
ABSTRACT
Introduction
The lungs possess many xenobiotic metabolizing enzymes which influence the pharmacokinetics and safety of inhaled medicines. Anticipating metabolism in the lungs provides an opportunity to optimize new inhaled medicines and overcome challenges in their development.
Areas covered
This article summarizes current knowledge on xenobiotic metabolizing enzymes in the lungs. The impact of metabolism on inhaled medicines is considered with examples of how this impacts small molecules, biologics and macromolecular formulation excipients. Methods for measuring and predicting xenobiotic lung metabolism are critically reviewed and the potential for metabolism to influence inhalation toxicology is acknowledged.
Expert opinion
Drugs can be optimized by molecular modification to (i) reduce systemic exposure using a ‘soft drug’ approach, (ii) improve bioavailability by resisting metabolism, or (iii) use a prodrug approach to overcome pharmacokinetic limitations. Drugs that are very labile in the lungs may require a protective formulation. Some drug carriers being investigated for PK-modification rely on lung enzymes to trigger drug release or biodegrade. Lung enzyme activity varies with age, race, smoking status, diet, drug exposure and preexisting lung disease. New experimental technologies to study lung metabolism include tissue engineered models, improved analytical capability and in silico models.
1. Introduction
Inhalation represents a major route of non-parenteral drug administration, alongside oral and dermal delivery. Inhalation may be chosen as the optimal route of delivery to target the site of drug action and achieve higher bioavailability in comparison to alternative routes. Bioavailability will be influenced by whether the drug is metabolized in the lungs. While the biotransformation of orally administered drugs is typically considered with regards to hepatic or gastrointestinal tract metabolism, the lungs represent a source of xenobiotic metabolizing enzymes that may significantly influence the pharmacokinetics of inhaled drugs, with implications for drug efficacy and safety.
This article summarizes current knowledge on xenobiotic metabolizing enzymes in the lungs and factors affecting metabolic activity. The impact of metabolism on inhaled medicines is considered with examples of how this impacts small molecules, biologics and formulation excipients. Methods for measuring and predicting xenobiotic lung metabolism are critically reviewed. The potential for metabolism to influence inhalation toxicology is also noted. The final section provides a perspective on the opportunities and challenges presented by lung metabolism for developers of inhaled medicines.
2. Xenobiotic metabolizing enzymes in the lungs
Despite being overshadowed by hepatic metabolism, the lungs contain many of the same xenobiotic metabolizing enzymes, as evidenced by gene expression and in vitro functional activity data [Citation1–3]. Knowledge of the presence (or absence) of functional metabolism pathways within the lungs is important due to the potential for biological activation or deactivation of inhaled drugs, which has implications on bioavailability, efficacy, local and systemic toxicity, clearance and overall pharmacokinetic profile [Citation4]. With regards to lung metabolism, more focus has historically been placed on Phase I metabolism than Phase II metabolism, perhaps because there is lower/negligible UDP-Glucoronyltransferase (UGT) expression in comparison to Cytochrome P450 (CYP) enzymes. Oesch and coworkers have extensively reviewed the drug metabolizing enzymes found within human lungs and compared data from numerous animal and human studies [Citation2], highlighting the importance of considering interspecies differences when studying pulmonary drug metabolism.
While there is sometimes limited evidence for functional protein expression of xenobiotic enzymes in the lungs, there are extensive mRNA data for a variety of key drug metabolizing enzymes including; CYP, CYP reductases, Flavin-dependent monooxygenases (FMO), alcohol dehydrogenases (ADH), NAD(P)H:quinone oxidoreductase (NQO), Hydrolases, Glutathione S-transferase (GST), UGT, Sulfotransferases (SULT) and N-Acetyltransferases (NAT) enzymes. Courcot and coworkers [Citation1] contributed substantially to characterizing this underlying transcriptional landscape and the relevance of commonly employed cell lines for drug metabolism studies by publishing gene expression profiles in several human cell lines compared with primary human lung tissue (both normal and cancer derived). The diverse range of enzymes shown to be expressed in the lungs is shown within .
Figure 1. Xenobiotic metabolizing enzymes expressed in human lungs. The most relevant enzymes based on expression/activity super(families) are highlighted in red with isoforms in blue. Enzymes expressed selectively in the lungs compared to other organs are asterisked, the enzymes that are most well established to be important to drug metabolism in the lung are underlined, italicized enzymes indicate that there is conflicting evidence for functional expression. CYP (Cytochromes P450), AHH (Aryl hydrocarbon hydroxylase), CES (Carboxyl esterase), FMO (Flavin-dependent monooxygenase), ADH (Alcohol dehydrogenase), ALDH (Aldehyde dehydrogenase), NQO (NAD(P)H quinone oxidoreductase), AKR (Aldo-keto reductase), EPHX (Epoxide hydrolase), GST (Glutathione-S-transferase), NAT (N-Acetyl transferase), SULT (Sulfotransferase) and UGT (UDP-glucuronosyl transferase)
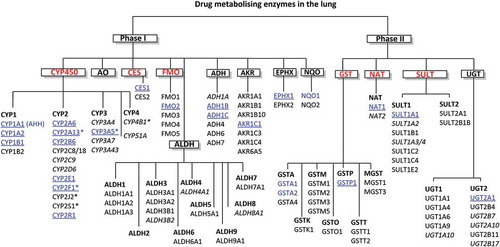
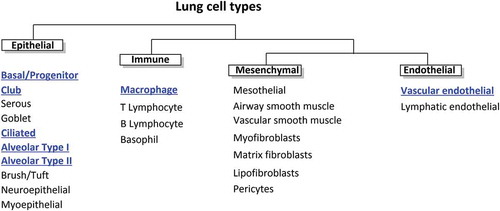
Club cells (formerly Clara cells) found in the bronchioles have been identified as the predominant pulmonary source of CYP enzymes within the bronchial region, but other bronchial and alveolar cells (particularly type II pneumocytes) have been shown in vitro to have significant metabolic capability [Citation5–7]. While several studies focus on enzymes obtained from lung homogenates, a plethora of in vitro studies have characterized the metabolic capacity of individual cell types ().
Figure 2. The different cell types of the lungs. Cells with significant xenobiotic metabolizing capacity are highlighted in blue
Phase I metabolizing enzymes have been the principal focus in respiratory metabolism studies rather than Phase II enzymes. The most relevant of the enzyme classes highlighted in and their potential role in drug metabolism are described in the following sections.
2.1. Phase I (CYP enzymes)
The key enzyme families responsible for Phase I metabolism are the CYP enzymes. These have received the most attention due to their ubiquitous expression in extrahepatic tissues, their key role in xenobiotic metabolism and their broad substrate selectivity. Several of the CYP enzymes are expressed in lung tissue, including but not limited to; CYP1A1, CYP1B1, CYP2B6, CYP2C8, CYP2C18, CYP2E1, CYP2F1, CYP2J2, CYP3A4, CYP3A5 [Citation1,Citation2]. Within the lungs, the widest range of CYP enzymes are expressed within the bronchiolar epithelium, primarily within club cells and ciliated columnar epithelial cells [Citation2,Citation3]. To a lesser extent, type I and II alveolar cells and alveolar macrophages have been shown to express some of these enzymes, such as CYP1A1, CYP1A2, CYP1B1 in in vitro and ex vivo models [Citation8–10]. However, some researchers have reported that not all CYP enzymes are likely to be functionally expressed in vivo [Citation11,Citation12], due to polymorphisms and nonfunctional variants. Studies using primary cells, lung cell lines and precision cut lung slices have confirmed CYP activity in the lungs which, while low relative to the liver, may be clinically significant for some inhaled drugs [Citation2].
Studies of CYP activity within the lungs have mostly focused on the implications for inhaled xenobiotics associated with respiratory disease, e.g. the bioactivation of the cigarette smoke associated polycyclic aromatic hydrocarbon benzo[a]pyrene by CYP1A1. However, several pharmaceutical drugs are also metabolized by CYP enzymes within the lungs. CYP3A4, an enzyme capable of metabolizing a wide range of exogenous substrates ranging from the inhaled β2 agonist salmeterol to the orally administered antiarrhythmic amiodarone, is expressed within the lungs with ~20% of the activity that is present within the liver [Citation13]. CYP3A5, which has greater expression than CYP3A4 within the lungs, has been shown to be induced in vitro by up to four-fold by nanomolar concentrations of commonly inhaled glucocorticoid drugs such as budesonide, beclomethasone dipropionate (BDP), and dexamethasone [Citation14]. While often regarded as nominal in comparison to the liver, basal expression of CYP enzymes in the lungs, or their potential induction under pathophysiological circumstances, means that CYP-mediated metabolism is of relevance to the fate of inhaled pharmaceuticals.
While the functional expression of other phase I metabolism enzymes such as FMO have been confirmed in human precision-cut lung slices [Citation15], they have received less attention than CYP enzymes. However, recently Rubin and coworkers showed higher enzyme activity for FMO than CYP enzymes with a small selection of substrates using human lung microsomes [Citation16]. Esterases represent an important class of phase I metabolism enzymes, due to their action on several inhaled pharmaceuticals such as BDP. High levels of carboxyl esterase 1 (CES1) mRNA have been found in lung parenchyma with Somers and coworkers not only finding a similar expression level of CES1 to that found in the liver, but showing comparable rates of BDP de-esterification in human lung parenchymal cells and hepatocytes [Citation17] .
2.2. Phase II (GST, UGT, SULT, NAT enzymes)
Except for UGTs, there is greater expression of phase II metabolism enzymes within the lungs than expression of CYP enzymes. Of these conjugating enzymes, one of the most widely studied families is the GST enzymes. Of the 22 GST class members, mRNA transcripts for at least 14 have been reported within human lung tissue, including GST alpha (GSTA1, GSTA2, GST3A4), kappa (GSTK1), mu (GSTM1, GSTM2, GSTM3, GSTM5), omega (GSTO1), pi (GSTP1), theta (GSTT1, GSTT2) and microsomal classes (MGST1, MGST3) [Citation18,Citation19]. GSTP1 has generally been reported to be the most abundantly expressed cytosolic GST protein in lung tissue [Citation20–22]. However, due to a lack of class-specific substrates/inhibitors for different classes, most studies report overall GST activity rather than the activity of individual classes. Nevertheless, GST activity is relatively high compared to other drug metabolizing enzymes within the lungs and based on enzyme activity studies is likely to contribute significantly to phase II metabolism in the lungs, along with NAT [Citation15,Citation23]. NAT1 and to a lesser extent NAT2 activity has been shown in the lungs with most research focused on either the metabolism of endogenous molecules, or inhaled toxicants along with genetic polymorphisms and the implications of this for cancer [Citation24–26]. SULT activity in primary lung parenchymal cells has been explored to a greater extent, and depending on the substrate appears to be both significant and comparable to that of human hepatocytes [Citation17].
While the presence of several types of UGT mRNA has been reported within the lungs, generally the catalytic activity is reported as very low, both based on freshly isolated human lung parenchymal cells and human precision cut lung slices [Citation15,Citation17]. Although Somers [Citation17] reported UGT2A1 expression in lung tissue to be higher than that of the liver, overall UGT expression and activity appears to be negligible compared to the liver and is likely overpredicted in rodent lung models [Citation15,Citation26]. In contrast, SULT activity within adult lungs may be higher and contribute more than UGTs to conjugative phase II metabolism, with Somers [Citation17] showing evidence of similar rates of sulfonation in the lung cells and hepatocytes.
3. Factors affecting metabolic activity
Several factors may affect the metabolic activity within the lungs and serve to alter the overall pulmonary metabolic capability of an individual. Genetic factors such as polymorphisms for specific enzymes may reduce or increase enzyme activity, as may environmental factors with the most widely studied example for the lungs being smoking, which has been shown to induce pulmonary CYP1A1/A2 expression together with other xenobiotic metabolizing enzymes [Citation27,Citation28]. Patient-specific factors such as age, sex or disease state have all been shown to influence the metabolic profile of individuals for xenobiotic metabolism within the lungs.
Drug–drug interactions may occur, for example, there is evidence of inhaled corticosteroids such as BDP inducing not only xenobiotic metabolizing enzymes such as UGT2B4 and UGT2B11, but also drug transporters such as P-glycoprotein (P-gp), Multidrug resistance-associated protein 1 (MRP1) and Multidrug resistance-associated protein 2 (MRP2) [Citation29]. Notably, the inhaled glucocorticoid fluticasone propionate (FP), which is metabolized by CYP3A4 has been shown to inactivate CYP3A5 by irreversible inhibition, and thereby may lead to drug–drug interactions, particularly as CYP3A5 is one of the predominant CYPs in the lungs [Citation30]. Additionally, while FP is an inhibitor of CYP3A5, it is a substrate of CYP3A4, and the polymorphism CYP3A4*22 which relates to decreased functional expression of the enzyme is associated with improved asthma control with FP [Citation31]. Although pulmonary CYP3A5 is an important CYP for lung metabolism, studies have found variable enzyme activity, likely due to genetic polymorphisms. While the CYP3A5*1 allele is responsible for the expression of active CYP3A5 and is common within sub-equatorial African populations, within Caucasian populations the homozygous genotype for CYP3A5*3 allele is common, with the latter allele expressing an inactive form of the enzyme [Citation32]. Thus, CYP3A5 mediated drug clearance within the lungs might be higher within certain populations, leading to drug insensitivity, or a reduced therapeutic benefit. With regards to this, the CYP3A5*3 polymorphism is relevant as this loss of function is associated with improved asthma control with inhaled BDP, likely relating to reduced deactivation rates [Citation32,Citation33]. Although CYP3A5 is induced by BDP [Citation14], this will be inconsequential in cases where the inactive form of CYP3A5 is expressed. Variability in CYP3A5 functional activity is important given that this enzyme has a substrate promiscuity that enables it to metabolize a wide range of drugs.
The extent of xenobiotic metabolism varies regionally within the lungs. Club cells, which are a predominant cell type involved in lung metabolism, are concentrated within the bronchial region where metabolism generally contributes more significantly to xenobiotic clearance compared to the peripheral region. Despite alveolar type II pneumocytes also expressing a large suite of important enzymes, as diffusion is more rapid in the alveolar region, a greater proportion of inhaled drug is likely to permeate and reach systemic circulation without being metabolized in the alveolar region of the lungs [Citation34]. Alveolar macrophages express CYP1B1, CYP2E1, CYP3A5, GSTP1, MGST3, phenylsulfotransferase and also UGTs [Citation35], although expression and activity is generally lower compared to bronchial epithelial cells. Similarly, vascular endothelial cells may also contribute to the metabolism of inhaled drugs in the lungs, but this is mostly minor compared to bronchiolar and alveolar epithelial cells [Citation2,Citation36].
Of relevance to nasally delivered drugs, the nasal epithelium also has a significant metabolic capability, not because of a significantly higher concentration of CYPs, but due to significantly elevated NADPH-cytochrome P450 reductase levels compared to the rest of the respiratory tract; reported as being four-fold higher than in the liver [Citation37]. Generally, studies have been limited to inhaled occupational exposure to various xenobiotics and/or inhaled recreational drugs such as cocaine [Citation38–40]. However, significant activity of CYPs, GSTs, esterases, aldehyde dehydrogenases and other enzymes in the nasal cavity may be of relevance to the development of future nasally inhaled medicines [Citation39,Citation40]. For example, oxidative and conjugative metabolism increase the susceptibility of some drugs to efflux transporters in the nasal cavity and reduce the bioavailability of intranasally administered drugs [Citation41].
The lower metabolic capability of the lungs compared to the liver may lead to local saturation of metabolism pathways following drug inhalation, which may affect the activity of a pro-drug or increase drug bioavailability. Permeability/retention are important considerations as a high lung absorptive clearance rate would limit time for metabolism to take place [Citation34]. Evidence in support of this contention includes the observation that metabolites are more frequently observed in lung slices, cells, homogenates and subcellular fractions than with the isolated perfused lung model where the parent compound may rapidly perfuse out of the system [Citation3]. Similarly to interactions in the gastrointestinal tract [Citation42], inhaled drugs may be subject to interplay between metabolism and active transport mechanisms that influence their overall pharmacokinetic profile.
4. Metabolism of inhaled medicines
Most research into the influence of drug metabolism on inhaled medicines has focused on the biotransformation of small molecules within the lungs. More recently, however, more diverse drug classes, requiring new formulations for inhaled medicines have increased attention on the metabolism of biologics and drug excipients.
4.1. Metabolism of inhaled small drug molecules
The significance of enzymatic activity in the lungs for inhaled drugs will depend on the temporal and spatial disposition of substrates determining availability to enzymes. Most studies of the metabolism of low molecular weight inhaled drugs have focussed on corticosteroids and β2 agonists studied using in vitro techniques, with the gold standard being the use of human tissue.
Using human precision cut lung slices, Nave and coworkers [Citation43] observed significant hydrolysis of BDP to beclometasone 17-monopropionate and beclometasone, ciclesonide to desisobutyryl-ciclesonide, the formation of fatty acid esters for budesonide and ciclesonide, while FP was not metabolized. Ciclesonide hydrolysis and fatty acid conjugation have been shown to occur in multiple in vitro models including precision cut lung slices, the type II like alveolar cell line A549, primary bronchial epithelial cells, and primary nasal epithelial cells [Citation44,Citation45]. There is little evidence of FP metabolism in the lungs, despite extensive metabolism via CYP3A4 in the liver [Citation43,Citation46]. Budesonide has been shown to undergo fatty acid conjugation within the lung, with oleate, linoleate, palmitate, palmitoleate and arachidonate metabolites having been identified [Citation17,Citation47]. The biotransformation of mometasone furoate to a minor epoxide metabolite has also been demonstrated [Citation48,Citation49].
The inhaled β2 agonist formoterol has been shown to be sulfated and glucuronidated to a minor extent in the lungs of at least some individuals (Somers et al., 2007). There is substantially more evidence of sulfation within the lung for the short acting β2 agonist salbutamol, with this occurring within human primary lung cells, human lung cytosol, and the bronchial cell line BEAS-2B [Citation17,Citation50]. Additionally, although now rarely used, the inhaled B2-agonists; isoprenaline, isoetharine and rimiterol have been shown to be methylated within the lungs by catechol-O-methyl transferase [Citation3,Citation51].
4.2. Inhaled prodrugs
The inhaled corticosteroids BDP and ciclesonide are examples of prodrugs delivered by inhalation. Researchers have previously described the advantages of prodrug strategy in the development of BDP [Citation52], which is metabolized by esterases to 17-beclomethasone monopropionate (BMP), the latter being the active metabolite which exhibits ~25-30 times greater binding affinity to the glucocorticoid receptor. BMP is further metabolized to beclomethasone, which has a binding affinity of a similar magnitude to BDP. This mechanism, shown in , is well described both in vivo and in vitro and as there is significant esterase activity within the lungs, it may be used to optimize the pharmacokinetic profile and bioavailability of future inhaled drugs.
Figure 3. (A) Metabolic pathway of beclomethasone dipropionate (BDP) to active metabolite beclomethasone 17-monopropionate (17-BMP), along with other metabolites assumed to have lower activity to the glucocorticoid receptor. Metabolism of parent compound is mediated by esterase and CYP3A enzymes within the lung epithelium. (B) Metabolic pathway of ciclesonide to active metabolite desisobutyryl-ciclesonide, along with the inactive fatty acid conjugate
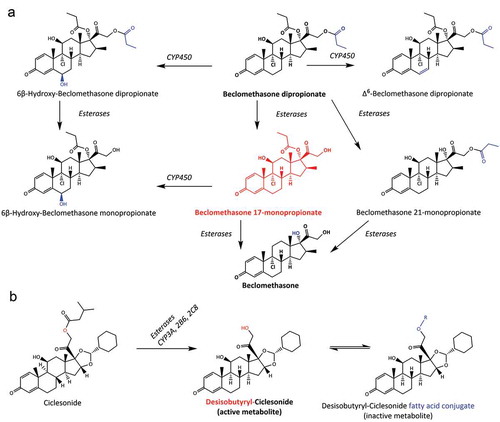
Ciclesonide is also a prodrug which is activated by metabolism after oral inhalation [Citation53]. Ciclesonide has been shown to be metabolized in vitro both in nasal mucosal cells and in various models (precision cut lung slices, primary bronchial epithelial cells, A549 type II alveolar cells), which are used to represent the lungs [Citation45,Citation54]. Similarly to BDP, ciclesonide acts as a pro-drug, with the active metabolite being deactivated by further metabolism to provide a ‘soft drug’ effect, i.e. limited systemic exposure due to pre-systemic metabolic clearance. Interestingly, while the fatty acid conjugates are inactive metabolites, as shown in , the reaction is reversible allowing them to serve as part of a ‘drug pool’ where the conjugate may be hydrolyzed to reform the active metabolite [Citation55]. This mechanism has significant implications on the pharmacokinetics resulting in prolonged action in the lungs and enables low dose frequency. A similar effect is reported for the inhaled glucocorticoid budesonide which forms reversible fatty acid esters including oleate, palmitate, linoleate, palmitoleate, and arachidonate conjugates [Citation47], leading to intracellular budesonide retention or ‘trapping’ [Citation56,Citation57].
In the design of soft drugs, mechanisms such as hydrolysis (rather than conjugation) are favored as they are less likely to become saturated. The strategy is employed for non-inhaled medicines, e.g. the inactive hydrocortisone metabolite (cortienic acid) was used as a starting point for the design of loteprednol etabonate, which is used in eyedrops, with the latter compound being a soft drug, which is metabolically inactivated in a single step [Citation52]. More recently, an approach to the development of novel soft drugs was described based on selective indazole ether-based glucocorticoid receptor modulators, with the goal of reducing systemic glucocorticoid activation by rapid elimination following a therapeutic effect after inhalation [Citation58]. Although soft drug design has not been used extensively for the development of inhaled medicines, it presents a significant opportunity to limit systemic bioavailability and thereby lower the risk of systemic adverse effects.
4.3. Drug candidates for inhaled administration
Lung metabolism is important when considering inhalation as a route of administration. An example of this is roflumilast, a long-acting bronchodilator pro-drug currently administered orally to treat COPD. At least some of the adverse gastrointestinal effects associated with the drug are likely to relate to the administration route and perhaps systemic exposure, and there is the potential that these may be reduced by inhaled administration. The active metabolite of roflumilast has been shown to be roflumilast N-oxide [Citation59,Citation60], which predominantly occurs via CYP1A2 and CYP3A4 metabolism within the liver, before reaching the site of action in the bronchioles via the systemic circulation [Citation61]. However, CYP1A2 has been shown to be present in the lungs, and while there is conflicting evidence for CYP3A4, a similar isoform CYP3A5 has also been shown to be present (although inactive in certain patient populations [Citation32]). Based on this evidence, there is the potential that roflumilast may also be activated if delivered directly to the lungs, albeit at a significantly slower rate. Inhalation of roflumilast has been shown to moderately improve asthma in a mouse model [Citation62] and roflumilast formulations for pulmonary drug delivery have been explored [Citation63].
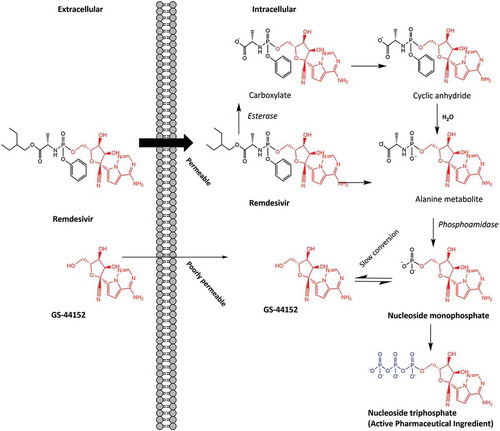
The potential to exploit lung metabolism with drugs currently not inhaled, as a means of optimizing drug delivery and efficacy was recently described for Remdesivir [Citation64]. Remdesivir is a broad-spectrum antiviral medication proposed as a treatment for COVID-19. While licensed for intravenous use, it has been suggested that a combination of inhaled and intravenous administration may improve clinical efficacy [Citation64]. Limited preclinical data for SARS-CoV and MERS-CoV provide some indication for the potential efficacy with COVID-19. Remdesivir is a prodrug with improved cell permeability compared to the active metabolite nucleoside monophosphate (Nuc-MP). The metabolic conversion is performed by intracellular hydrolases (), before further biotransformation to the active metabolite nucleoside triphosphate (Nuc-TP), which is negatively charged and therefore trapped intracellularly allowing accumulation of ~20-40 µM in cells such as peripheral blood mononuclear cells (PBMCs). While this may provide some clinical efficacy against COVID-19, it also means limited concentrations of the active metabolite reach the lung tissue [Citation65,Citation66]. To achieve an optimal efficacious dose, dosing via the pulmonary route may be required, with a projected daily inhalation dose of ~52.5 mg delivered by nebulizer or dry powder inhalation [Citation64]. However, the parent nucleoside GS-441,524 (containing a hydroxyl group instead of the monophosphate of Nuc-MP) has been suggested as potentially favorable over remdesivir due to its similar potency and simpler synthesis for mass production [Citation67]. It has also been suggested that while remdesivir is intended to act as a prodrug, the majority is prematurely metabolized to GS-441,524 and then to Nuc-MP, and therefore it is GS-441,524 which should be used to treat COVID-19 via inhaled administration [Citation67,Citation68].
Figure 4. Remdesivir and GS-44,152 metabolism to nucleoside triphosphate with antiviral properties as a potential treatment for COVID-19
4.4. Metabolism of biologic drugs and excipients
The bioavailability of biologic drugs delivered to the lungs is limited by enzymatic degradation. The lungs possess peptidase and protease activity [Citation69,Citation70] and it has been demonstrated experimentally that the systemic bioavailability of a number of peptide and protein drugs is enhanced by protease inhibitors [Citation71–74]. Interestingly, some inhaled therapies are based on modifying metabolism in the lungs. Pulmozyme® (dornase alfa) is an inhaled enzyme-based therapy containing recombinant human deoxyribonuclease I (DNASE1) which acts by hydrolyzing DNA present in the excess mucus of cystic fibrosis patients, alleviating some symptoms associated with the increased viscosity of their airway mucus [Citation75]. DNASE1 is expressed at a low level within the lungs and while the recombinant enzyme is susceptible to degradation by lung proteases, PEGylation of the N-terminal may make it protease resistant, while also preserving its enzymatic activity [Citation76]. In addition to cystic fibrosis, research has focused on the potential benefits for emphysema prevention [Citation77]. Inhaled α1-antitrypsin formulations have also been tested in clinical trials [Citation78,Citation79], and although some trials have highlighted limitations in efficacy, certain patient subgroups may benefit [Citation80,Citation81].
As well as therapeutic entities, biological macromolecules have been investigated as excipients for use in carrier formulations to protect labile drugs [Citation82], modify pharmacokinetic profiles to promote lung retention [Citation74] or target drug delivery [Citation83]. For example, drugs have been formulated to be bound to larger macromolecules that do not readily permeate the epithelial barrier; an approach commonly applied for oral delivery [Citation84]. Woods and coworkers recently identified and quantified predominant proteases within the human lungs to develop four in vitro models [Citation74], permitting an initial prediction of the extent to which albumin bound nanoparticles may be degraded as part of a sustained drug release formulation strategy following inhalation. An advantage of this method for optimizing the release of inhaled medicines, based on inherent lung metabolism, is not only that the pharmacokinetic profile may be adapted to provide an increased therapeutic benefit, but also that as human serum albumin is naturally present within the airway surface liquid (ASL) at relatively high concentrations (~8.8 mg/ml) [Citation85], the potential for adverse effects associated with this excipient are reduced.
Lung metabolism is also important for the biocompatibility and to avoid accumulation of other natural or endogenous macromolecules that have been investigated as drug carriers. Sodium hyaluronate has been investigated both for its therapeutic potential and as a drug carrier of inhaled medicines [Citation83]. Controlled drug release in the lungs, using sodium hyaluronate as a drug carrier, is mediated by its mucoadhesive properties that delay drug particle clearance by mucociliary clearance and the presence of hyaluronidase enzymes in human airway epithelial cells which ensure its degradation. This has been explored as a method of sustained insulin release, following pulmonary delivery [Citation86]. Other naturally occurring carbohydrate polymers have also been proposed as respirable microparticle drug carriers such as konjac glucomannan which is biodegradable by β-mannosidase [Citation87] and chitosan degradation which is biodegradable by lysozyme [Citation88].
5. Measuring and predicting xenobiotic metabolism within the lungs
There are several methods that may be used to measure xenobiotic metabolism within the lungs. Except for a few studies utilizing intravenous injections of radiolabeled test compounds in combination with positron emission scanning (PET), there are currently a lack of methods to measure in vivo metabolic capability within the human lungs [Citation89,Citation90]. Furthermore, this approach has not been widely applied to inhaled pharmaceuticals and has focused on metabolic trapping within tumor tissue. Although there are currently limited options for measuring in situ metabolism of inhaled drugs within the human lungs, there are several methods which rely on either ex vivo animal models or in vitro human/animal models. Prediction of metabolism may be performed using in silico methods based on ex vivo or in vitro data.
5.1. In vitro and ex vivo methods for measuring xenobiotic lung metabolism
Due to the complexity of assessing human pulmonary drug metabolism, the main technique used are in vivo animal models, in vitro and ex vivo methods. While animal models raise the issue of in vitro to in vivo (clinical) relevance they do provide considerable advantages for elucidating metabolic pathways. In decreasing order of complexity, the use of isolated perfused lung models, human precision cut lung slices, primary cells, cell lines, S9 fraction, microsomal or cytosolic fractions and recombinant enzymes have all been used to assess drug metabolism within the lungs, as summarized in . While extensive research has been conducted with regards to characterization of the gene expression profiles for xenobiotic metabolizing enzymes [Citation1], less attention has been paid to the challenges and opportunities for optimizing metabolism of inhaled drugs for which studies on enzyme activity are needed. Precision cut lung slices are perhaps the most representative in vitro model of the in vivo reality, due to the conservation of 3D lung structure, morphology and cellular diversity, which better reflects the spectrum enzyme expression and functionality compared to other models. However, due to limitations with obtaining tissue and experimental complexity, primary cells are more commonly used. Cell lines such as A549 (an alveolar epithelial type II like cell) and BEAS-2B (a bronchial epithelial cell line), provide advantages over primary tissue models, such as constrained genetic variation, the potential to be cultured continuously, lower costs and greater ease of use in high throughput assays. However, cell lines may suffer from alterations in enzyme expression and in some cases exhibit lower enzyme activity than primary cells, whereby the variety and extent of lung metabolism may be underestimated. The use of immortalized cell lines that are non-cancer derived may be more likely to display a normal phenotype with regards to enzyme expression in particular cell types of interest. For example, the immortalized bronchial cell line BEAS-2B that has been shown to more closely represent the genes expressed in primary bronchial tissue compared to cancer-derived cell lines, and the immortalized alveolar type I cell line TT1 that was shown to be a more sensitive in vitro model than the cancer-derived A549 alveolar type II cell line [Citation1,Citation8].
Table 1. Advantages and disadvantages of different methods used to measure xenobiotic metabolism in the lungs. The models are listed in order of decreasing complexity and ability to replicate the xenobiotic exposure to biotransforming enzymes that would be produced by inhalation
Subcellular fractions represent several advantages for studying lung metabolism and are one of the most common in vitro methods for studying hepatic drug metabolism. The S9 fraction is typically the supernatant of tissue homogenate that has been centrifuged at 9000 g to remove cell debris, nuclei and plasma membranes, and recover cytosolic or microsomal enzymes that may be used to study drug metabolism. The S9 fraction may be further purified to extract microsomal enzymes such as CYP or UGT enzymes, or cytosolic enzymes such as NATs or SULTs. While enzyme cofactors such as NADPH, UDPGA or PAPS need to be added alongside the drug of interest, this provides the advantage of being able to focus exclusively on Phase I or Phase II metabolism, or using S9 fractions to assess the total extent of metabolism.
Additional advantages of using subcellular fractions are that they may be frozen and stored easily compared to tissue samples, are more suited for high throughput kinetic studies, and typically have reduced biological variability between experiments. However, in addition to subcellular fractions not being representative of the in vivo situation, especially due to the addition of excess enzyme cofactors, a commonly overlooked consideration is the effect of macromolecular crowding on enzymatic reactions. Diluted subcellular fractions do not mimic the high concentrations of proteins within the cell, where ‘crowding’ has the effect of reducing the available solvent volume, and the low solvent volume subsequently impairs enzyme-substrate encounters such that the rates of enzymatic reactions in dilute in vitro solutions may be over 10-fold higher than those in vivo [Citation91–93]. In addition, degradation of enzymes may occur in fractions obtained from lysed cells and produce differences in enzyme kinetics between whole cells and subcellular fractions.
In comparison to whole cell models where the substrate must gain intracellular access, subcellular fractions may overpredict metabolic clearance rates due to the substrate-enzyme complexes being readily formed in the absence of structural barriers. This has been shown in the case of salbutamol sulfation, where metabolic clearance of salbutamol is much lower in intact bronchial epithelial cells than in human lung cytosol [Citation94]. However, for the purpose of metabolite identification subcellular fractions have the advantage of allowing metabolites to be rapidly formed. Another consideration when extrapolating metabolism data from simple lung homogenates is the potential to underpredict metabolism, as the homogenate is diluted with less-metabolically active cells compared to areas with high localized metabolic capacity in the bronchial epithelium. The spatial distribution of xenobiotic metabolizing capacity is especially relevant to predicting site-dependent metabolite concentration based on lung surface area.
Data from in vitro models may also be compared with ex vivo models, such as isolated perfused lungs, especially to relate the rate of lung metabolism to the intrinsic clearance of inhaled drugs [Citation16]. In all the in vitro methods, enzyme kinetics studies may be performed to give estimates on the rate of metabolism for a particular inhaled drug, which may then be used independently, or in physiologically based pharmacokinetic (PBPK) modeling where the pharmacokinetics of the parent drug and metabolite(s) may be predicted.
5.2. In silico methods for predicting xenobiotic lung metabolism
While there are a variety of methods for predicting drug metabolism in silico, very few have been applied to the assessment of lung metabolism specifically, due to models focusing primarily on hepatic metabolism. Existing in silico prediction models such as Meteor Nexus (Lhasa Limited) may be used to predict the metabolic fate of a range of chemicals based on the molecular structure and existing data for similar compounds for which metabolism data exists. Similarly, ADMET Predictor® (SimulationsPlus) provides predictions of human liver microsomal clearance, likely CYP and UGT enzyme kinetic contributions, going further to suggest which CYP or UGT isoform may be primarily responsible for the enzymatic reaction. An inherent challenge with these methods is that the accurate prediction of the metabolites formed, which is often based on training sets from existing molecules and may often be less predictive of unique or novel structures. Additionally, many predictive software packages focus primarily on CYP metabolism, followed by UGT metabolism, which is based on the enzymes of greatest relevance to hepatic drug metabolism. Software packages such as Meteor Nexus (Lhasa Limited) may predict a wider range of potential metabolites including those formed by esterase hydrolysis or conjugative reactions by GST, NAT, or SULT enzymes, which may be of more relevance to the lungs. However, by including a more exhaustive list of potential reactions, there is the risk of generating a significant number of false positives in terms of metabolites that would not be formed experimentally.
There may be opportunities in future to adapt in silico methods based on existing data on pulmonary metabolic activity to optimize this approach for inhaled drugs. A challenge to this might be incorporating predictions of drug deposition within the lungs, as differing metabolic enzyme activities within discrete regions of the lungs (e.g. large bronchial airways, small bronchial airways and the alveolar region) are likely to interplay with the permeability rates of the parent drug and its metabolites [Citation34]. Lung metabolism related to regional drug deposition when administered by aerosol is particularly important as unlike hepatic metabolism where 80–90% of the organ volume consists of a single cell type responsible for metabolism, there are at least 40 different lung cell types, with varying prevalence and function dependent on lung region [Citation16]. This approach, highlighted in , remains a significant challenge and would necessitate combining methodology from existing software, such as those used for inhaled deposition prediction, metabolism prediction and pharmacokinetic prediction of respiratory bioavailability, to allow the prediction and elucidation of the metabolic fate of inhaled drugs. Due to the rapidity of in silico methods relative to in vitro approaches, this could provide an expedited initial screen of inhaled drug metabolism to guide in vitro human or in vivo animal experimentation.
Figure 5. Diagram showing potential strategy for optimizing inhaled drug development with regards to lung metabolism
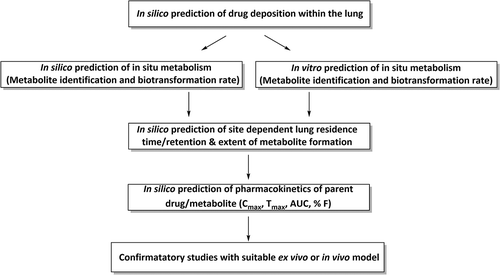
A holistic approach that focuses on metabolite identification, metabolic stability screening, biotransformation rate (relating to drug deposition in the lungs), varying PK profiles of metabolite and parent drug, would represent a significant leap forward in in silico prediction and the optimization of inhaled drug development. The scale of the challenge should not be underestimated as there are significant technical hurdles associated with accurately measuring local metabolism in the lungs. Nonetheless, prediction of metabolism is useful before investing in preliminary in vitro and confirmatory in vivo studies (which would typically be collected within the drug development process), and may be of value for rational development of inhaled ‘soft drugs’, prodrugs or drugs formulated in enzymatically dependent controlled release systems. By combining in silico deposition data with predicted or experimentally confirmed metabolism data, estimates could be made for site-dependent concentrations of active/inactive metabolites within the lung epithelium. Using this approach requires caution for the extrapolation of metabolism data from simple lung homogenates as described above.
The adaptability of an appropriate in silico model would also allow for the influence of genetic or environmental differences affecting the metabolism of inhaled drugs to be investigated, e.g. enzyme polymorphisms, or the induction of xenobiotic metabolizing enzymes in smokers or through drug–drug interactions as described in Section 3 [Citation29,Citation95,Citation96]. Ehrmann and coworkers [Citation97] recently described how such innovations alongside more established preclinical models for pulmonary drug delivery research may be applied as a strategy to optimize inhaled drug development. The review reinforces the requirement to use both in silico and in vitro approaches in parallel, with careful consideration of both the structural-functional characteristics of the lungs and the implications of aerosol deposition.
6. Inhalation toxicology
Metabolism in the lungs may generate metabolites with greater toxicity than the inhaled parent molecule. It is important that metabolites are considered when interpreting adverse effects of inhaled xenobiotics and that in vitro systems for inhalation toxicology incorporate metabolic capability to ensure that effects of metabolites are detected. Most research into biotransformation of xenobiotics into toxic metabolites within the lungs has focused on chemicals associated with smoking [Citation7,Citation98], environmental exposure to air pollutants [Citation99,Citation100] and occupational exposure to anthropogenic aerosols such as pesticides [Citation101,Citation102]. Although metabolism within the lungs often serves to reduce potential toxicity and increase the hydrophilicity and systemic clearance of the compound, oxidative CYP-catalyzed reactions within the lungs can lead to bioactivation of pro-carcinogens and other organic toxicants [Citation98]. Phase I metabolism within the lungs may lead to the formation of potentially reactive metabolites including electrophilic species, which rather than being directly toxic may react with endogenous molecules to form adducts; an example of this is the biotransformation of pro-toxicants to reactive epoxides, which may then generate DNA adducts potentially causing genotoxicity. Similarly, glucuronidation by UGTs as part of Phase II metabolism may create reactive acyl glucuronide conjugates that subsequently form protein adducts, which may act as haptens to illicit an immune response that may appear as a Type III hypersensitivity, or an idiosyncratic adverse reaction. While this process has been shown to have significance for drug induced liver injury and hepatic drug hypersensitivity, less research has been performed with a focus on pulmonary risks. Gram [Citation103] reviewed the effects of chemically reactive intermediates and potential pulmonary toxicity but this did not include any inhaled drugs and focused primarily on histological changes after exposure to a range of non-therapeutic xenobiotics. Similarly, naphthalene toxicity in the lung and nasal mucosa has been shown to be mediated by CYP2A13/2F1 in a humanized mouse model [Citation104]. The use of human or humanized models, is of great value for overcoming the potential interspecies differences in inhalation toxicology studies. An example of this is 4-ipomeanol, a pulmonary ‘pro-toxin’ which within the club cells of rodent lungs is metabolically activated by CYP4B1, while this selective CYP-mediated activation and subsequent toxicity from the metabolites occurs within the human liver, human lung bioactivation is minimal [Citation105]. The CYP4B1 biotransformation of 4-ipomeanol to reactive electrophilic intermediates that may covalently bind tissue macromolecules, has been shown to be marginal within the human lung, as despite selective expression of CYP4B1 within lung tissue, in comparison to several rodent and non-rodent animal models, human lung CYP4B1 is unable to effectively metabolize 4-ipomeanol due to the replacement of proline with serine at the 427 position of the enzyme [Citation106].
Foth [Citation107] highlights the potential for lung metabolism from specific cell types to cause the bioactivation of toxicants, in addition to lung-specific toxicity due to accumulation. The relative lack of data for metabolism of inhaled medicines directly causing local respiratory toxicity, is likely due to the success of in vitro and in vivo preclinical studies at removing unsuitable drug candidates, especially as rodent metabolism in the lungs typically exceeds that within human lungs. However, the metabolic capability of human lungs to produce toxic or reactive metabolites is sufficient that it could contribute to lung-specific toxicity and should be a consideration for the development of inhaled pharmaceuticals [Citation108]. The contribution of metabolic clearance may be crucial for some inhaled medicines in preventing toxicity associated with the accumulation of chronically administered drug or excipients in the lungs.
7. Expert opinion
The lungs are a complex organ with over 40 cell types in different regions to which drugs may be delivered by the bronchial or pulmonary blood circulation, or inhalation. The regio-spatial location of metabolizing enzymes at sub-cellular, cellular, tissue and airway generation level will determine access to drugs, requiring different models to evaluate drug metabolism. Sequestration of drugs in lung cells or compartments as a result of their physicochemical properties or active transport determines access to metabolizing enzymes, local concentrations and duration of exposure.
Lung metabolism offers opportunities for inhaled drug and formulation design by:
7.1. Safer inhaled drug design
Inhalation delivers drug directly to lungs where target sites are located, providing efficacy at doses that are much lower than those required if delivered systemically. A favorable metabolic profile can enhance safety by reducing off-target exposure if drug is deactivated before reaching the systemic circulation, e.g. ‘soft’ inhaled corticosteroids.
7.2. Pharmacokinetic optimization of drugs
Drugs may be modified to be metabolism resistant in the lungs and improve bioavailability,
A prodrug approach can be used to overcome pharmacokinetic limitations imposed by unfavorable physicochemical drug properties. Prodrug strategies involve molecular modification to facilitate access to the site of action where the drug undergoes enzymatically driven activation.
7.3. Protective formulations for labile molecules
Formulation approaches may be designed for drugs that are so labile in the lungs that their efficacy is impaired by significant metabolic clearance before reaching their target. e.g. nucleic acid therapeutics.
7.4. Biodegradable formulations for PK-modification
Drug carrier formulations under investigation for drug targeting or controlled drug release require a mechanism of enzymatic biodegradation to avoid drug carrier accumulation. Examples include protein carriers such as albumin which is degraded by proteases and carbohydrate carriers such as chitosan or konjac glucomannan which are degraded by lysozyme and β-mannosidase, respectively.
Opportunities and challenges for understanding drug metabolism in the lungs include:
7.5. Development and adoption of new technologies
Biological test systems: there are opportunities for more extensive use of human lung S9 fractions and precision cut lung slices and development of newer cell lines, e.g. club cell lines. Cell lines and primary cell models require benchmarking for similarity to normal lung cells. Newer systems which are still evolving include microfluidic models or micropatterned co-cultures of the type being developed for studying other organ systems [Citation109].
Experimental methodology: improved and standardized methods to ensure that substrates access enzymes in a biorelevant manner.
Analytical methods: more sensitive or insightful methods for application to metabolism studies, e.g. mass spectrometry imaging [Citation110,Citation111].
It is hoped that new technologies for measuring lung metabolism in situ will reduce reliance on animal methods and improve translation from preclinical to clinical settings by avoiding species differences in drug metabolism.
7.5.1. In silico modeling
Mechanistic modeling to simulate the effect of metabolism on inhaled drug PK is challenging, but potentially a powerful method for optimizing inhaled drugs and understanding bioactivation of airborne toxicants. Current PBPK models for inhalation exposure do not include metabolism. There is scope to develop a framework for use of complementary suites of ex vivo methods and interfacing experimental and computational modeling.
7.6. Population variability, including lung disease
Understand and model metabolic variations with age, race, smoking, diet and drug exposure as a step toward: (i) personalized or stratified medicine for therapies for which lung metabolism is a significant determinant of target site exposure, and (ii) identification of metabolism-related risk factors for susceptibility to airborne pollutants or disease.
Article highlights
Optimizing inhaled drugs requires consideration of drug metabolism.
A summary of current knowledge regarding Phase I and II metabolism in the lungs, how this influences the efficacy and safety of inhaled drugs and the variability in enzymatic activity arising from genetic, physiologic, and environmental factors.
Examples that highlight challenges and opportunities for the development of inhaled medicines that are subject to drug metabolism in the lungs.
Understanding and predicting metabolism of inhaled drugs presents opportunities to optimize inhaled medicines. For example, improved efficacy and safety, or more selective lung exposure to drugs due to their pharmacokinetic profiles.
This box summarizes key points contained in the article.
Declaration of interest
E Bäckström is a full-time employee of AstraZeneca, which develops and markets inhaled treatments for respiratory diseases. AstraZeneca reviewed the publication, without influencing the opinions of the authors, to ensure medical and scientific accuracy, and the protection of intellectual property. IS Mudway was partly funded by the National Institute for Health Research Health Protection Research Units (NIHR HPRU) in Environmental Exposures and Health and Chemical and Radiation Threats and Hazards at Imperial College in partnership with Public Health England (PHE). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care or Public Health England. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- Courcot E, Leclerc J, Lafitte -J-J, et al. Xenobiotic metabolism and disposition in human lung cell models: comparison with in vivo expression profiles. Drug Metab Dispos. 2012;40(10):1953.
- Oesch F, Fabian E, Landsiedel R. Xenobiotica-metabolizing enzymes in the lung of experimental animals, man and in human lung models. Arch Toxicol. 2019;93(12):3419–3489.
- Taylor G. The absorption and metabolism of xenobiotics in the lung. J Advanced Drug Delivery Reviews. 1990;5(1–2):37–61.
- Olsson B, Bondesson E, Borgström L, et al. Pulmonary drug metabolism, clearance, and absorption. In: Smyth HDC, Hickey AJ, editors. Controlled pulmonary drug delivery. New York: Springer New York; 2011. p. 21–50.
- Devereux TR, Domin BA, Philpot RM. Xenobiotic metabolism by isolated pulmonary cells. Pharmacol Ther. 1989;41(1):243–256. 1989/01/01/
- Foster KA, Oster CG, Mayer MM, et al. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp Cell Res. 1998;243(2):359–366. 1998/09/15/
- Hukkanen J, Pelkonen O, Hakkola J, et al. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol. 2002;32(5):391–411. 2002/01/01
- Jarvis IW, Enlo‐Scott Z, Nagy E, et al. Genotoxicity of fine and coarse fraction ambient particulate matter in immortalised normal (TT1) and cancer‐derived (A549) alveolar epithelial cells. Environmental Molecular Mutagenesis. 2018;59(4):290–301.
- Piipari R, Savela K, Nurminen T, et al. Expression of CYP1A1, CYP1B1 and CYP3A, and polycyclic aromatic hydrocarbon‐DNA adduct formation in bronchoalveolar macrophages of smokers and non‐smokers. Int J Cancer. 2000;86(5):610–616.
- Thum T, Erpenbeck VJ, Moeller J, et al. Expression of xenobiotic metabolizing enzymes in different lung compartments of smokers and nonsmokers. Environ Health Perspect. 2006;114(11):1655–1661.
- Bernauer U, Heinrich-Hirsch B, Tönnies M, et al. Characterisation of the xenobiotic-metabolizing Cytochrome P450 expression pattern in human lung tissue by immunochemical and activity determination. Toxicol Lett. 2006;164(3):278–288. 2006/07/15/
- Tournel G, Cauffiez C, Billaut-Laden I, et al. Molecular analysis of the CYP2F1 gene: identification of a frequent non-functional allelic variant. Mutat Res. 2007;617(1):79–89. 2007/04/01/
- Anttila S, Hukkanen J, Hakkola J, et al. Expression and localization of CYP3A4 and CYP3A5 in human lung. Am J Respir Cell Mol Biol. 1997 Mar;16(3):242–249. PubMed PMID: 9070608; eng.
- Hukkanen J, Väisänen T, Lassila A, et al. Regulation of CYP3A5 by glucocorticoids and cigarette smoke in human lung-derived cells. J Pharmacol Exp Ther. 2003;304(2):745.
- Yildiz Y, Gareth W, Markus W, et al. Comparison of rat and human pulmonary metabolism using Precision-cut Lung Slices (PCLS). Drug Metab Lett. 2019;13(1):53–63.
- Rubin K, Ewing P, Bäckström E, et al. Pulmonary metabolism of substrates for key drug-metabolizing enzymes by human alveolar type II cells, human and rat lung microsomes, and the isolated perfused rat lung model. Pharmaceutics. 2020;12(2):117.
- Somers GI, Lindsay N, Lowdon BM, et al. A Comparison of the expression and metabolizing activities of phase I and II enzymes in freshly isolated human lung parenchymal cells and cryopreserved human hepatocytes. Drug Metab Dispos. 2007;35(10):1797.
- Van Dyck E, Nazarov PV, Muller A, et al. Bronchial airway gene expression in smokers with lung or head and neck cancer. Cancer Med. 2014;3(2):322–336. 2014/04/01
- Zuo WL, Shenoy SA, Li S, et al. Ontogeny and biology of human small airway epithelial club cells. Am J Respir Crit Care Med. 2018 Dec 1;198(11):1375–1388. PubMed PMID: 29874100; PubMed Central PMCID: PMCPMC6290945. eng.
- Howie AF, Forrester LM, Glancey MJ, et al. Glutathione S-transferase and glutathione peroxidase expression in normal and tumour human tissues. Carcinogenesis. 1990 Mar;11(3):451–458. PubMed PMID: 2311189; eng.
- Wang Y, Spitz MR, Schabath MB, et al. Association between glutathione S-transferase p1 polymorphisms and lung cancer risk in Caucasians: a case-control study. Lung Cancer. 2003 Apr;40(1):25–32. PubMed PMID: 12660004; eng.
- Lewis SJ, Cherry NM, Niven RM, et al. GSTM1, GSTT1 and GSTP1 polymorphisms and lung cancer risk. Cancer Lett. 2002;180(2):165–171.
- Pacifici GM, Franchi M, Bencini C, et al. Tissue distribution of drug-metabolizing enzymes in humans. Xenobiotica. 1988;18(7):849–856. 1988/01/01
- Chen C-H. Genetic variations and polymorphisms of metabolic enzymes. In: Xenobiotic metabolic enzymes: bioactivation and antioxidant defenser. Springe, Cham; 2020. p. 155–168.
- Conway LP, Rendo V, Correia MS, et al. Unexpected acetylation of endogenous aliphatic amines by Arylamine N‐Acetyltransferase NAT2. In: Angewandte chemie international edition. 2020;59(34):14342–14346.
- Zhang JY, Fen Wang Y, Prakash C. Xenobiotic-metabolizing enzymes in human lung. Curr Drug Metab. 2006;7(8):939–948.
- Jamaludin J, Marlin N, Wood H, et al. Evaluating the impact of genotype on the relationship between impaired lung growth and chronic exposure to traffic derived pollutants. Eur Respir J. 2013;42(Suppl 57):P3623.
- Tekpli X, Zienolddiny S, Skaug V, et al. DNA methylation of the CYP1A1 enhancer is associated with smoking‐induced genetic alterations in human lung. Int J Cancer. 2012;131(7):1509–1516.
- Kuzuya Y, Adachi T, Hara H, et al. Induction of drug‐metabolizing enzymes and transporters in human bronchial epithelial cells by beclomethasone dipropionate. IUBMB Life. 2004;56(6):355–359.
- Murai T, Reilly CA, Ward RM, et al. The inhaled glucocorticoid fluticasone propionate efficiently inactivates cytochrome P450 3A5, a predominant lung P450 Enzyme. Chem Res Toxicol. 2010;23(8):1356–1364. 2010/08/16
- Stockmann C, Fassl B, Gaedigk R, et al. Fluticasone propionate pharmacogenetics: CYP3A4• 22 polymorphism and pediatric asthma control. J Pediatr. 2013;162(6):1222–1227.e2. 2013/06/01/
- Roberts JK, Moore CD, Ward RM, et al. Metabolism of beclomethasone dipropionate by cytochrome P450 3A enzymes. J Pharmacol Exp Ther. 2013;345(2):308–316. PubMed PMID: 23512537; eng.
- Stockmann C, Reilly CA, Fassl B, et al. Effect of CYP3A5• 3 on asthma control among children treated with inhaled beclomethasone. J Allergy Clin Immunol. 2015;136(2):505–507.
- Enlo-Scott Z, Swedrowska M, Forbes B. Epithelial permeability and drug absorption in the lungs. In: Inhaled Medicines. Elsevier; 2021. p. 267–299.
- Gundert-Remy U, Bernauer U, Blömeke B, et al. Extrahepatic metabolism at the body’s internal–external interfaces. Drug Metab Rev. 2014;46(3):291–324. 2014/08/01
- Zeldin DC, Foley J, Ma J, et al. CYP2J subfamily P450s in the lung: expression, localization, and potential functional significance. Mol Pharmacol. 1996;50(5):1111–1117.
- Sarkar MA. Drug metabolism in the nasal mucosa. Pharm Res. 1992;9(1):1–9. 1992/01/01
- Brittebo EB. Metabolism-dependent activation and toxicity of chemicals in nasal glands. Mutat Res. 1997;380(1):61–75. 1997/10/31/
- Dahl AR, Hadley WM. Nasal cavity enzymes involved in xenobiotic metabolism: effects on the toxicity of inhalants. Crit Rev Toxicol. 1991;21(5):345–372. 1991/01/01
- Reed C. Drug metabolism in the nasal cavity: relevance to toxicology. Drug Metab Rev. 1993;25(1–2):173–205.
- Oliveira P, Fortuna A, Alves G, et al. Drug-metabolizing enzymes and efflux transporters in nasal epithelium: influence on the bioavailability of intranasally administered drugs. Curr Drug Metab. 2016;17(7):628–647.
- Wu C-Y, Benet LZ. Predicting drug disposition via application of BCS: transport/Absorption/Elimination Interplay and Development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. 2005/01/01
- Nave R, Fisher R, McCracken N. In vitro metabolism of beclomethasone dipropionate, budesonide, ciclesonide, and fluticasone propionate in human lung precision-cut tissue slices. Respir Res. 2007;8(1):65.
- Nonaka T, Nave R, McCracken N, et al. Ciclesonide uptake and metabolism in human alveolar type II epithelial cells (A549). BMC Pharmacol. 2007;7(1):12. 2007/09/27
- Nave R, McCracken N. Metabolism of ciclesonide in the upper and lower airways: review of available data. J Asthma Allergy. 2008;1:11–18. PubMed PMID: 21436981; eng.
- Pearce RE, Leeder JS, Kearns GL. BIOTRANSFORMATION OF FLUTICASONE: IN VITRO CHARACTERIZATION. Drug Metab Dispos. 2006;34(6):1035.
- Tunek A, Sjödin K, Hallström G. Reversible formation of fatty acid esters of budesonide, an antiasthma glucocorticoid, in human lung and liver microsomes. Drug Metab Dispos. 1997;25(11):1311–1317.
- Sahasranaman S, Issar M, Hochhaus G. METABOLISM OF MOMETASONE FUROATE AND BIOLOGICAL ACTIVITY OF THE METABOLITES. Drug Metab Dispos. 2006;34(2):225.
- Teng XW, Cutler DJ, Davies NM. Mometasone furoate degradation and metabolism in human biological fluids and tissues. Biopharm Drug Dispos. 2003;24(8):321–333. 2003/11/01
- Eaton EA, Walle UK, Wilson HM, et al. Stereoselective sulphate conjugation of salbutamol by human lung and bronchial epithelial cells. Br J Clin Pharmacol. 1996;41(3):201–206. 1996/03//. PubMed PMID: 8866919; eng.
- Evans ME, Shenfield GM, Thomas N, et al. The pharmacokinetics of rimiterol in man. Xenobiotica. 1974;4(11):681–692.
- Winkler J, Hochhaus G, Derendorf H. How the lung handles drugs: pharmacokinetics and pharmacodynamics of inhaled corticosteroids. Proc Am Thorac Soc. 2004;1(4):356–363.
- Mukker JK, Singh RSP, Derendorf H. Ciclesonide: a pro-soft drug approach for mitigation of side effects of inhaled corticosteroids. J Pharm Sci. 2016;105(9):2509–2514. 2016/09/01/
- Sato H, Nave R, Nonaka T, et al. In vitro metabolism of ciclesonide in human nasal epithelial cells. Biopharm Drug Dispos. 2007;28(1):43–50. 2007/01/01
- Nave R, Meyer W, Fuhst R, et al. Formation of fatty acid conjugates of ciclesonide active metabolite in the rat lung after 4-week inhalation of ciclesonide. Pulm Pharmacol Ther. 2005;18(6):390–396. 2005/12/01/
- Wieslander E, Delander E-L, Järkelid L, et al. Pharmacologic importance of the reversible fatty acid conjugation of budesonide studied in a rat cell line in vitro. Am J Respir Cell Mol Biol. 1998;19(3):477–484. PubMed PMID: 9730876
- Borchard G, Cassará ML, Roemelé PE, et al. Transport and local metabolism of budesonide and fluticasone propionate in a human bronchial epithelial cell line (Calu‐3). J Pharm Sci. 2002;91(6):1561–1567.
- Hemmerling M, Nilsson S, Edman K, et al. Selective nonsteroidal glucocorticoid receptor modulators for the inhaled treatment of pulmonary diseases. J Med Chem. 2017;60(20):8591–8605. 2017/10/26
- Nassr N, Huennemeyer A, Herzog R, et al. Effects of rifampicin on the pharmacokinetics of roflumilast and roflumilast N‐oxide in healthy subjects. Br J Clin Pharmacol. 2009;68(4):580–587.
- Huennemeyer A, Hauns B, David M, et al. Pharmacokinetics and safety of Roflumilast, a once-daily, oral, selective PDE4 inhibitor, and its active metabolite Roflumilast N-oxide in healthy subjects. J Allergy Clin Immunol. 2004;113(2, Supplement):S222. 2004/02/01/
- Lahu G, Nassr N, Hünnemeyer A. Pharmacokinetic evaluation of roflumilast. Expert Opin Drug Metab Toxicol. 2011;7(12):1577–1591. 2011/12/01
- Murad HA, Habib HS, Rafeeq MM, et al. Co-inhalation of roflumilast, rather than formoterol, with fluticasone more effectively improves asthma in asthmatic mice. Experimental Biology and Medicine. 2017;242(5):516–526.
- Éy S, Amaro MI, De Almeida GS, et al. Development of a new formulation of roflumilast for pulmonary drug delivery to treat inflammatory lung conditions. Int J Pharm. 2018;550(1):89–99. 2018/10/25/
- Sun D. Remdesivir for Treatment of COVID-19: combination of pulmonary and iv administration may offer aditional benefit. Aaps J. 2020;22(4):1–6.
- Sheahan TP, Sims AC, Graham RL, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9(396):396.
- Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381–385.
- Yan V, Muller F. Advantages of the parent nucleoside GS-441524 over remdesivir for Covid-19 treatment. ACS Med Chem Lett. 2020;11(7):1361–1366.
- Yang K. What do we know about remdesivir drug interactions? Clin Transl Sci. 2020;13(5):842–844.
- Baginski L, Tachon G, Falson F, et al. Reverse transcription polymerase chain reaction (RT-PCR) analysis of proteolytic enzymes in cultures of human respiratory epithelial cells. Journal of Aerosol Medicine and Pulmonary Drug Delivery. 2011;24(2):89–101.
- Forbes B, Wilson C, Gumbleton M. Temporal dependence of ectopeptidase expression in alveolar epithelial cell culture: implications for study of peptide absorption. Int J Pharm. 1999;180(2):225–234.
- Morita T, Yamamoto A, Takakura Y, et al. Improvement of the pulmonary absorption of (Asu 1, 7)-eel calcitonin by various protease inhibitors in rats. Pharm Res. 1994;11(6):909–913.
- Kobayashi S, Kondo S, Juni K. Study on pulmonary delivery of salmon calcitonin in rats: effects of protease inhibitors and absorption enhancers. Pharm Res. 1994;11(9):1239–1243.
- Hussain A, Arnold JJ, Khan MA, et al. Absorption enhancers in pulmonary protein delivery. J Control Release. 2004;94(1):15–24.
- Woods A, Andrian T, Sharp G, et al. Development of new in vitro models of lung protease activity for investigating stability of inhaled biological therapies and drug delivery systems. Eur J Pharm Biopharm. 2020;146:64–72. 2020/01/01/
- Frederiksen B, Pressler T, Hansen A, et al. Effect of aerosolized rhDNase (Pulmozyme®) on pulmonary colonization in patients with cystic fibrosis. Acta Paediatrica. 2006;95(9):1070–1074.
- Guichard MJ, Leal T, Vanbever R. PEGylation, an approach for improving the pulmonary delivery of biopharmaceuticals. Curr Opin Colloid Interface Sci. 2017;31:43–50. 2017/09/01/
- King PT, Sharma R, O’Sullivan KM, et al. Deoxyribonuclease 1 reduces pathogenic effects of cigarette smoke exposure in the lung. Sci Rep. 2017;7(1):12128. 2017/09/21
- Griese M, Scheuch G. Delivery of Alpha-1 antitrypsin to airways. Ann Am Thorac Soc. 2016;13(Supplement 4):S346–S351.
- Brantly M, Stocks J, Rouhani F, et al. Inhaled alpha-1-antitrypsin restores lower respiratory tract protease-anti-protease homeostasis and reduces inflammation in alpha-1 antitrypsin deficient individuals: a phase 2 clinical study using inhaled kamada-api. C74. ADVANCES IN TRANSLATIONAL COPD: American Thoracic Society. 2017;A7677–A7677.
- Barrecheguren M, Miravitlles M. Treatment with inhaled α1-antitrypsin: a square peg in a round hole? Eur Respir J. 2019;54(5):1901894.
- Stolk J, Tov N, Chapman KR, et al. Efficacy and safety of inhaled α1-antitrypsin in patients with severe α1-antitrypsin deficiency and frequent exacerbations of COPD. Eur Respir J. 2019;54(5):5.
- Morales JO, Fathe KR, Brunaugh A, et al. Challenges and future prospects for the delivery of biologics: oral mucosal, pulmonary, and transdermal routes. Aaps J. 2017;19(3):652–668. 2017/05/01
- Martinelli F, Balducci AG, Kumar A, et al. Engineered sodium hyaluronate respirable dry powders for pulmonary drug delivery. Int J Pharm. 2017;517(1):286–295. 2017/01/30/
- Mantaj J, Abu-Shams T, Enlo-Scott Z, et al. Role of the basement membrane as an intestinal barrier to absorption of macromolecules and nanoparticles. Mol Pharm. 2018;15(12):5802–5808. 2018/12/03
- Kumar A, Terakosolphan W, Hassoun M, et al. A biocompatible synthetic lung fluid based on human respiratory tract lining fluid composition. Pharm Res. 2017;34(12):2454–2465.
- Surendrakumar K, Martyn GP, Hodgers ECM, et al. Sustained release of insulin from sodium hyaluronate based dry powder formulations after pulmonary delivery to beagle dogs. J Control Release. 2003;91(3):385–394. 2003/09/04/
- Guerreiro F, Pontes JF, Am RDC, et al. Spray-drying of konjac glucomannan to produce microparticles for an application as antitubercular drug carriers. Powder Technol. 2019;342:246–252. 2019/01/15/
- Islam N, Dmour I, Taha MO. Degradability of chitosan micro/nanoparticles for pulmonary drug delivery. Heliyon. 2019;5(5):e01684. 2019/05/01/
- Juhász C, Muzik O, Lu X, et al. Quantification of tryptophan transport and metabolism in lung tumors using PET. J Nucl Med. 2009 Mar;50(3):356–363. PubMed PMID: 19223408; PubMed Central PMCID: PMCPMC2784997. eng.
- Faubert B, Li KY, Cai L, et al. Lactate metabolism in human lung tumors. Cell. 2017;171(2):358–371.e9. 2017/10/05/
- Mittal S, Chowhan RK, Singh LR. Macromolecular crowding: macromolecules friend or foe. Biochim Biophys Acta Gen Subj. 2015;1850(9):1822–1831. 2015/09/01/
- Silverstein TP, Slade K. Effects of macromolecular crowding on biochemical systems. J Chem Educ. 2019;96(11):2476–2487. 2019/11/12
- Kim JS, Yethiraj A. Effect of macromolecular crowding on reaction rates: a computational and theoretical study. Biophys J. 2009;96(4):1333–1340. 2009/02/18/
- Ward JK, Dow J, Dallow N, et al. Enantiomeric disposition of inhaled, intravenous and oral racemic-salbutamol in man — no evidence of enantioselective lung metabolism. Br J Clin Pharmacol. 2000;49(1):15–22. 2000/01/01
- Aridgides DS, Mellinger DL, Armstrong DA, et al. Functional and metabolic impairment in cigarette smoke-exposed macrophages is tied to oxidative stress. Sci Rep. 2019;9(1):1–11.
- Hussain T, Al-Attas OS, Al-Daghri NM, et al. Induction of CYP1A1, CYP1A2, CYP1B1, increased oxidative stress and inflammation in the lung and liver tissues of rats exposed to incense smoke. Molecular and Cellular Biochemistry. 2014;391(1–2):127–136.
- Ehrmann S, Schmid O, Darquenne C, et al. Innovative preclinical models for pulmonary drug delivery research. Expert Opin Drug Deliv. 2020;17(4):463–478. 2020/04/02
- Castell JV, Teresa Donato M, Gómez-Lechón MJ. Metabolism and bioactivation of toxicants in the lung. The in vitro cellular approach. Exp Toxicol Pathol. 2005;57:189–204. 2005/07/22/
- Cohen GM. Pulmonary metabolism of foreign compounds: its role in metabolic activation. Environ Health Perspect. 1990;85:31–41.
- Okona-Mensah KB, Battershill J, Boobis A, et al. An approach to investigating the importance of high potency polycyclic aromatic hydrocarbons (PAHs) in the induction of lung cancer by air pollution. Food Chem Toxicol. 2005;43(7):1103–1116. 2005/07/01/
- Dahl AR, Gerde P. Uptake and metabolism of toxicants in the respiratory tract. Environ Health Perspect. 1994;102(suppl 11):67–70.
- Chapter HE. 38 - metabolism of pesticides. In: Krieger R, editor. Hayes’ Handbook of Pesticide Toxicology (Third Edition). New York: Academic Press; 2010. p. 893–921.
- Gram TE. Chemically reactive intermediates and pulmonary xenobiotic toxicity. Pharmacol Rev. 1997;49(4):297.
- Li L, Carratt S, Hartog M, et al. Human CYP2A13 and CYP2F1 mediate naphthalene toxicity in the lung and nasal mucosa of CYP2A13/2F1-humanized mice. Environ Health Perspect. 2017;125(6):067004.
- Parkinson OT, Teitelbaum AM, Whittington D, et al. Species differences in microsomal oxidation and glucuronidation of 4-ipomeanol: relationship to target organ toxicity. Drug Metab Dispos. 2016;44(10):1598–1602. PubMed PMID: 27468999; eng.
- Wiek C, Schmidt EM, Roellecke K, et al. Identification of amino acid determinants in CYP4B1 for optimal catalytic processing of 4-ipomeanol. Biochem J. 2015;465(1):103–114. PubMed PMID: 25247810; eng.
- Foth H. Role of the lung in accumulation and metabolism of xenobiotic compounds — implications for chemically induced toxicity. Crit Rev Toxicol. 1995;25(2):165–205. 1995/01/01
- Owen K. Regulatory toxicology considerations for the development of inhaled pharmaceuticals. Drug Chem Toxicol. 2013;36(1):109–118. 2013/01/01
- Seo J, Huh D. Chapter 7 - Microphysiological models of human organs: a case study on microengineered lung-on-a-chip systems. In: Borenstein JT, Tandon V, Tao SL, editors. Microfluidic cell culture systems. Second. Elsevier.187–208. 2019.
- Yamamoto E, Taquahashi Y, Kuwagata M, et al. Visualizing the spatial localization of ciclesonide and its metabolites in rat lungs after inhalation of 1-μm aerosol of ciclesonide by desorption electrospray ionization-time of flight mass spectrometry imaging. Int J Pharm. 2021;595:120241. 2021/02/15/
- Dexter A, Steven RT, Patel A, et al. Imaging drugs, metabolites and biomarkers in rodent lung: a DESI MS strategy for the evaluation of drug-induced lipidosis. Anal Bioanal Chem. 2019;411(30):8023–8032. 2019/12/01