
ABSTRACT
Introduction
The N-acetylation polymorphism has been the subject of comprehensive reviews describing the role of arylamine N-acetyltransferase 2 (NAT2) in the metabolism of numerous aromatic amine and hydrazine drugs.
Areas covered
We describe and review data that more clearly defines the effects of NAT2 haplotypes and genotypes on the expression of acetylator phenotype towards selected drugs within human hepatocytes in vitro, within human hepatocyte cultures in situ, and clinical measures such as bioavailability, plasma metabolic ratios of parent to N-acetyl metabolite, elimination rate constants and plasma half-life, and/or clearance determinations in human subjects. We review several drugs (isoniazid, hydralazine, sulfamethazine, amifampridine, procainamide, sulfasalazine, amonafide and metamizole) for which NAT2 phenotype-guided therapy may be important. The value of pharmacogenomics-guided isoniazid therapy for the prevention and treatment of tuberculosis is presented as a paradigm for NAT2 phenotype-dependent dosing strategies.
Expert opinion
Studies in human subjects and cryopreserved human hepatocytes show evidence for rapid, intermediate and slow acetylator phenotypes, with further data suggesting genetic heterogeneity within the slow acetylator phenotype. Incorporation of more robust NAT2 genotype/phenotypes relationships, including genetic heterogeneity within the slow acetylator phenotype, should lead to further advancements in both health outcomes and cost benefit for prevention and treatment of tuberculosis.
1. Introduction
Arylamine N-acetyltransferase 1 and N-acetyltransferase 2 (NAT2) are both subject to genetic polymorphisms in humans [Citation1]. NAT2 is expressed primarily in liver and the GI tract [Citation2] and is responsible for the N-acetylation polymorphism described in human populations [Citation3–6]. Since NAT1 is widely expressed in extrahepatic tissues [Citation7], differences between NAT2 rapid and slow acetylator phenotypes in the metabolism, efficacy, and/or toxicity of arylamine and hydrazine drugs are most likely observed after oral rather than parenteral administration. Early studies showed excellent correlations between NAT2 genotype and phenotype assessed with probe substrate caffeine [Citation8,Citation9]. With respect to drugs used clinically, studies assessing isoniazid [Citation10], dapsone [Citation11], procainamide [Citation12], sulfamethazine, also known as sulfadimidine [Citation13,Citation14], sulfapyridine (byproduct of sulfasalazine) [Citation14] and hydralazine [Citation15,Citation16] metabolism in human subjects exhibited bimodal distributions described as rapid (sometimes termed fast) and slow acetylators. Subsequent reviews provide more detailed listing of therapeutic agents subject to the N-acetylation polymorphism resulting from NAT2 metabolism [Citation3–6,Citation17,Citation18].
Numerous single nucleotide polymorphisms (SNPs) in the coding exon of the NAT2 gene, inherited as NAT2 haplotypes and genotypes, confer rapid, intermediate, and slow acetylator phenotypes that modify the metabolism of drugs and carcinogens [Citation4,Citation19,Citation20]. An updated listing of NAT2 alleles, signature allele clusters, SNPs and corresponding phenotypes is shown in , modified from information published previously [Citation4,Citation21]. The frequency of the NAT2 SNPs, haplotypes, and genotypes associated with slow acetylator phenotype vary significantly across various racial and ethnic groups [Citation22,Citation23]. The frequencies of NAT2 haplotypes NAT2*5, NAT2*6, NAT2*10, NAT2*12, NAT2*14, NAT2*17 and NAT2*18 among individuals from African individuals, America, East Asia, Europe, and South Asia are clearly illustrated in a recent review [Citation5] using data available at http://www.internationalgenome.org. A few noteworthy slow acetylator haplotypes that vary substantially include the NAT2*5 cluster which is present in nearly 50% of Europeans but less than 5% of East Asia individuals, the NAT2*7 cluster present in nearly 20% of East Asia individuals but less than 5% of European or African individuals and the NAT2*14 cluster present in nearly 10% of African and American populations, but negligible in East and South Asia and Europe.
Table 1. NAT2 SNPs and their corresponding alleles/haplotypesa
Although numerous studies report relationships between NAT2 genotype with drug toxicity or disease risk, problems and misinterpretations that can result from investigations that assess these associations between NAT2 genotype and drug toxicity or disease risk in the absence of an assessment of NAT2 phenotype was recently reviewed [Citation5]. A subsequent review [Citation6] covering many of the same drugs concluded that although slow NAT2 acetylators seem to be more at risk from certain side-effect of drugs that undergo N-acetylation, many studies are tentative and equivocal associations which await confirmatory evidence of the effects of the NAT2 polymorphism at the phenotypic level that measures the effects of NAT2 polymorphism on substrate turnover and metabolic transformation. Questions concerning the strength and importance of observed associations of adverse events related to the phenotype/genotype remain somewhat ambiguous and inconsistent due in part to inference of phenotype based on NAT2 genotype. The optimum number and identity of NAT2 SNPs necessary for accurate inference of NAT2 phenotype clearly differs with ethnicity and race of the population investigated [Citation21,Citation25,Citation26]. This is one of the most relevant factors that hamper the use of NAT2 genotyping in clinical practice despite the fact that more than 50 years have elapsed since the first identification of rapid and slow acetylator phenotypes. Current methods of assessing NAT2 phenotype require administration of a probe substrate or drug, and the development of more rapid and less invasive methods for determination of NAT2 phenotype would be quite valuable, a topic we discuss later in this review.
A prior review published in this journal [Citation20] focused on the effects of individual single nucleotide polymorphisms on NAT2 expression and activity. In this focused review, data is presented and reviewed that more clearly defines the effects of NAT2 haplotypes and genotypes on the expression of acetylator phenotype towards selected drugs within human hepatocytes in vitro, within human hepatocyte cultures in situ, and clinical measures such as bioavailability (area under the plasma versus time curve), plasma metabolic ratios of parent to N-acetyl metabolite, plasma half-life, and/or clearance determinations in human subjects. As these relationships are better defined and understood, we include drugs for which NAT2 phenotype-guided therapy has been proposed. We conclude with a more detailed discussion of the value of pharmacogenomics-guided isoniazid therapy for the prevention and treatment of tuberculosis (TB) as a paradigm for NAT2 phenotype-dependent dosing strategies to reduce risk of adverse reactions, increase therapeutic efficacy, reduce costs and improve patient care and disease prevention.
2. Isoniazid (INH)
The role of NAT2 and its genetic polymorphism in the metabolism and pharmacokinetic profile of INH has been reviewed [Citation4,Citation27]. INH N-acetylation in human subjects clearly differs with respect to rapid, intermediate and slow acetylator NAT2 genotypes with respect to plasma half-life, bioavailability (area under the curve), plasma metabolic ratio of INH to N-acetyl-INH, and clearance (). The NAT2 genotype dependent pharmacokinetic parameters measured in human subjects have been confirmed by measurement of INH N-acetylation both in vitro [Citation28] and in situ () in cryopreserved human hepatocytes. The extensive use of INH for the treatment and prevention of TB is compromised by INH-induced hepatotoxicity and liver failure. A study conducted in Brazil found that the incidence of INH-induced hepatotoxicity differed among rapid (2.9%), intermediate (9.8%), and slow (22%) NAT2 acetylators [Citation29]. Meta-analysis studies report slow acetylators were significantly more likely to experience hepatotoxicity from INH treatment for tuberculosis than rapid acetylators [Citation30,Citation31].
Figure 1. INH pharmacokinetics in subjects with rapid, intermediate, and slow NAT2 acetylator genotype
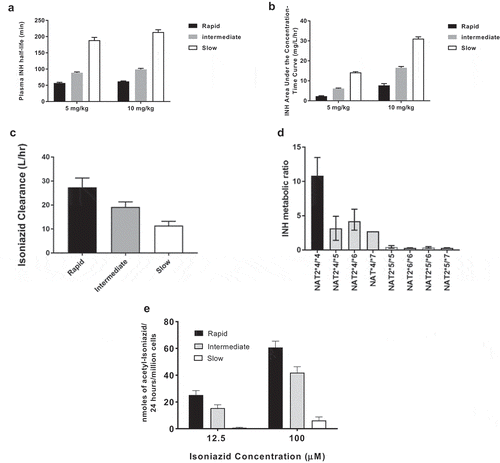
Other studies have shown an effect of NAT2 genotype on INH efficacy [Citation35,Citation36]. One randomized trial of pharmacogenomics-guided INH therapy found a significant reduction in hepatotoxicity in slow acetylators who were randomized to a lower dose of INH, as well as a reduction in 2-month culture positivity in NAT2 rapid acetylators who received a higher dose [Citation37]. Meta-analyses found that rapid acetylators are at increased risk of treatment failure, relapse, and drug resistance [Citation38]. Several studies have proposed pharmacogenomics-guided INH therapy for TB [Citation29,Citation38–43].
3. Hydralazine (HYD)
HYD has been used for decades in the treatment of hypertension and is indicated in the long-term therapy of essential hypertension, in the short-term therapy of pregnancy-induced hypertension and eclampsia, and in the therapy of hypertensive crisis [Citation44,Citation45]. As recently reviewed [Citation46], adverse effects of HYD include headache, reflex tachycardia, and/or angina pectoris. Another far less common adverse effect is a systemic lupus erythematosus (SLE)-like syndrome which although infrequent, has potential serious and long-term consequences. There is substantial evidence supporting the correlation between NAT2 acetylator phenotype or genotype and urine and plasma hydralazine concentrations (reviewed in [Citation46]). HYD treatment for hypertension and resistant hypertension is modified by acetylator phenotype following oral administration [Citation15, Citation16, Citation47]. In early studies in patients with hypertension [Citation48], HYD plasma concentrations varied as much as 15-fold among individuals reflecting an ‘acetylator index’ in the N-acetylation of SMZ. Differences in HYD metabolism in the rapid and slow acetylators varied with dose, the greatest difference being after a 200‐mg (100 mg twice daily) dose and became less clearly bimodal at lower doses, with overlap between phenotypes occurring at doses of 100 mg (50 mg twice daily) or less [Citation47]. DNA samples from 169 patients with resistant hypertension treated with hydralazine were identified as rapid (12.4%), intermediate (38.5%) and slow (35.5%) acetylators (13.6% indeterminate) and only slow acetylators had significant blood pressure reductions although they also had a higher incidence of adverse effects [Citation49]. Recommendations for NAT2 acetylator phenotype-guided HYD treatment for hypertension and resistant hypertension have been proposed [Citation46]. Rapid and intermediate acetylators with resistant hypertension are recommended a 50–100% higher initial dose of HDZ with a total daily dose limit of 300 mg. In contrast, slow acetylators are expected to experience increased hydralazine levels on standard dosing protocols and thus increased efficacy and/or adverse effects, and clinicians should exhibit caution with HYD daily doses of 200 mg or higher.
HYD is currently in clinical trials in combination with valproate for epigenetic cancer therapy [reviewed in Citation50]. In a 2011 study, HYD plasma levels were evaluated in 26 healthy volunteers after a single oral dose of 182 mg of a controlled-release HYD tablet [Citation51]. HYD area under the curve plasma levels were 2.2-fold higher in slow than in rapid acetylators, which subsequently has been used as rationale for prescribing hydralazine doses 2.2-fold lower in slow (83 mg) than rapid (182 mg) acetylators. This dose adjustment yielded similar plasma levels of HYD in rapid and slow acetylators in several studies [Citation51–53]. They appear to be the experimental basis for clinical trials using a HYD dosing strategy of 83 mg in slow and 183 mg in rapid acetylators (either alone or in combination with other drugs such as valproic acid) for cancer treatment.
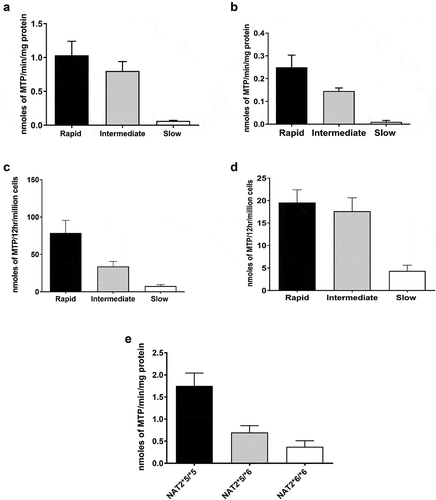
NAT2 genotype-dependent N-acetylation of HYD has been documented in human hepatocytes both in vitro and in situ at multiple doses (). Confirming the earlier findings in patients [Citation47], the effect of NAT2 genotype on N-acetylation of HDZ was more pronounced at higher concentrations. Data in cryopreserved human hepatocytes further support that hydralazine efficacy and safety could be improved by NAT2 genotype-dependent dosing strategies. More specific dosing strategies such as those described above, could facilitate maximal therapeutic benefit from hydralazine.
Figure 2. HYD N-acetyltransferase activities in vitro in cryopreserved human hepatocytes from rapid, intermediate and slow NAT2 acetylators
4. Sulfamethazine (sulfadimidine)
Sulfamethazine (SMZ), also identified as sulfadimidine, is a sulfonamide antibiotic that has historically been effectively used as a probe drug for assessment of NAT2 acetylator phenotype [Citation13,Citation14]. Although it is primarily used for veterinary indications at present, we include it here because of fairly extensive data obtained in human hepatocytes as well as in human subjects. Although early studies in human subjects provided metabolism data in serum and urine that appeared bimodal [Citation14], more recent studies provide clear evidence for acetylator genotype-dependent N-acetylation of sulfamethazine. As illustrated in , SMZ N-acetyltransferase activities and N-acetylation in situ in cultures of cryopreserved human hepatocytes clearly differ with rapid, intermediate or slow acetylator genotype. This finding is also confirmed when measuring SMZ/N-acetyl-SMZ metabolic ratios in plasma of human subjects from two populations which differ markedly in the frequency of rapid and slow NAT2 acetylator phenotype. Thus, the frequency of slow NAT2 phenotype is much more frequent in Mexico [Citation55] and the frequency of rapid NAT2 phenotype is much more frequent in China [Citation56]. Nevertheless, the metabolic ratios of SMZ to N-acetyl-SMZ in plasma of subjects from Mexico (both healthy and those with cancer) as well as in China (healthy) are nearly identical in rapid, intermediate, and slow acetylator phenotypes ()).
Figure 3. (a) SMZ N-acetyltransferase catalytic activities in cryopreserved human hepatocyte samples
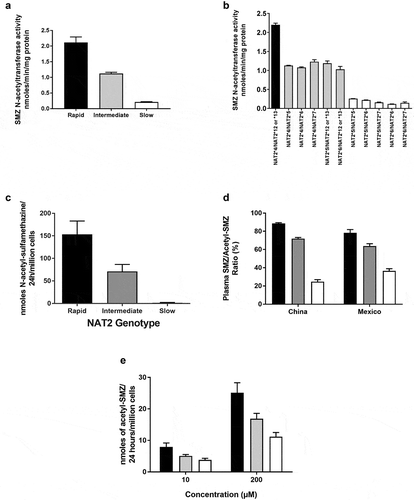
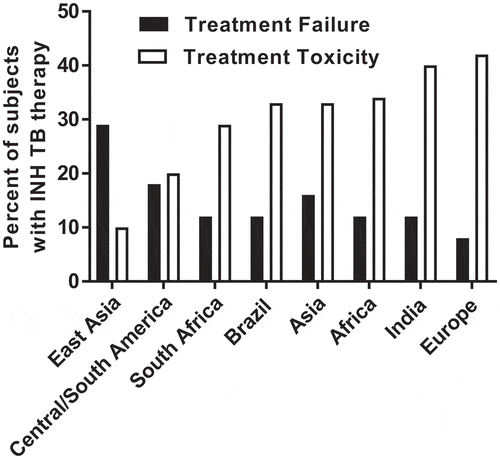
Figure 4. Estimated frequency of treatment failure and toxicity of INH treatment for tuberculosis in populations across the world
5. Amifampridine
Amifampyridine is marketed for the symptomatic treatment of Lambert-Eaton myasthenic syndrome in adults [Citation60]. Amifampridine is metabolized by NAT2 to 3-N-acetyl-amifampridine, which is considered as an inactive metabolite. Although investigations to assess three phenotypes (rapid, intermediate, and slow) have not to our knowledge been explored, significant differences between rapid and slow acetylators have been reported in numerous pharmacokinetic parameters for both amifampridine and 3-N-acetyl-amifampridine (). In addition, treatment-emergent adverse events from amifampridine were more frequent in slow acetylators [Citation61]. The United States of America Food and Drug Administration presently lists an NAT2 acetylator phenotype dosing strategy for amifampridine (https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations). In slow acetylators, the lowest recommended dose of 15 mg/day should be used with monitoring for adverse effects.
Table 2. Amifampridine pharmacokinetic parameters in rapid and slow NAT2 acetylators
6. Procainamide
Procainamide has been used in the treatment of cardiovascular disorders including arrythmias. An adverse effect of procainamide is the production of antinuclear antibodies described as a drug-induced lupus like syndrome. The duration of therapy required to induce antibodies in 50% of slow (11) and rapid (9) acetylators was 2.9 and 7.3 months and the median total dose that produced antibodies was 1.5 g per kilogram and 6.1 g per kilogram in slow and rapid NAT2 acetylators, respectively [Citation62]. Retrospective studies of patients in whom procainamide lupus had developed revealed that the duration of therapy required for induction in 14 slow and seven rapid acetylators was 12 ± 5 and 48 ± 22 months respectively (P < 0.002). The N‐acetylprocainamide/procainamide ratio in urinary excretion was 0.60 ± 0.17 (mean ± SD) for those with NAT2*4/*4, 0.37 ± 0.06 for NAT2*4/*6A, 0.40 ± 0.03 for NAT2*4/*7B, and 0.17 for NAT2*6A/*7B [Citation63].
7. Sulfasalazine
Sulfasalazine is used to treat rheumatoid arthritis and other autoimmune diseases, such as ankylosing spondylitis, Crohn’s disease, and ulcerative colitis [Citation4]. It is a combination of 5-aminosalicyclic acid and sulfapyridine linked together by an azo bond. Sulfasalazine consists of two components, 5-aminosalicylate and sulfapyridine (SP). SP is one of the first drugs or metabolites shown to be subject to the N-acetylation polymorphism [Citation64] and NAT2 genotype-related differences in sulfasalazine pharmacokinetic parameters, efficacy, and/or adverse effects are thought to be mediated via SP.
NAT2 genotype modified the pharmacokinetics, efficacy, and incidence of adverse reactions of sulfasalazine in patients treated for rheumatoid arthritis. NAT2 genotypes were determined and the plasma concentration ratios of sulfapyridine (SP) to N-acetyl- sulfapyridine (SP/AcSP) and the efficacy of sulfasalazine (p < 0.05) were significantly different among rapid, intermediate and slow acetylator phenotypes [Citation65]. Subsequent studies reported a gene dose response in AcSP/SP AUC ratios (mean ± SD) of 3.1 ± 0.6, 1.9 ± 0.5, and 0.5 ± 0.4 in rapid, intermediate, and slow acetylators, respectively which differed significantly (p < 0.0001) among the three phenotypes [Citation66]. AcSP Cmax also exhibits a gene dose-response with levels of 12.67 ± 3.32, 9.07 ± 2.29 and 4.22 ± 0.93 mg/l, in rapid, intermediate, and slow acetylator genotypes, respectively [Citation67]. Although variable results have been reported in small individual studies regarding the role of adverse effects in sulfasalazine adverse effects, nine cohort studies involving 1,077 patients were recently reviewed for an effect of NAT2 phenotype [Citation68]. This analysis [Citation68] reported a fold increase in overall adverse drug reactions (OR 3.37, 95% CI: 1.43 to 7.93; p = 0.005), nearly a fold discontinuation due to overall adverse drug reactions (OR 2.89, 95% CI: 1.72 to 4.86; p < 0.0001), and a five-fold increase in dose-related adverse drug reactions (OR 5.20, 95% CI: 2.44 to 11.08; p < 0.0001) in slow compared with rapid and intermediate acetylators suggesting that NAT2 genotyping may be useful to predict the occurrence of adverse drug reactions during sulfasalazine treatment.
8. Amonafide
Amonafide is marketed in the treatment of various cancers. It is N-acetylated by NAT2 and the N-acetylated metabolite is active and causes myelosuppression. Plasma N-acetyl-amonafide levels were measured 45 min and 24 hr after administration of amonafide at a dose of 300 mg/m2 over 60 min. Plasma levels of N-acetyl-amonafide were substantially higher in slow than rapid acetylators both 45 min (585 vs 130 ng/ml) and 24 hr (112 vs 11 ng/ml) after amonafide administration [Citation69]. At standard dosing schedules, rapid acetylators experienced higher incidence of myelosuppression suggesting it is mediated by N-acetyl-amonafide [Citation69,Citation70]. In subsequent studies, amonafide has minimized toxicity in rapid acetylators when adopting acetylator phenotype-dependent dosing [Citation71,Citation72].
9. Metamizole
As described in a comprehensive review [Citation73], metamizole is a non-steroidal antinflammatory in use for nearly a century for its analgesic, antipyretic, and spasmolytic properties. Metamizole was extensively applied worldwide until incidences of hypersensitivity leading to agranulocytosis led to its ban in a number of countries including the USA. Nevertheless, metamizole is still available in many countries worldwide either by prescription or over the counter for indications such as pain due to cancer, colic postoperative, headache, or acute injuries. A recent review found it as efficacious as 60 mg oral morphine/day in the treatment of cancer pain [Citation74]. In countries where metamizole is sold over the counter, the drug is used as a common self‐medication and is commonly used for chronic pain. Metamizole also is widely used by immigrants, even in countries where the substance is banned such as among Latino families in the United States. A single‐site analysis in a US urban pediatric hospital revealed that over one‐third of all Spanish‐speaking Latino families had used metamizole [Citation74,Citation75]. In addition, metamizole is increasingly used as an adulterant in illicit drugs [Citation73].
Metamizole is a prodrug. After oral administration, it is rapidly hydrolyzed in the gastric juice first to 4-methyl-amino-antipyrine, and then to multiple metabolites including 4-amino-antipyrine, which is further metabolized to 4-acetyl-amino-antipyrine catalyzed by NAT2. An acetylation ratio (4-acetyl-amino-anipyrine/4-amino-antipyrine) was reported as (mean ± SD) 16.0 ± 10.1 in homozygous rapid NAT2*4/*4 acetylators, 10.7 ± 8.6 and 11.1 ± 7.1 in heterozygous intermediate NAT2*4/*5 and NAT2*4/*6 acetylators, and 6.83 ± 3.91 and 5.19 ± 3.68 in homozygous slow NAT2*5/*5 and NAT2*6/*6 acetylators [Citation76]. In a subsequent study [Citation77], slow acetylator NAT2 phenotype was associated with an increased risk of developing selective hypersensitivity to metamizole [odds ratio = 2.17 (95% CI 1.44–3.27); p = 0.00016], particularly anaphylaxis [odds ratio = 4.77 (95% CI 2.28–9.98); p = 0.000006]. In contrast, agranulocytosis from metamizole is rare with no data suggesting that it is related to NAT2 phenotype. Thus both metamizole metabolism [Citation76] and toxicity [Citation77] were NAT2 genotype-dependent. Slow NAT2 acetylator phenotype also has been associated with the incidence of infant leukemia in mothers exposed to metamizole during pregnancy particularly when both mother and child were slow NAT2 acetylators [Citation78].
10. Aliphatic amine drugs subject to the NAT2 acetylation polymorphism
Most if not all of the drugs identified as subject to NAT2 genetic polymorphism are aromatic amine or hydrazine drugs. Some of them (such as clonazepam) are metabolized to an aromatic amine metabolite which is then subject to NAT2 genetic polymorphism [Citation79]. A recent new study [Citation80] provides data suggesting that NAT2 substrate specificity may also extend to drugs with aliphatic amine functional groups. Using cell-based and in vitro assays, they report acetylation of several endogenous metabolites and major drugs that have not previously been described as substrates for NAT2. The study [Citation80] investigated eight representative drugs possessing aliphatic amines to determine whether they are acetylated by NAT2. The calcium channel blocker amlodipine, the serotonin-norepinephrine inhibitor duloxetine, the beta blockers nebivolol and carvedilol were found to be N-acetylated by NAT2. These drugs are used to treat and prevent prevalent conditions such as hypertension, stroke, fibromyalgia, anxiety, and depression and over 20% of the 200 most prescribed drugs in the USA contain aliphatic amines, representing almost 900 million prescriptions [Citation80]. This is a new and unconfirmed report which did not assess the effects of acetylator phenotype on metabolism, efficacy, or toxicity. Nevertheless, the finding may substantially expand utilization of NAT2 acetylator phenotype assessments in pharmacogenomics-guided therapy.
11. Genetic heterogeneity within the slow acetylator phenotype
Early [Citation8,Citation9] and more recent [Citation81,Citation82] studies investigating the metabolism of an acetylator phenotype probe (caffeine) provided data consistent with the existence of a very slow acetylator phenotype. HYD ()) and SMZ ()) N-acetylation rates within the slow acetylator phenotype follow the rank order of NAT2*5B/*5B > NAT2*5B/*6A > NAT2*6A/NAT2*6A in cryopreserved human hepatocytes. A recent review and meta-analysis [Citation39] included 18 studies with 822 cases of anti-tuberculosis drug-induced liver injury and 4630 controls. A more robust association was noted with liver injury in ultra-slow NAT2 acetylators (OR: 3.60; 95% CI: 2.30–5.63) compared to all NAT2 slow acetylators (OR: 2.80; 95% CI: 2.20–3.57). Following administration of metamizole, homozygous NAT2*6 slow acetylators displayed significantly lower 4-acetyl-amino-antipyrene recovery and lower acetylation ratio than homozygous NAT2*5 slow acetylators [Citation76]. NAT2*7B also has been linked to very slow acetylator phenotype, but this phenotype is likely substrate-dependent [Citation83].
12. Pharmacogenomic-guided isoniazid therapy for treatment of TB
Tuberculosis (TB) is a communicable disease that is a major cause of ill health in about 10 million people [Citation84]. The current COVID-19 pandemic is exacerbating the effect of TB on certain populations. In China, India, and Indonesia, national TB case detection dropped by 20%, 75%, and 68%, respectively, following dates of strict national lockdown implementation that is predicted to cause an increase in 2020 TB related deaths – up to 380,000 deaths [Citation85]. Prevalence of TB is heaviest in less developed countries with limited access to laboratory testing. It is no coincidence that the areas of the world predicted to be most affected by the social and economic consequences of COVID-19 are also the areas with the highest TB burden [Citation86]
Isoniazid is widely used for treatment and prevention of TB [Citation4,Citation87]. Pharmacogenomic-guided INH therapy for TB is proposed [Citation88] due to differences in metabolism summarized in this article. Due to the lack of laboratory testing availability in areas with heaviest use of isoniazid for TB, having access to a simple, cost effect point of care (POC) test in the clinic would provide the patient immediate access to the correct dose, avoiding adverse effects while maximizing treatment efficacy. Knowing the correct dose of isoniazid at the first visit could also reduce frequency of in-office visits, which are being limited during the current coronavirus pandemic. The ability to prescribe the correct dose may also allow this affordable medication to be used more widely for prevention in at risk populations by mitigating risk of adverse events. Saunders et al [Citation86] suggests national tuberculosis programs could use locally derived, simple risk stratification tools to focus interventions such as active case finding to provide preventive treatment to highest risk households. Knowing the correct preventative dose would again reduce frequency of in-office visits during this current pandemic. This strategy would help reduce mortality rates of COVID exacerbated TB.
Fortunately, timely diagnosis and treatment with first-line antibiotics, including INH can be effective in the treatment and prevention of TB disease. Nevertheless, INH toxicity and treatment failure in some patients leads to additional healthcare costs, morbidity and mortality. As shown in , standard weight-based INH therapy for TB results in over 40% of patients experiencing treatment toxicity and over 30% of patients experiencing treatment failure in populations across the world. Using a robust analysis model of TB treatment costs, Rens and coworkers [Citation89] document that pharmacogenomics-guided therapy is highly cost-effective even with conservative estimates of its impact on INH-induced liver injury and response to therapy.
13. Expert opinion
The adoption of clinical pharmacogenomic testing in general has been slow for many reasons including ambiguous or conflicting study results, lack of consensus guidelines, lack of insurance coverage, and reports with unclear interpretations. However, the field is gaining momentum with large academic medical centers generating real-world data. The Clinical Pharmacogenetics Implementation Consortium of the National Institutes of Health’s Pharmacogenomics Research Network (http://www.pgrn.org) and the Pharmacogenomics Knowledge Base (PharmGKB, http://www.pharmgkb.org) provides peer-reviewed, updated, evidence-based, freely accessible guidelines for gene/drug pairs to facilitate the translation of pharmacogenomic knowledge from bench to bedside [Citation90]. As evidence continues to demonstrate benefits of genetic testing, more insurance companies and other payers are covering costs.
Haga has summarized the challenges of development and implementation of POCTs related to pharmacogenetic testing [Citation91]. One of the major issues is provider training. Providers need to be knowledgeable of pharmacogenomics in general and have the skill to perform the test. Several reports have described providers’ limited knowledge and/or experience with pharmacogenetic testing and interpretation. In some regions, another potential roadblock to routine use is the need to connect results from POCT to a patient’s electronic medical record. Another potential roadblock is cost. Many POCTs have a higher per test cost compared to traditional lab tests in addition to the cost and time associated with submission to the FDA for clearance. POCTs are required to obtain FDA clearance while many molecular tests performed in a laboratory are considered laboratory developed tests and are not required to have FDA clearance [Citation91]. Despite these potential roadblocks, there is a lot of effort being put forth in developing these types of tests. Combining interest in pharmacogenomic tests with continuous technology development will likely drive development of these tests in the near future.
The United States Food and Drug Administration (FDA) publishes tables identifying Pharmacogenetic Associations (https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations) that it has evaluated and believes there is sufficient scientific evidence to suggest that subgroups of patients with certain genetic variants, or genetic variant-inferred phenotypes are likely to have altered drug metabolism, and in certain cases, differential therapeutic effects, including differences in risks of adverse events. Amifampridine is listed in the first table for which the data support therapeutic management recommendations while isoniazid and sulfasalazine are listed in a second table of for which the data indicate a potential impact on safety or response. HDZ is included in a third table for which the data demonstrate a potential impact on pharmacokinetic properties only. It is likely that studies such as those reviewed here will expand the listing including updated classifications, particularly for isoniazid treatment of TB.
The body of evidence surrounding the relationship between NAT2 genotype and the effect on patient outcomes is compelling and should drive development for everyday clinical use. The accumulated data on NAT2 clinical significance substantiates the need to replace standard weight-based therapy practices with pharmacogenomics-guided therapies. It has demonstrated economic benefit, reduced adverse effects, and increased efficacy. Before this can go into practice, however, standardization is needed with clear guidelines for treatment.
In the future, it should be the standard of care for physicians and other members of the health care team to use pharmacogenomic information to guide treatment for improved success and decreased adverse effects. NAT2 genotyping should be among those soon widely used pharmacogenomic tools. Incorporation of more robust NAT2 genotype/phenotypes relationships, including genetic heterogeneity within the slow acetylator phenotype should lead to further advancements in both health outcomes and cost benefit of TB treatment. The ultimate goal would be to simultaneously offer NAT2 genotyping as a companion diagnostic. One area of particular need is a point of care test used in conjunction with INH and TB treatment. TB is a major global healthcare concern – it is one of the top 10 causes of death and the leading cause of death from a single infectious agent. The current COVID-19 pandemic is also exacerbating the effect of TB on certain populations. Prevalence of TB is heaviest in less developed countries with limited access to laboratory testing. Of greatest benefit would be point of care testing to enable the patient immediate access to the correct dose to avoid adverse effects while maximizing treatment efficacy. The major needs for this to be adopted are development of a cost-effective point of care and clear guidelines for treatment.
Article highlights
The effects of the N-acetylation polymorphism on metabolism, efficacy, and/or toxicity of numerous drugs is described, including:
isoniazid
hydralazine
sulfamethazine
amifampridine
procainamide
sulfasalazine
amonafide
metamizole
Studies in human subjects and cryopreserved human hepatocytes show evidence for rapid, intermediate and slow acetylator phenotypes, with further data suggesting genetic heterogeneity within the slow acetylator phenotype.
The current COVID-19 pandemic is exacerbating effects of tuberculosis in many countries.
Point of care testing for NAT2 phenotype/genotype and providing dose guidance could improve safety and efficacy of isoniazid for tuberculosis prevention and treatment.
More robust methods for assessing N-acetylation genotype and/or phenotype should lead to further advancements in health outcomes and cost benefits.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Laurieri N, Sim E, editors. Arylamine N-acetyltransferases in health and disease: from pharmacogenetics to drug discovery and diagnostics. Singapore: World Scientific Publishing; 2018. ISBN: 978-981-3232-00-6.
- Husain A, Zhang X, Doll MA, et al. Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantitation of NAT2-specific mRNA in human tissues. Drug Metab Dispos. 2007 May;35(5):721–727.
- Weber WW, Hein DW. N-acetylation pharmacogenetics. Pharmacol Rev. 1985 Mar;37(1):25–79.
- McDonagh EM, Boukouvala S, Aklillu E, et al. PharmGKB summary: very important pharmacogene information for N-acetyltransferase 2. Pharmacogenet Genomics. 2014 Aug;24(8):409–425.
- Agundez JA, Garcia-Martin E. Human arylamine N-acetyltransferases type 2: phenotype correlation with genotype-A clinical perspective. In: Laurieri N, Sim E, editors. Chapter 1.3, Arylamine N-acetyltransferases in health and disease: from pharmacogenetics to drug discovery and diagnostics. Singapore: World Scientific Publishing; 2018. ISBN: 978-981-3232-00-6.
- Mitchell SC. N-acetyltransferase: the practical consequences of polymorphic activity in man. Xenobiotica. 2020 Jan;50(1):77–91.
- Husain A, Zhang X, Doll MA, et al. Functional analysis of the human N-acetyltransferase 1 major promoter: quantitation of tissue expression and identification of critical sequence elements. Drug Metab Dispos. 2007 Sept;35(9):1649–1656.
- Cascorbi I, Drakoulis N, Brockmöller J, et al. Arylamine N-acetyltransferase (NAT2) mutations and their allelic linkage in unrelated Caucasian individuals: correlation with phenotypic activity. Am J Hum Genet. 1995 Sept;57(3):581–592.
- Cascorbi I, Brockmöller J, Mrozikiewicz PM, et al. Arylamine N-acetyltransferase activity in man. Drug Metab Rev. 1999 May;31(2):489–502.
- Evans DA, Manley KA, McKusick VA. Genetic control of isoniazid metabolism in man. Br Med J. 1960 Aug 13;2(5197):485–491.
- Gelber R, Peters JH, Gordon GR, et al. The polymorphic acetylation of dapsone in man. Clin Pharmacol Ther. 1971 Mar–Apr;12(2):225–238.
- Campbell W, Tilstone WJ, Lawson DH, et al. Acetylator phenotype and the clinical pharmacology of slow-release procainamide. Br J Clin Pharmacol. 1976 Dec;3(6):1023–1026.
- Evans DA, White TA. Human acetylation polymorphism. J Lab Clin Med. 1964 Mar;63:394–403.
- Evans DA. An improved and simplified method of detecting the acetylator phenotype. J Med Genet. 1969 Dec;6(4):405–407.
- Timbrell JA, Harland SJ, Facchini V. Polymorphic acetylation of hydralazine. Clin Pharmacol Ther. 1980 Sept;28(3):350–355.
- Facchini V, Timbrell JA. Further evidence for an acetylator phenotype difference in the metabolism of hydralazine in man. Br J Clin Pharmacol. 1981 Apr;11(4):345–351.
- Clark DW. Genetically determined variability in acetylation and oxidation. Therapeutic implications. Drugs. 1985 Apr;29(4):342–375.
- Relling MV. Polymorphic drug metabolism. Clin Pharm. 1989 Dec;8(12):852–863.
- Hein DW, Doll MA, Fretland AJ, et al. Molecular genetics and epidemiology of the NAT1 and NAT2 acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000 Jan;9(1):29–42.
- Hein DW. N-acetyltransferase SNPs: emerging concepts serve as a paradigm for understanding complexities of personalized medicine. Expert Opin Drug Metab Toxicol. 2009 Apr;5(4):353–366.
- Hein DW, Doll MA. Accuracy of various human NAT2 SNP genotyping panels to infer rapid, intermediate and slow acetylator phenotypes. Pharmacogenomics. 2012 Jan;13(1):31–41.
- Hein DW. N-acetyltransferase 2 genetic polymorphism: effects of carcinogen and haplotype on urinary bladder cancer risk. Oncogene. 2006 Mar 13;25(11):1649–1658.
- Walker K, Ginsberg G, Hattis D, et al. Genetic polymorphism in N-acetyltransferase (NAT): population distribution of NAT1 and NAT2 activity. J Toxicol Env Heal B. 2009;12(5–6):440–472.
- Hein DW, Doll MA. Catalytic properties and heat stabilities of novel recombinant human N-acetyltransferase 2 allozymes support existence of genetic heterogeneity within the slow acetylator phenotype. Arch Toxicol. 2017 Aug;91(8):2827–2835.
- Deitz AC, Rothman N, Rebbeck TR, et al. Impact of misclassification in genotype-exposure interaction studies: example of N-acetyltransferase 2 (NAT2), smoking, and bladder cancer. Cancer Epidemiol Biomarkers Prev. 2004 Sept;13(9):1543–1546.
- Selinski S, Blaszkewicz M, Lehmann ML, et al. Genotyping NAT2 with only two SNPs (rs1041983 and rs1801280) outperforms the tagging SNP rs1495741 and is equivalent to the conventional 7-SNP NAT2 genotype. Pharmacogenet Genomics. 2011 Oct;21(10):673–678.
- Weber WW, Hein DW. Clinical pharmacokinetics of isoniazid. Clin Pharmacokinet. 1979;4:401–422.
- Doll MA, Salazar-González RA, Bodduluri S, et al. Arylamine N-acetyltransferase 2 genotype-dependent N-acetylation of isoniazid in cryopreserved human hepatocytes. Acta Pharm Sin B. 2017 Jul 7;4:517–522.
- Teixeira RL, Morato RG, Cabello PH, et al. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Memorias Do Instituto Oswaldo Cruz. 2011 Sept;106(6):716–724.
- Richardson M, Kirkham J, Dwan K, et al. NAT2 variants and toxicity related to anti-tuberculosis agents: a systematic review and meta-analysis. Int J Tuberc Lung Dis. 2019 Mar 1;23(3):293–305.
- Khan S, Mandal RK, Elasbali AM, et al. Pharmacogenetic association between NAT2 gene polymorphisms and isoniazid induced hepatotoxicity: trial sequence meta-analysis as evidence. Biosci Rep. 2019 Jan 31;39:1.
- Parkin DP, Vandenplas S, Botha FJ, et al. Trimodality of isoniazid elimination: phenotype and genotype in patients with tuberculosis. Am J Respir Crit Care Med. 1997 May;155(5):1717–1722.
- Huerta-García A, Medellín-Garibay S, Ortiz-Álvarez A, et al. Population pharmacokinetics of isoniazid and dose recommendations in Mexican patients with tuberculosis. Int J Clin Pharm. 2020. DOI:10.1007/s11096-020-01086-1.
- Smith CA, Wadelius M, Gough AC, et al. A simplified assay for the arylamine N-acetyltransferase 2 polymorphism validated by phenotyping with isoniazid. J Med Genet. 1997 Sept;34(9):758–760.
- Donald PR, Parkin DP, Seifart HI, et al. The influence of dose and N-acetyltransferase-2 (NAT2) genotype and phenotype on the pharmacokinetics and pharmacodynamics of isoniazid. Eur J Clin Pharmacol. 2007 July;63(7):633–639.
- Pasipanodya JG, Srivastava S, Gumbo T. Meta-analysis of clinical studies supports the pharmacokinetic variability hypothesis for acquired drug resistance and failure of antituberculosis therapy. Clin Infect Dis. 2012 July;55(2):169–177.
- Azuma J, Ohno M, Kubota R, et al. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: a randomized controlled trial for pharmacogenetics-based therapy. Eur J Clin Pharmacol. 2013 May;69(5):1091–1101.
- Jung JA, Kim TE, Lee H, et al. A proposal for an individualized pharmacogenetic-guided isoniazid dosage regimen for patients with tuberculosis. Drug Des Devel Ther. 2015;9:5433–5438.
- Suvichapanich S, Fukunaga K, Zahroh H, et al. NAT2 ultra-slow acetylator and risk of anti-tuberculosis drug-induced liver injury: a genotype-based meta-analysis. Pharmacogenet Genomics. 2018 July;28(7):167–176.
- Matsumoto T, Ohno M, Azuma J. Future of pharmacogenetics-based therapy for tuberculosis. Pharmacogenomics. 2014 Apr;15(5):601–607.
- Choi R, Jeong BH, Koh WJ, et al. Recommendations for optimizing tuberculosis treatment: therapeutic drug monitoring, pharmacogenetics, and nutritional status considerations. Ann Lab Med. 2017 Mar;37(2):97–107.
- Motta I, Calcagno A, Bonora S. Pharmacokinetics and pharmacogenetics of anti-tubercular drugs: a tool for treatment optimization? Expert Opin Drug Metab Toxicol. 2018 Jan;14(1):59–82.
- Jing W, Zong Z, Tang B, et al. Population pharmacokinetic analysis of isoniazid among pulmonary tuberculosis patients from China. Antimicrob Agents Chemother. 2020 Feb 21;64:3.
- Cohn JN, McInnes GT, Shepherd AM. Direct-acting vasodilators. J Clin Hypertens (Greenwich, CT). 2011 Sept;13(9):690–692.
- Han LW, Ryu RJ, Cusumano M, et al. Effect of N-acetyltransferase 2 genotype on the pharmacokinetics of hydralazine during pregnancy. J Clin Pharmacol. 2019 Dec;59(12):1678–1689.
- Collins KS, Raviele ALJ, Elchynski AL, et al. Genotype-guided hydralazine therapy. Am J Nephrol. 2020;14:1–13.
- Timbrell JA, Harland SJ, Facchini V. Effect of dose on acetylator phenotype distribution of hydralazine. Clin Pharmacol Ther. 1981 Mar;29(3):337–343.
- Shepherd AM, McNay JL, Ludden TM, et al. Plasma concentration and acetylator phenotype determine response to oral hydralazine. Hypertens. 1981 Sept-Oct;3(5):580–585.
- Spinasse LB, Santos AR, Suffys PN, et al. Different phenotypes of the NAT2 gene influences hydralazine antihypertensive response in patients with resistant hypertension. Pharmacogenomics. 2014 Feb;15(2):169–178.
- Dueñas-Gonzalez A, Coronel J, Cetina L, et al. Hydralazine-valproate: a repositioned drug combination for the epigenetic therapy of cancer. Expert Opin Drug Metab Toxicol. 2014 Oct;10(10):1433–1444.
- Gonzalez-Fierro A, Vasquez-Bahena D, Taja-Chayeb L, et al. Pharmacokinetics of hydralazine, an antihypertensive and DNA-demethylating agent, using controlled-release formulations designed for use in dosing schedules based on the acetylator phenotype. Int J Clin Pharmacol Ther. 2011 Aug;49(8):519–524.
- Arce C, Pérez-Plasencia C, González-Fierro A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One. 2006 Dec 20;1(1):e98.
- Garcés-Eisele SJ, Cedillo-Carvallo B, Reyes-Núñez V, et al. Genetic selection of volunteers and concomitant dose adjustment leads to comparable hydralazine/valproate exposure. J Clin Pharm Ther. 2014 Aug;39(4):368–375.
- Allen CE, Doll MA, Hein DW. N-acetyltransferase 2 genotype-dependent N-acetylation of hydralazine in human hepatocytes. Drug Metab Dispos. 2017 Dec;45(12):1276–1281.
- Taja-Chayeb L, González-Fierro A, Miguez-Muñoz C, et al. Acetylator status and N-acetyltransferase 2 gene polymorphisms; phenotype-genotype correlation with the sulfamethazine test. Pharmacogenet Genomics. 2011 Dec;21(12):894–901.
- Chen B, Zhang WX, Cai WM. The influence of various genotypes on the metabolic activity of NAT2 in a Chinese population. Eur J Clin Pharmacol. 2006 May;62(5):355–359.
- Doll MA, Zang Y, Moeller T, et al. Codominant expression of N-acetylation and O-acetylation activities catalyzed by N-acetyltransferase 2 in human hepatocytes. J Pharmacol Exp Ther. 2010 Aug;334(2):540–544.
- Habil MR, Doll MA, Hein DW. N-acetyltransferase 2 acetylator genotype-dependent N-acetylation of 4-aminobiphenyl in cryopreserved human hepatocytes. Pharmacogenet Genomics. 2020 Apr;30(3):61–65.
- Doll MA, Hein DW. Genetic heterogeneity among slow acetylator N-acetyltransferase 2 phenotypes in cryopreserved human hepatocytes. Arch Toxicol. 2017 July;91(7):2655–2661.
- Mantegazza R. Amifampridine tablets for the treatment of lambert-eaton myasthenic syndrome. Expert Rev Clin Pharmacol. 2019 Nov;12(11):1013–1018.
- Haroldsen PE, Garovoy MR, Musson DG, et al. Genetic variation in aryl N-acetyltransferase results in significant differences in the pharmacokinetic and safety profiles of amifampridine (3,4-diaminopyridine) phosphate. Pharmacol Res Perspect. 2015 Feb;3(1):e00099.
- Woosley RL, Drayer DE, Reidenberg MM, et al. Effect of acetylator phenotype on the rate at which procainamide induces antinuclear antibodies and the lupus syndrome. N Engl J Med. 1978 May 25;298(21):1157–1159.
- Okumura K, Kita T, Chikazawa S, et al. Genotyping of N-acetylation polymorphism and correlation with procainamide metabolism. Clin Pharmacol Ther. 1997 May;61(5):509–517.
- Schröder H, Evans DA. The polymorphic acetylation of sulphapyridine in man. J Med Genet. 1972 June;9(2):168–171.
- Kumagai S, Komada F, Kita T, et al. N-acetyltransferase 2 genotype-related efficacy of sulfasalazine in patients with rheumatoid arthritis. Pharm Res. 2004 Feb;21(2):324–329.
- Yamasaki Y, Ieiri I, Kusuhara H, et al. Pharmacogenetic characterization of sulfasalazine disposition based on NAT2 and ABCG2 (BCRP) gene polymorphisms in humans. Clin Pharmacol Ther. 2008 July;84(1):95–103.
- Ma JJ, Liu CG, Li JH, et al. Effects of NAT2 polymorphism on SASP pharmacokinetics in Chinese population. Clin Chim Acta. 2009 Sept;407(1–2):30–35.
- Yee J, Kim SM, Han JM, et al. The association between NAT2 acetylator status and adverse drug reactions of sulfasalazine: a systematic review and meta-analysis. Sci Rep. 2020 Feb 27;10(1):3658.
- Ratain MJ, Mick R, Berezin F, et al. Paradoxical relationship between acetylator phenotype and amonafide toxicity. Clin Pharmacol Ther. 1991 Nov;50(5 Pt 1):573–579.
- Ratain MJ, Mick R, Berezin F, et al. Phase I study of amonafide dosing based on acetylator phenotype. Cancer Res. 1993 May 15;53(10 Suppl):2304–2308.
- Ratain MJ, Mick R, Janisch L, et al. Individualized dosing of amonafide based on a pharmacodynamic model incorporating acetylator phenotype and gender. Pharmacogenetics. 1996 Feb;6(1):93–101.
- Innocenti F, Iyer L, Ratain MJ. Pharmacogenetics of anticancer agents: lessons from amonafide and irinotecan. Drug Metab Dispos. 2001 Apr;29(4 Pt 2):596–600.
- Lutz M. Metamizole (dipyrone) and the liver: a review of the literature. J Clin Pharmacol. 2019 Nov;59(11):1433–1442.
- Bonkowsky JL, Frazer JK, Buchi KF, et al. Metamizole use by Latino immigrants: a common and potentially harmful home remedy. Pediatrics. 2002 June;109(6):e98.
- Garcia S, Canoniero M, Lopes G, et al. Metamizole use among Hispanics in Miami: report of a survey conducted in a primary care setting. South Med J. 2006 Sept;99(9):924–926.
- Martínez C, Andreu I, Amo G, et al. Gender and functional CYP2C and NAT2 polymorphisms determine the metabolic profile of metamizole. Biochem Pharmacol. 2014 Dec 1;92(3):457–466.
- García-Martín E, Esguevillas G, Blanca-López N, et al. Genetic determinants of metamizole metabolism modify the risk of developing anaphylaxis. Pharmacogenet Genomics. 2015 Sept;25(9):462–464.
- Zanrosso CW, Emerenciano M, Gonçalves BA, et al. N-acetyltransferase 2 polymorphisms and susceptibility to infant leukemia with maternal exposure to dipyrone during pregnancy. Cancer Epidemiol Biomarkers Prev. 2010 Dec;19(12):3037–3043.
- Olivera M, Martínez C, Gervasini G, et al. Effect of common NAT2 variant alleles in the acetylation of the major clonazepam metabolite, 7-aminoclonazepam. Drug Metab Lett. 2007 Jan 1;1:3–5.
- Conway LP, Rendo V, Correia MSP, et al. Unexpected acetylation of endogenous aliphatic amines by arylamine N-acetyltransferase NAT2. Angew Chem. 2020;59(34):14342–14346.
- Ruiz JD, Martínez C, Anderson K, et al. The differential effect of NAT2 variant alleles permits refinement in phenotype inference and identifies a very slow acetylation genotype. PLoS One. 2012;7(9):e44629.
- Selinski S, Blaszkewicz M, Ickstadt K, et al. Refinement of the prediction of N-acetyltransferase 2 (NAT2) phenotypes with respect to enzyme activity and urinary bladder cancer risk. Arch Toxicol. 2013 Dec;87(12):2129–2139.
- Zang Y, Doll MA, Zhao S, et al. Functional characterization of single-nucleotide polymorphisms and haplotypes of human N-acetyltransferase 2. Carcinogenesis. 2007 Aug;28(8):1665–1671.
- World Health Organization Global Tuberculosis Executive Summary. 2019 [cited 2020 May 12]. https://www.who.int/tb/publications/global_report/tb19_Exec_Sum_12Nov2019.pdf?ua=1
- Glaziou P. Predicted impact of the COVID-19 pandemic on global tuberculosis deaths in 2020. medRxiv. 2020. 2020.04.28.20079582. DOI:10.1101/2020.04.28.20079582
- Saunders MJ, Evans CA. COVID-19, tuberculosis and poverty: preventing a perfect storm. Eur Respir J. 2020 July;56(1). PubMed PMID: 32444399; PubMed Central PMCID: PMCPMC7243392. DOI:10.1183/13993003.01348-2020
- Villarino ME, Scott NA, Weis SE, et al. Treatment for preventing tuberculosis in children and adolescents: a randomized clinical trial of a 3-month, 12-dose regimen of a combination of rifapentine and isoniazid. JAMA Pediatr. 2015 Mar;169(3):247–255. PubMed PMID: 25580725; PubMed Central PMCID: PMCPMC6624831.
- Kinzig-Schippers M, Tomalik-Scharte D, Jetter A, et al. Should we use N-acetyltransferase type 2 genotyping to personalize isoniazid doses? Antimicrob Agents Chemother. 2005 May;49(5):1733–1738.
- Rens NE, Uyl-de Groot CA, Goldhaber-Fiebert JD, et al. Cost-effectiveness of a pharmacogenomic test for stratified isoniazid dosing in treatment of active tuberculosis. Clin Infect Dis. 2020 Jan 6:ciz1212. DOI:10.1093/cid/ciz1212. Epub ahead of print. PMID: 31905381.
- Relling MV, Klein TE. CPIC: clinical pharmacogenetics implementation consortium of the pharmacogenomics research network. Clin Pharmacol Ther. 2011 Mar;89(3):464–467.
- Haga SB. Challenges of development and implementation of point of care pharmacogenetic testing. Expert Rev Mol Diagn. 2016 Sept;16(9):949–960. PubMed PMID: 27402403; PubMed Central PMCID: PMCPMC6709578.