, AUCR values with the antiretroviral regimens were calculated using Equation (1). Published data for [I], fu, and dose [Citation55,Citation56], as well as different scenarios for fractions metabolized via CYP1A1 and CYP3A4 (please refer to section Estimation of fraction metabolized (fm) for CYP1A1 and CYP3A4 in Materials and Methods) were used for AUCR calculation without implementation of TDI and are summarized in , , and Tables S1–S5. Estimated AUCRs were highest with 1.06–4.10 for abacavir/dolutegravir/lamivudine (TRIUMEQ®), followed by 1.03–2.52 for emtricitabine/rilpivirine/tenofovir disoproxil (COMPLERA®), 1.07–2.44 for elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil (STRIBILD®), 1.18–2.42 for PI-based regimens, and 1.02–2.25 for efavirenz/emtricitabine/tenofovir disoproxil (ATRIPLA®). For comparison, estimated AUCR values for ketoconazole were 1.08–5.96 and for clarithromycin, 1.09–1.57.
Table 5. Effect of antiretroviral drugs on the metabolic clearance of riociguat in human hepatocytes.
Table 4. Comparison of the predicted AUCR ranges of riociguat with HIV treatments based on in vitro experiments in recombinant CYP1A1 and CYP3A4, as well as in human hepatocytes with observed AUCR ranges of riociguat in vivo.
Figure 1. Predicted increase in riociguat exposure in vivo based on in vitro data after co-incubation of antiretroviral regimens with riociguat in comparison to AUCR observed in clinical DDI study.
3.2. Human hepatocyte study
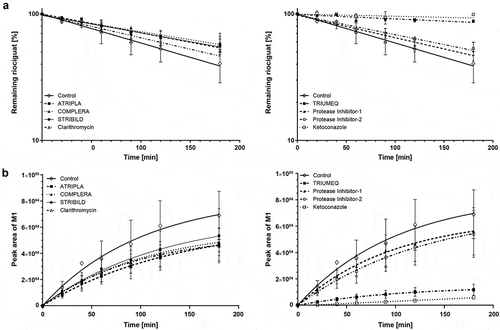
The effect of antiretroviral regimens as cocktail incubations using concentrations of the single components close to their described Cmax [Citation55,Citation56] on intrinsic clearance of riociguat in human hepatocytes in vitro is presented in and . In both donors, CLint was reduced substantially in the presence of ketoconazole, whereas clarithromycin had little effect. Abacavir/dolutegravir/lamivudine (TRIUMEQ®) was associated with substantial reductions in CLint comparable to the effect of ketoconazole. Smaller reductions were seen with the other antiretroviral regimens in the following order: emtricitabine/rilpivirine/tenofovir disoproxil (COMPLERA®), efavirenz/emtricitabine/tenofovir disoproxil (ATRIPLA®), elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil (STRIBILD®), and PI-based regimens ().
Figure 2. Change in intrinsic clearances of riociguat (a) and formation of metabolite M1 (b) in the presence of HIV treatments, ketoconazole and clarithromycin in one donor of human hepatocytes exhibiting CYP1A1 and CYP3A4 activity.
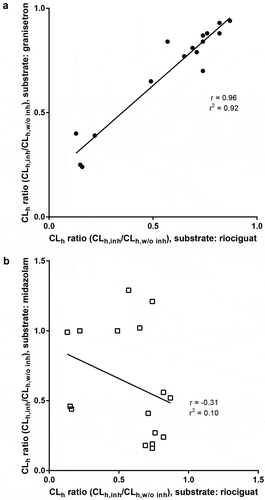
The estimated AUCR for riociguat based on calculated blood clearance (CLh) and fm,total from human mass balance study (Equation (4)) was highest with 1.33–3.84 for abacavir/dolutegravir/lamivudine (TRIUMEQ®), whereas AUCRs of 1.12–1.77 for emtricitabine/rilpivirine/tenofovir disoproxil (COMPLERA®), 1.09–1.58 for efavirenz/emtricitabine/tenofovir disoproxil (ATRIPLA®), 1.09–1.36 for elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil (STRIBILD®), and 1.06–1.33 for the PI regimens were predicted to have small effects on riociguat exposure in vivo (, ; Table S6). Estimated AUCR values for ketoconazole and clarithromycin were 1.37–3.57 and 1.04–1.18, respectively. CLh ratios with the respective antiretroviral regimens as cocktail incubations were also calculated for granisetron and midazolam (Table S7). There was a clear correlation of the CLh ratios for granisetron and riociguat (r = 0.96; r2 = 0.92), but not for midazolam and riociguat (r = – 0.31; r2 = 0.10) (), suggesting that CYP1A1 is predominantly metabolizing riociguat to M1. Inhibitory effects of antiretroviral agents on M1 formation correlated well with decreased riociguat clearance ().
Figure 3. Correlation between CLh ratios (with and without inhibitor) of riociguat with (a) granisetron, and (b) midazolam, after incubation with antiretroviral regimens, ketoconazole, and clarithromycin in human hepatocytes.
4. Discussion
The present in vitro studies evaluated the inhibitory potential of antiretroviral drugs and their combination regimens on the formation of metabolite M1 as a major metabolic clearance pathway of riociguat using recombinant CYP1A1 or CYP3A4, as well as human hepatocytes, at concentrations near to clinical plasma levels for these agents. Metabolite M1 is by a factor of 3 to 10 less pharmacologically active than riociguat and its plasma exposure in patients with PAH is about half of that of riociguat, hence its overall contribution to the PD effect is limited [Citation57]. Therefore, this in vitro-in vivo correlation focused only on the change in riociguat exposure in the presence of the antiretroviral combination regimens. The two studies were consistent in predicting the largest effect on riociguat exposure to be with abacavir/dolutegravir/lamivudine (components of TRIUMEQ®) followed by emtricitabine/rilpivirine/tenofovir disoproxil (components of COMPLERA®), efavirenz/emtricitabine/tenofovir disoproxil (components of ATRIPLA®), elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil (components of STRIBILD®), and the two additional PI regimens.
The slight differences in the calculated AUCR ranges from both in vitro systems may be because hepatocytes represent a more complex system, well reflecting the various metabolic pathways of the liver, while recombinant enzymes are more specific for tested enzymes at the expense of less biologic complexity. Furthermore, the inhibitory potency with recombinant CYP1A1 and CYP3A4 was assessed on IC50 values of the single components of antiretroviral agents by analyzing M1 formation, whereas in human hepatocytes the change in intrinsic clearance of riociguat after incubation with a cocktail of components at defined concentrations near Cmax of the antiretroviral agents was analyzed. The predicted magnitude of the AUCR of riociguat might be slightly underestimated based on the data with human hepatocytes if the in vivo concentration of tested inhibitors at the site of the metabolizing enzymes is higher than the tested Cmax value (e.g. unbound liver inlet concentration) or if the fm,total value in humans is higher than predicted since the data were calculated only from four subjects in the human mass balance study. The calculated AUCRs correlated with the broad range of observed AUCRs and results from a relatively high range of fm,total obtained from human mass balance study and a variable content of CYP1A1 in hepatocytes including the rather unlikely lower limit of fm,CYP1A1 = 0.0.
Focusing on the inhibitory potency on CYP3A4 in hepatocytes using midazolam as a sensitive CYP3A4 substrate, results of this study revealed that the significant decrease in clearance of midazolam with fixed-dose antiretroviral combinations containing strong CYP3A4 inhibitors cobicistat, ritonavir, atazanavir, and darunavir, as well as clarithromycin, and ketoconazole is consistent with previous clinical studies (Table S7) [Citation16,Citation58–Citation61]. Based on the published clinical literature [Citation16], no DDIs were expected with CYP3A substrates for abacavir/dolutegravir/lamivudine (components of TRIUMEQ®) or emtricitabine/rilpivirine/tenofovir disoproxil (components of COMPLERA®), which is consistent with the in vitro data observed with recombinant CYP3A4 and in human hepatocytes using midazolam as a substrate in the present study.
There are very limited data available on the effect of antiretroviral agents on CYP1A1 enzyme activity. While there are some in vitro gene expression data to suggest that efavirenz is a potential inducer of CYP1A1 [Citation62], the in vivo effect is unknown. To date, no dedicated clinical drug–drug antiretroviral interaction studies with granisetron, a sensitive CYP1A1 substrate, have been performed. In the present study, we found the most potent inhibition of CYP1A1 with ketoconazole, rilpivirine (a component of COMPLERA®), abacavir (a component of TRIUMEQ®), ritonavir (component of PIs), and efavirenz (component of ATRIPLA®). Based on the exposure, the greatest inhibitory effect expressed as [I]/Ki on CYP1A1 was estimated for abacavir and ketoconazole followed by rilpivirine and efavirenz (Table S1). The broad range of AUCR values resulted from the various predicted fm values for CYP1A1. Data obtained in human hepatocytes in the present study clearly indicate that riociguat clearance ratio correlates well with that of granisetron, a marker for CYP1A1 activity [Citation39], but not with midazolam clearance ratio, a CYP3A index substrate (). These in vitro data together with investigations showing highly variable CYP1A1 expression levels in human liver microsomes [Citation39] confirmed that CYP1A1 seems to be responsible for the observed moderate-to-high inter-individual PK variability in healthy volunteers [Citation29].
The predicted AUCR values from these in vitro studies were generally consistent with those obtained in a clinical DDI study in HIV-infected adults on stable fixed-dose antiretroviral regimens (, ). Individuals receiving TRIUMEQ® had an almost 3-fold increase in riociguat exposure compared with riociguat alone [Citation36]. However, some differences in riociguat AUCR values between the in vitro studies and the clinical DDI study may be related to observed TDIs, which were not included in the calculations. For example, the study with human hepatocytes underestimated the magnitude of the AUC increase of riociguat in vivo with elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil (STRIBILD®, predicted AUCR in vitro, 1.09–1.36; in vivo, 2.06 [90% CI: 1.24–3.44]). In contrast, the in vitro studies with recombinant enzymes overestimated the magnitude of riociguat AUC increase in vivo with efavirenz/emtricitabine/tenofovir disoproxil (ATRIPLA®). This may reflect the reported moderate inductive and activation effect of efavirenz on CYP3A4 [Citation19].
In conclusion, the findings of these in vitro studies, together with data from clinical DDI studies, confirm that CYP1A1 is the predominant enzyme metabolizing riociguat to M1, and therefore potent in vivo CYP1A1 inhibitors are expected to have a more pronounced impact on change in riociguat exposure than potent CYP3A inhibitors. Since a lot of the components of antiretroviral regimens are inhibitors of CYP3A4 and not CYP1A1, these findings explain and support the partially unexpected results of the clinical DDI study in HIV-infected patients where the greatest increase in riociguat exposure was observed with co-administration of TRIUMEQ® containing the potent CYP1A1 inhibitor abacavir.
Authorship contributions
Participated in research design: NA Jungmann, D Lang, M Gerisch.
Conducted experiments: NA Jungmann, D Lang, M Gerisch.
Contributed new reagents or analytic tools: NA Jungmann, D Lang, M Gerisch.
Performed data analysis: NA Jungmann, D Lang, M Gerisch.
Wrote or contributed to the writing of the manuscript: NA Jungmann, D Lang, M Gerisch, S Saleh, D van der Mey.
Declaration of interest
All authors of this manuscript are employees of Bayer AG. D Lang, M Gerisch and D van der Mey have stock in Bayer AG but are not paid in stock or stock options. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Riociguat_ARV_biotransformation_SUPPL_MAT.docx
Download MS Word (78.9 KB)Acknowledgments
Medical writing assistance was provided by Adelphi Communications Ltd, funded by Bayer AG, Germany.
The authors thank Torsten Löffler, Klaudia Schulz, and Franziska Weigner for technical support, and Dr. Corina Becker, Dr. Armin Kern, Dr. Martin Radtke, and Dr. Wolfgang Mueck for scientific assistance.
Supplemental material
The supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Sitbon O, Lascoux-Combe C, Delfraissy JF, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177(1):108–113.
- Newman JH, Hemnes AR. Pulmonary hypertension. In: Schraufnagel DA, editor. Breathing in America: diseases, progress, and hope. New York, NY: ATS; 2010. p. 175–184.
- McLaughlin VV, Gaine SP, Howard LS, et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D73–D81.
- Galiè N, Humbert M, Vachiery JL, et al. ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC) International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46(4):903–975.
- Günthard HF, Saag MS, Benson CA, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2016 recommendations of the international antiviral society-USA panel. JAMA. 2016 Jul 12;316(2):191–210.
- Janssen PA, Lewi PJ, Arnold E, et al. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, rilpivirine). J Med Chem. 2005;48(6):1901–1909.
- Weiss J, Haefeli WE. Potential of the novel antiretroviral drug rilpivirine to modulate the expression and function of drug transporters and drug-metabolising enzymes in vitro. Int J Antimicrob Agents. 2013;41(5):484–487.
- Ramanathan S, Mathias AA, German P, et al. Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin Pharmacokinet. 2011;50(4):229–244.
- Zembruski NC, Buchel G, Jodicke L, et al. Potential of novel antiretrovirals to modulate expression and function of drug transporters in vitro. J Antimicrob Chemother. 2011;66(4):802–812.
- Ernest C 2nd., Hall SD, Jones DR. Mechanism-based inactivation of CYP3A by HIV protease inhibitors. J Pharmacol Exp Ther. 2005;312(2):583–591.
- Kirby BJ, Collier AC, Kharasch ED, et al. Complex drug interactions of HIV protease inhibitors 1: inactivation, induction, and inhibition of cytochrome P450 3A by ritonavir or nelfinavir. Drug Metab Dispos. 2011;39(6):1070–1078.
- Kearney BP, Flaherty JF, Shah J. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin Pharmacokinet. 2004;43(9):595–612.
- Svard J, Spiers JP, Mulcahy F, et al. Nuclear receptor-mediated induction of CYP450 by antiretrovirals: functional consequences of NR1I2 (PXR) polymorphisms and differential prevalence in whites and sub-Saharan Africans. J Acquir Immune Defic Syndr. 2010;55(5):536–549.
- Reese MJ, Savina PM, Generaux GT, et al. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos. 2013;41(2):353–361.
- Johnson MA, Moore KH, Yuen GJ, et al. Clinical pharmacokinetics of lamivudine. Clin Pharmacokinet. 1999;36(1):41–66.
- AIDS Info. Panel on antiretroviral guidelines for adults and adolescents. Guidelines for the use of antiretroviral agents in adults and adolescents living with HIV. Department of Health and Human Services. 2018 [cited 2018 Oct 19]. Available from: https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf
- Mikus G, Heinrich T, Bodigheimer J, et al. Semisimultaneous midazolam administration to evaluate the time course of CYP3A activation by a single oral dose of efavirenz. J Clin Pharmacol. 2017;57(7):899–905.
- Faucette SR, Zhang TC, Moore R, et al. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320(1):72–80.
- Hariparsad N, Nallani SC, Sane RS, et al. Induction of CYP3A4 by efavirenz in primary human hepatocytes: comparison with rifampin and phenobarbital. J Clin Pharmacol. 2004;44(11):1273–1281.
- Pfizer Revatio. EU summary of product characteristics. 2010 [cited 2018 Oct 19]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000638/WC500055840.pdf
- Pfizer Revatio. US prescribing information. 2014 [cited 2018 Oct 19]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021845s011,022473s004,0203109s002lbl.pdf
- Eli-Lilly Adcirca. EU summary of product characteristics. 2013 [cited 2018 Oct 19]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001021/WC500032789.pdf
- Eli-Lilly Adcirca. Prescribing information. 2017 [cited 2018 Oct 19]. Available from: http://pi.lilly.com/us/adcirca-pi.pdf
- Actelion Tracleer. Highlights of prescribing information. 2015 [cited 2018 Oct 19]. Available from: http://www.tracleer.com/assets/PDFs/Tracleer_Full_Prescribing_Information.pdf
- Actelion. Summary of product characteristics: tracleer. 2016 [cited 2018 Oct 19]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000401/WC500041597.pdf
- Actelion. Opsumit (Macitentan) EU summary of product characteristics. 2017 [cited 2018 Oct 19]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002697/WC500160899.pdf
- Actelion. Opsumit (Macitentan) US Prescribing Information. 2017 [cited 2018 Oct 19]. Available from: http://opsumit.com/opsumit-prescribing-information.pdf
- Bayer AG. Adempas summary of product characteristics: 0.5 g, 1 mg, 1.5 mg, 2 mg and 2.5 mg film-coated tablets. 2018 [cited 2018 Oct 19]. Available from: https://www.ema.europa.eu/en/documents/product-information/adempas-epar-product-information_en.pdf
- Frey R, Becker C, Saleh S, et al. Clinical pharmacokinetic and pharmacodynamic profile of riociguat. Clin Pharmacokinet. 2018;57(6):647–661.
- Lang D, Radtke M, Bairlein M. Highly variable expression of CYP1A1 in human liver and impact on pharmacokinetics of riociguat and granisetron in humans. Chem Res Toxicol. 2019;32(6):1115–1122.
- Saleh S, Becker C, Frey R, et al. Population pharmacokinetics of single-dose riociguat in patients with renal or hepatic impairment. Pulm Circ. 2016;6(Suppl 1):S75–S85.
- Frey R, Becker C, Unger S, et al. Assessment of the effects of hepatic impairment and smoking on the pharmacokinetics of a single oral dose of the soluble guanylate cyclase stimulator riociguat (BAY 63-2521). Pulm Circ. 2016;6(Suppl 1):S5–S14.
- Frey R, Becker C, Unger S, et al. Assessment of the effects of renal impairment and smoking on the pharmacokinetics of a single oral dose of the soluble guanylate cyclase stimulator riociguat (BAY 63-2521). Pulm Circ. 2016;6(Suppl 1):S15–S26.
- Czekaj P, Wiaderkiewicz A, Florek E, et al. Tobacco smoke-dependent changes in cytochrome P450 1A1, 1A2, and 2E1 protein expressions in fetuses, newborns, pregnant rats, and human placenta. Arch Toxicol. 2005;79(1):13–24.
- McLemore TL, Adelberg S, Liu MC, et al. Expression of CYP1A1 gene in patients with lung cancer: evidence for cigarette smoke-induced gene expression in normal lung tissue and for altered gene regulation in primary pulmonary carcinomas. J Natl Cancer Inst. 1990;82(16):1333–1339.
- DeJesus E, Saleh S, Cheng S, et al. Pharmacokinetic interaction of riociguat and antiretroviral combination regimens in HIV-1-infected adults. Pulm Circ. 2019;9(2):2045894019848644.
- Hinton LK, Galetin A, Houston JB. Multiple inhibition mechanisms and prediction of drug-drug interactions: status of metabolism and transporter models as exemplified by gemfibrozil-drug interactions. Pharm Res. 2008;25(5):1063–1074.
- Hallifax D, Houston JB. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab Dispos. 2006;34(4):724–726.
- Nakamura H, Ariyoshi N, Okada K, et al. CYP1A1 is a major enzyme responsible for the metabolism of granisetron in human liver microsomes. Curr Drug Metab. 2005;6(5):469–480.
- Kronbach T, Mathys D, Umeno M, et al. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol. 1989;36(1):89–96.
- Lu C, Li P, Gallegos R, et al. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos. 2006;34(9):1600–1605.
- Mohutsky MA, Chien JY, Ring BJ, et al. Predictions of the in vivo clearance of drugs from rate of loss using human liver microsomes for phase I and phase II biotransformations. Pharm Res. 2006;23(4):654–662.
- Wilson ZE, Rostami-Hodjegan A, Burn JL, et al. Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br J Clin Pharmacol. 2003;56(4):433–440.
- Molina DK, DiMaio VJ. Normal organ weights in men: part II-the brain, lungs, liver, spleen, and kidneys. Am J Forensic Med Pathol. 2012;33(4):368–372.
- Yang J, Jamei M, Yeo KR, et al. Misuse of the well-stirred model of hepatic drug clearance. Drug Metab Dispos. 2007;35(3):501–502.
- Rowland M, Matin SB. Kinetics of drug-drug interactions. J Pharmacokinet Biopharm. 1973;1(6):553–567.
- Ito K, Iwatsubo T, Kanamitsu S, et al. Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev. 1998;50(3):387–412.
- Becker C, Frey R, Unger S, et al. Pharmacokinetic interaction of riociguat with ketoconazole, clarithromycin, and midazolam. Pulm Circ. 2016;6(Suppl 1):S49–S57.
- Gurley BJ, Swain A, Barone GW, et al. Effect of goldenseal (Hydrastis canadensis) and kava kava (Piper methysticum) supplementation on digoxin pharmacokinetics in humans. Drug Metab Dispos. 2007;35(2):240–245.
- Prueksaritanont T, Tatosian DA, Chu X, et al. Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin Pharmacol Ther. 2017;101(4):519–530.
- Larsen UL, Hyldahl Olesen L, Guldborg Nyvold C, et al. Human intestinal P-glycoprotein activity estimated by the model substrate digoxin. Scand J Clin Lab Invest. 2007;67(2):123–134.
- Cooper KJ, Martin PD, Dane AL, et al. Lack of effect of ketoconazole on the pharmacokinetics of rosuvastatin in healthy subjects. Br J Clin Pharmacol. 2003;55(1):94–99.
- Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681–696.
- Loue C, Tod M. Reliability and extension of quantitative prediction of CYP3A4-mediated drug interactions based on clinical data. Aaps J. 2014;16(6):1309–1320.
- PharmaPendium. 2018 [cited 2018 Oct 19]. Available from: https://pharmapendium.com/#/login
- Drug Interaction Database Program. 2018 [cited 2018 Oct 19]. Available from: https://www.druginteractioninfo.org/
- Bayer AG. Adempas (riociguat tablets): EU summary of product characteristics. 2017 [cited 2018 Oct 19]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002737/WC500165034.pdf
- Quinney SK, Haehner BD, Rhoades MB, et al. Interaction between midazolam and clarithromycin in the elderly. Br J Clin Pharmacol. 2008;65(1):98–109.
- Gorski JC, Jones DR, Haehner-Daniels BD, et al. The contribution of intestinal and hepatic CYP3A to the interaction between midazolam and clarithromycin. Clin Pharmacol Ther. 1998;64(2):133–143.
- Lam YW, Alfaro CL, Ereshefsky L, et al. Pharmacokinetic and pharmacodynamic interactions of oral midazolam with ketoconazole, fluoxetine, fluvoxamine, and nefazodone. J Clin Pharmacol. 2003;43(11):1274–1282.
- Liu B, Crewe HK, Ozdemir M, et al. The absorption kinetics of ketoconazole plays a major role in explaining the reported variability in the level of interaction with midazolam: interplay between formulation and inhibition of gut wall and liver metabolism. Biopharm Drug Dispos. 2017;38(3):260–270.
- Gomez-Sucerquia LJ, Blas-Garcia A, Marti-Cabrera M, et al. Profile of stress and toxicity gene expression in human hepatic cells treated with efavirenz. Antiviral Res. 2012;94(3):232–241.