
ABSTRACT
Introduction
Artemisinin-based combination therapies (ACTs) are recommended first-line antimalarials for uncomplicated Plasmodium falciparum malaria. Pharmacokinetic/pharmacodynamic variation associated with ACT drugs and their effect is documented. It is accepted to an extent that inter-individual variation is genetically driven, and should be explored for optimized antimalarial use.
Areas covered
We provide an update on the pharmacogenetics of ACT antimalarial disposition. Beyond presently used antimalarials, we also refer to information available for the most notable next-generation drugs under development. The bibliographic approach was based on multiple Boolean searches on PubMed covering all recent publications since our previous review.
Expert opinion
The last 10 years have witnessed an increase in our knowledge of ACT pharmacogenetics, including the first clear examples of its contribution as an exacerbating factor for drug–drug interactions. This knowledge gap is still large and is likely to widen as a new wave of antimalarial drug is looming, with few studies addressing their pharmacogenetics. Clinically useful pharmacogenetic markers are still not available, in particular, from an individual precision medicine perspective. A better understanding of the genetic makeup of target populations can be valuable for aiding decisions on mass drug administration implementation concerning region-specific antimalarial drug and dosage options.
1. Introduction
In 2011, a comprehensive review on the pharmacogenetics associated with artemisinin-based combination therapy (ACT) was published [Citation1]. In the intervening 10 years, the scenario of malaria treatment has developed in several ways. Some examples are the implementation of new strategies (e.g. Seasonal Malaria Chemotherapy and Intermittent Preventive Treatment in Pregnancy), the significant progress in malaria elimination efforts, which drive new concerns on transmission interruption, the proposal of more complex triple ACTs opening the possibility of new adverse events situations, the enrichment and progression of the pipeline of new antimalarials [Citation2] including new ACTs, and the inescapable shadow of the ongoing Covid-19 pandemic.
Entering the third decade of the 21st Century, and as a follow-up of the previous review, we herein offer an updated overview of the field. Most of the presently available data point to polymorphisms in hepatic cytochrome P450s (CYPs) as the major source of pharmacogenetic variation in the disposition of antimalarial drugs. To this phase I metabolism factors, some data have been recently added on phase II enzymes (especially Uridine 5’-diphospho-glucuronosyltransferases (UDPGTs)), as well as for the expected involvement of transport systems, namely members of the ATP Binding Cassette (ABC) superfamily.
ACT has proved to be a resilient strategy against the development of parasite resistance. Albeit Plasmodium falciparum has scored early victories, namely in Southeast (SE) Asia, against artesunate-mefloquine [Citation3] and more recently handling dihydroartemisinin-piperaquine [Citation4], the fact is that in Africa – where most of the global malaria burden is located – ACT is still highly efficacious. The future importance of pharmacogenetics in ACT success might rise at a point when resistance emerges, shrinking therapeutic windows and potentially forcing changes to higher dosing or more complex combinations. Such developments will drive another issue – compliance. Lack of adherence to treatment is a recurrent issue in malaria control [Citation5,Citation6]. The 3-day ACT course is frequently considered cumbersome, a reality further composed by the fact that the most used ACT, artemether-lumefantrine, involves the taking of six instead of three doses, the second of them (T8h) being often in the middle of the night [Citation5]. An increase in adverse side effects, including mild ones can further decrease compliance, to a certain extent opening a door for resistance development, potentially generating a positive feedback on ever increasing dosing needed. Pharmacogenetics is likely to be a sound contributor for dose-dependent secondary events, as suggested by recent reports [Citation7].
In the last two decades, malaria elimination efforts have been successful on significantly decreasing malaria transmission in large regions. During this period of time previously certified malaria-free countries have avoided re-establishment of malaria transmission [Citation8]. But the progress has slowed down in some critical aspects. Albeit the reduction on malaria case incidence has been significant from 80/1000 in 2000 to 58/1000 cases in 2015, the progression has stalled, 2019 registering 57/1000. In particular in Africa, the age profile of the disease is also changing, likely through decreases in natural immune protection. Malaria incidence is expanding beyond under-five children, toward older subjects, as premunition develops later and later due to lowered malaria exposure. Again, less natural protection might need tailoring the treatment toward higher dosing, especially when aiming on single-dose treatments [Citation9]. In these scenarios, personalizing drug administration might be an option to be considered, as the number of patients reduces, while the likely gravity of their conditions increases.
As in its previous iteration [Citation1], this review will be mainly organized by focusing on each major antimalarial drug. presents a summary of the main genes involved in Absorption, Distribution, Metabolism and Excretion (ADME genes) of currently administrated ACT drugs.
Figure 1. Schematic simplified view of the metabolic routes of antimalarials, including transporters (localized in PM) and drug-metabolizing enzymes (localized in ERM). Metabolic routes are indicated by arrows and distribution and elimination goes from the lower to upper part of the Figure. This visual compilation is based at several levels of evidence, including in vitro and in vivo data. The former includes cell free approaches (e.g [Citation141].), cellular systems (e.g [Citation147].), as well as in vivo data as drug–drug interactions (e.g [Citation47].) and pharmacogenetic marker/phenotype associations (e.g [Citation91].). In some specific cases, phase III transporters were assumed due to their known functional characteristics (e.g. the transport of phase II hydrophilic conjugates by ABCC2/MRP2). Note that the denomination ‘M1’ and ‘M2’ are just routine nomenclature for peaks in mass spectrometry analysis. As such, the M1 and M2 compounds referred are different between reactions – e.g. the M2 precursor of QNMs from the CYP1A1 and CYP1B1 action over AQ and DEAQ is not the same compound as the M2 associated with the action of CYP2C8 on PPQ. (abbreviations: PM: plasma membrane of the hepatocyte, ERM: endoplasm reticulum membrane of the hepatocyte).
![Figure 1. Schematic simplified view of the metabolic routes of antimalarials, including transporters (localized in PM) and drug-metabolizing enzymes (localized in ERM). Metabolic routes are indicated by arrows and distribution and elimination goes from the lower to upper part of the Figure. This visual compilation is based at several levels of evidence, including in vitro and in vivo data. The former includes cell free approaches (e.g [Citation141].), cellular systems (e.g [Citation147].), as well as in vivo data as drug–drug interactions (e.g [Citation47].) and pharmacogenetic marker/phenotype associations (e.g [Citation91].). In some specific cases, phase III transporters were assumed due to their known functional characteristics (e.g. the transport of phase II hydrophilic conjugates by ABCC2/MRP2). Note that the denomination ‘M1’ and ‘M2’ are just routine nomenclature for peaks in mass spectrometry analysis. As such, the M1 and M2 compounds referred are different between reactions – e.g. the M2 precursor of QNMs from the CYP1A1 and CYP1B1 action over AQ and DEAQ is not the same compound as the M2 associated with the action of CYP2C8 on PPQ. (abbreviations: PM: plasma membrane of the hepatocyte, ERM: endoplasm reticulum membrane of the hepatocyte).](/cms/asset/70132220-e307-458f-9ba9-0beb59b15b42/iemt_a_2049235_f0001_oc.jpg)
The present review was based on a systematic literature search between January 2021, and April 2021 of published articles present at PubMed and, sporadically, Google Scholar as a broader engine until June 2021, covering all the recent publications after our previous review. Exceptionally, Clarivate:Web of Science Book Citation Index and Google Scholar were used for exploring other potentially interesting documents. This included PhD thesis and documents from internationally recognized organizations (e.g. WHO), for which we provide web sites, when available. Meeting communications were not considered as sufficiently robust from a peer review perspective. We only exceptionally included information from animal models. The basic approach involved Multiple Boolean searches including the large number of combinations between the antimalarial drug designations with general (e.g. CYP) or specific enzyme/transporter/receptor names (e.g. G6PD), as well as pharmacokinetics and pharmacodynamics associated terms.
2. Artemisinin-based combination therapy and beyond
During the 1980s and 1990s, P. falciparum resistance to the mainstay chloroquine (CQ) and mefloquine (MQ) monotherapies has led to the progressive proposal and adoption of drug combination regimens [Citation10]. The validity of this approach has been to a certain extent previously shown upon the blending of antifolate drugs, namely adding sulfadoxine to pyrimethamine (SP), due to the rapid development of resistance against the latter [Citation11]. Albeit initially successful on preventing the rapid rise of pyrimethamine resistance, the parasite was ultimately able to develop resistance to this weak combination [Citation12]. This was to a significant extent due to the limited scope of action of these two related drugs, both targeting the same metabolic pathway. It became evident that the next generation of combinations had to include drugs with very different modes of action. Conceptually introduced in the late 1980s, ACT fits these objectives [Citation13]. These combinations are based on the premise that the powerful and rapid pharmacodynamic effect of the fast acting/fast eliminated artemisinin derivative (ART) component leads to a decisive reduction in parasitemia during the first hours of treatment. This decreases the burden faced by the partner drug not only in simple parasite numbers, but also chrono-pharmacologically: such lower parasitemias would only be available for the partner drug when already acting alone, significantly later in the therapy course. At such a downstream point in time the partner drug blood concentration would be much lower, and less effective. For more details on the ACT concept refer to the previous review [Citation1], as well as to specifically focused references [Citation14]. ACT has been a decisive public health success story [Citation8]. Presently, the major available ACTs are: artesunate blend with mefloquine (ATS-MQ), amodiaquine (ATS-AQ) or pyronaridine (ATS-PYR), dihydroartemisinin with piperaquine (DHA-PPQ), artesunate-sulfadoxine-pyrimethamine (ATS-SP), and the de facto global mainstay, Artemether-Lumefantrine (AL). Though a potentially valuable option, the artesunate-atovaquone-proguanil combination will not be considered in this review, as it has essentially only been used exploratory.
Albeit still largely effective, full efficacy of ACT is anyway under pressure by the parasite. Resistance to the established ATS-MQ [Citation3,Citation15] and more recently introduced DHA-PPQ [Citation16,Citation17] is entrenched in SE Asia. As for ATS-AQ, the capacity of the parasite to evade AQ action has been long known [Citation18–20]. Most worrying, signals of the capacity of the parasite to handle the global mainstay artemether-lumefantrine have been accumulating, particularly in Africa [Citation21–23].
Avoiding the trap of prolonged expansion of resistance witnessed with chloroquine in the 20th Century [Citation24], a comprehensive pipeline of new antimalarials and combinations is now emerging, for example, KAF156 (Ganaplacide) [Citation25], KAE609 (Cipargamine) [Citation26], Artefenomel, (OZ439), Arterolane (OZ227), and Fosmidomycin, all already at least at phase 2b trialing [Citation27]. Additionally, there is also a trend toward recovering old compounds, most notably the XIX Century methylene blue, the first ever synthetic drug [Citation28]. The objective is to keep an ACT-level of high efficacy with combinations showing more balanced pharmacokinetic characteristics between partner drugs, opening the possibility of more compact routines, down to two, and even one-day administration courses (‘single encounter treatment’) [Citation9,Citation25,Citation29,Citation30]. Additionally, some of these new combinations are able to handle liver forms of the parasites, as well as gametocytes. The latter, leading to transmission interruption capacities, which is key in the perspective of current elimination efforts [Citation31].
3. ACT: The artemisinin component
In the last 10 years no new artemisinin derivative has been introduced to ACT. The three mainstream ARTs available for ACT are still the water-soluble Artesunate (ATS), the more lipophilic Artemether (ATM), and dihydroartemisinin (DHA, Artenimol) (), the latter representing simultaneously the main phase I active metabolite of both ATS and ATM, and an available drug on its own merit.
Figure 2. Drugs chemical structures. List of compounds referred in the present review. Adapted from PubChem.ncbi.
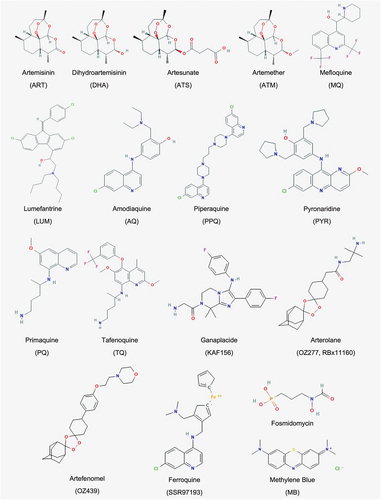
3.1. Artesunate and dihydroartemisinin
ATS is the semi-synthetic artemisinin derivative of choice for most ACTs, being blend with MQ, amodiaquine, and, lately with pyroniridine, the latter constituting the most recently approved ACT [Citation32]. In some regions, for example, in Western Asia, ATS is also still combined with SP, determining a more fragile ACT due to P. falciparum well-established capacity to rapidly develop intense resistance against these antifolates. Importantly, ATS is also presently the de facto global first choice for the management of severe malaria.
Hepatic biotransformation of ATS is fast, with an average half-life of conversion to DHA of ca. 15 minutes. ATS swift metabolism makes the study of its inter-individual PK variations particularly challenging. Nevertheless, significant inter-individual variation in PK parameters, including nominal exposure (Area Under the Curve, AUC0-∞) has been robustly documented [Citation33].
The influence of this variation in therapeutic success is unclear, but it is likely to be more noticeable in the context of P. falciparum ATS (and DHA) resistance infections. Of more concern is the narrowness of the ATS therapeutic window. Preliminarily studies before Artesunate-MQ implementation in Thailand showed that a 5-day course of escalating daily doses of ATS (2 to 4 mg/Kg, oral or intravenous) – especially the higher ones – as being associated with a worrisome, albeit recoverable, >20% reduction in neutrophils [Citation34].
The emergence in SE Asia of more tolerant P. falciparum parasites against artemisinin [Citation35,Citation36], raised the possibility of increasing the clinical dosing of ATS. New studies, now with the canonical 7-day ATS monotherapy, confirmed and expanded the toxicity observations from 20 years before [Citation37]. An increase in ATS dose from the basal 2–4 mg/kg to 6 mg/kg was linked with a marked increase in risk of neutropenia, enough to halt the trial. Of the six patients experiencing neutropenia, five were in the higher dose group, and one from the 4 mg/kg group. All had significantly higher AUC and Cmax levels. The precise nature of this dose-dependency was not clear, but one cannot exclude the contribution of some specific pharmacogenetic characteristics of the affected group. The narrow nature of the window prompts the question: can a regular 4 mg/kg dose lead to a 6 mg/kg-like exposure in individuals with pharmacogenetic-driven low ATS metabolism?
Microsome-based studies have pointed for CYP2A6 as the major cytochrome P450 involved in the conversion of ATS toward DHA, with a likely secondary contribution of CYP2B6 [Citation38,Citation39]. CYP2A6 harbors significant Open Reading Frame (ORF) polymorphism, with several alleles leading to reduced enzyme activity (e.g. 2A6/2A7 gene conversion (*12A), V365M (*17), K476R (*21)) or its total absence (e.g. L160H (*2), G479V (*5)), for example, through gene deletion (*4). Scarce information is available concerning associations between CYP2A6 polymorphism and artesunate adverse events. A small (24 patients) study in Malaysia showed an increased incidence of adverse events among CYP2A6*1B carriers upon treatment with ATS-AQ [Citation40]. Concerning treatment efficacy, Phompradit et al. (2014) [Citation41] studied the frequencies of CYP2A6 (*1A, *1B, *4C, *2, *3 and *6), CYP2B6 (*1-*9) in 71 Burmese uncomplicated malaria patients under ATS-MQ treatment. The standout observation was the significantly decreased adequate clinical and parasitological response (ACPR) of CYP2B6*9/*9 carriers. Albeit interesting, these observations need to be followed up by more robust investigations, in particular, as the P. falciparum infections were not analyzed for the presence of mutations in artemisinin and MQ resistance markers. Single Nucleotide Polymorphisms (SNPs) in the parasite K13 (providing artemisinin partial resistance) gene, as well as pfmdr1 duplications might have a significant influence on the role of pharmacogenetics on treatment efficacy.
The phase I metabolite DHA undergoes phase II conjugation, performed by the UDP-glucuronosyltransferases UGT1A9 and UGT2B7 into several inactive metabolites [Citation42,Citation43]. In the aforementioned studies among Burmese malaria patients, the UGT1A9 (*1, *4, and *5) were not found to influence individual DHA exposure. Glucuronidated Phase II conjugates are ultimately excreted through the biliary route [Citation43,Citation44]. In the case of DHA, the hepatobiliary membrane located ATP-Binding Cassette (ABC) transporter MRP2/ABCC2 is likely to be involved in the elimination route of its relatively hydrophilic glucuronide metabolites [Citation45].
Finally, a recent targeted NGS analysis of a large panel of pharmacogenetic markers among 50 Gabonese children under severe malaria treatment with ATS did not find any significant associations between CYP2A6, CYP2B6, UGT1A9, UGT2B7, ABCB1, and ABCC2 polymorphisms with relevant clinical phenotypes, albeit detecting several alleles linked to decreased or absent phase I and II enzyme activity [Citation46]. At the presently used ACT-based artesunate dosing for malaria treatment (up to 4 mg/kg), there is no major evidence supporting the patient pharmacogenetics as a significant efficacy and safety modifier. However, for higher doses commanding frequent adverse events such factors might be of importance. The scarcity of studies exploring higher doses preclude further conclusions for now. This might change in the future, in particular, upon the repurposing of artesunate for other applications, namely cancer, where higher exposures are justifiable in terms of cost-benefit balance.
3.2. Artemether
Artemether (ATM) is mainly used in combination with LUM. AL has been the most administered antimalarial worldwide in the last two decades, having been adopted by the vast majority of the National Malaria Control Programs [Citation47] in Africa. ATM was initially investigated as a mono-therapy agent. As with ATS, the highest doses tested (4 mg/kg) led to significant but reversible neutropenic effect in a significant fraction of the studied volunteers [Citation34]. With AL, ATM is administered six times in three days, following the established treatment schedule. The combination has proven to be remarkably safe, with only very rare adverse events having been registered. As with ATS, ATM is rapidly metabolized toward DHA, in its case mainly by CYP3A4/5, with secondary roles having been suggested for CYP1A2 and CYP2B6 [Citation48,Citation49]. To date no studies have been published on artemether monotherapy pharmacogenetics. The few available reports concern AL by Staehli-Hodel et al. [Citation50], whom did not find associations between ATM elimination and the individual status for the CYP2B6*6, CYP3A4*1b and CYP3A5*3 alleles among Tanzanian subjects.
Pharmacogenetic associations with potential ART adverse events have not been investigated in depth, deserving future attention, especially in the context of potential changes in ACT treatment leading to increased exposures, as recently suggested with triple-drug therapies (ART component + two partner drugs) [Citation51] and extended administration courses [Citation52].
Artemisinin derivatives have been shown the potential to influence their own – and other drugs disposition, probably through the Human Pregnane X Receptor (PXR/NR112)/Constitutive Androstane Receptor (CAR/NR113) activation circuit [Citation53,Citation54]. These nuclear receptors operate as xeno-sensors in the cell cytoplasm. When binding to specific xenobiotics, like therapeutic drugs, these proteins become active transcription factors, which typically induce the expression of genes coding for drug metabolism enzymes and transport proteins, affecting the disposition of the triggering ligand. Functionally relevant polymorphisms in these proteins are able to potentiate interactions affecting pharmacokinetic profiles [Citation55,Citation56].
4. ACT: the long half-life partners
4.1. Mefloquine
Mefloquine (MQ), an amino alcohol quinoline () was developed during the 1960s and 1970s in the United States, during the large program of antimalarial drug discovery, originally established during WWII and later carried over to the Vietnam War. Registered in the late 1970s, MQ was introduced at national scale in the Thai national malaria program during the 1980s, as a key tool for managing chloroquine resistant P. falciparum. Initially applied as a 2-dose monotherapy, it soon started being used in combination with SP. As parasite resistance pressure built-up, it evolved toward the first widely used ACT: mefloquine plus artesunate (ATS), in a two-dose regimen.
The regimen has been further updated to the presently regular oral administration dosing for the treatment of uncomplicated malaria in the context of ACT, comprising 25 mg/kg MQ (+12 mg/kg of ATS) in three days. MQ pharmacokinetics parameters can vary significantly between individuals, albeit with some dependency on the regimen type. AUC0-∞ has a range of 2 to 7-fold, with 2–6-fold for Cmax, and 2–4-fold for the characteristically long (>15 days) T1/2 [Citation15,Citation57–66]. It is conceivable that part of this variation is associated with individual pharmacogenetic variation.
MQ is extensively metabolized toward a major carboxy-mefloquine metabolite, and to lesser extent, hydroxyl-mefloquine. These pathways are mainly catalyzed by CYP3A4, as supported by in vitro (microsome systems) data [Citation67] and drug interaction pharmacokinetic studies [Citation68,Citation69].
It has been long assumed that there is a significant CYP3A4 pharmacogenetics contribution in inter-individual drug response [Citation70]. Unfortunately, the exact nature of its mechanism is still unclear. The large majority of CYP3A4 ORF polymorphisms are considered rare (i.e. <1%), albeit some being known to have significant consequences on the patient drug metabolism status [Citation71–73]. The most studied CYP3A4 SNP is the CYP3A4*1b allele, a relatively frequent change located in the gene 5’ proximal regulatory region [Citation74], proposed to influence the gene transcription activity [Citation75]. It is present in significant prevalence among African populations [Citation50,Citation76–78]. The importance of CYP3A4*1b has been controversial with some reports pointing for its potential as a modulating factor, with many others not finding significant correlations [Citation79]. In the case of MQ, no significant associations were found between individual MQ pharmacokinetics and their CYP3A4*1b status among 68 Cambodian patients treated with 3 doses of ATS-MQ [Citation50].
MQ long elimination half-life prompted its early adoption as a prophylactic agent, commanding significantly larger exposures. MQ has been well known for its associated adverse events, in particular, of neuropsychiatric nature. These are likely related with the capacity of this antimalarial to cross the blood-brain barrier (BBB), of which protection function is dependent on the action of membrane drug transporters, namely P-glycoprotein (Pgp)/ABCB1, a member of the ABC (ATP-binding cassette) superfamily. MQ is a likely Pgp substrate [Citation80]. Following this premise, Aarnoudse et al. [Citation81] found a positive association between the canonical lower activity MDR1 1236 T/2777 T/3435 T SNP haplotype and psychiatric grade secondary events upon prophylaxis. This is consistent with the hypothesis of a higher BBB permeability for MQ among these subjects, and hence a decreased protection of the central nervous system (CNS). The effect was mainly observed among women, a result still without a clear explanation. It is also conceivable that in cases of a large range of Cmax values (e.g. >5-fold [Citation62]), a differential Pgp/MDR1 activity in the gut is involved; this is a hypothesis still in need of exploration.
Other efflux pumps may play a role in MQ disposition, including the ABC proteins ABCC1/MRP1 and ABCC4/MRP4 – both significantly polymorphic – leading to a range of transport capabilities. From the perspective of malaria, it is to note that both are well represented at the erythrocyte plasma membrane [Citation82], opening the possibility of pharmacogenetics-driven differences in intra-cellular MQ accumulation [Citation83], and as such access to parasite drug targets.
Finally, as observed for some artemisinin derivatives, carboxy-mefloquine – MQ main metabolite – has been shown in vitro to be a strong PXR/NR1I2 agonist, leading to the significantly increased expression of several cytochrome P450s, e.g. CYP2B6, which bio-transforms artemether [Citation84]. Considering the wide polymorphism of PXR/NR1I2 [Citation85], and recent observations of pharmacogenetic-driven HIV drug/antimalarial interactions [Citation86], the influence of specific nuclear receptor pharmacogenetic profiles on carboxy-mefloquine driven drug–drug interactions deserves further studies.
4.2. Lumefantrine
Lumefantrine (LUM, initially referred as benflumetol, ) in combination with artemether (ATM) (120 mg LUM + 20 mg ATM) represents the most frequently used antimalarial in the world, with more than one billion doses administered since its global introduction during the first decade of the Century [Citation87]. LUM is a lipophilic amino-alcohol quinoline drug, with a slow absorption, highly influenced by the concomitant ingestion of a fatty meal. Albeit shorter than in the case of MQ, it presents a reasonably long terminal half-life with significant variability (T1/2 ~ 2–6 days), and clear inter-individual differences in AUC0-∞ [Citation88]. These have been described even in studies with fat content, where a large part of absorption variability is hence controlled [Citation89]. Considering D7 LUM blood levels as a proxy of treatment outcome [Citation90], field data shows a 10–50-fold range in these concentrations [Citation91–93].
LUM is eliminated at the hepatic levels, mainly untransformed. The metabolized fraction results from the action of CYP3A4/5, generating desbutylbenflumetol (DBB), the only until now identified and studied metabolite [Citation94]. DBB represents approximately 1% of the total exposure (AUC0-∞ DBB / AUC0-∞ LUM) [Citation95], but its longer half-life (T1/2 > 6 days) [Citation96] guarantees an increase in this ratio along time. DBB is also a significantly more powerful antimalarial, (IC50 4–5-fold higher), able to synergistically interact with LUM [Citation97,Citation98]. These characteristics prompted a possible importance of DBB in LUM-based treatments, supported by a preliminary observation of its significant influence on D28 recurrence rates [Citation99], and ACPR outcome among pregnant (a status frequently leading to lower LUM metabolism) [Citation100]. Further consistent with the involvement of CYP3A, interaction studies with ketoconazole have shown a mild but significant increase in AUC0-∞ as well as Cmax [Citation49]. These data reinforce the possible importance of the patient CYP3A pharmacogenetic status for AL therapy.
Staheli-Hodel et al. [Citation50] found no influence of the aforementioned CYP3A4*1B, as well as the splice defect carrying CYP3A5*3 on the pharmacokinetic parameters of 150 Tanzanian malaria patients treated with AL. Also, a study in 78 patients from Tanzania attempted to associate D7 LUM blood levels and CYP3A4*1B, *1G and *22 status, with no positive links found [Citation101]. Interestingly, the author found a notable 14% prevalence of CYP3A4*22, an allele previously reported to be associated with decreased protein expression [Citation102] and enzyme activity [Citation103]. Similarly, treatment outcomes were also not influenced by CYP3A4*1B in a Ghanaian AL efficacy trial, as all patients were classified as wild-type [Citation104]. Similar observations were reported in an Angolan study, where further analysis of the CYP3A4*3 and CYP3A5*3 alleles did not produce significant associations [Citation105]. It is to note that CYP3A4 is a relatively low information pharmacogenetic marker, due to the importance of environmental factors, combined with the scattered nature of its SNP diversity. These characteristics make the understanding of the contribution of CYP3A4 genetic variations particularly challenging.
Further concerning the polymorphism of CYP3A5, Mutagonda et al. [Citation106] have reported in Tanzania that CYP3A5*1/*1 was associated to significantly decreased LUM D7 concentrations after regular 3-day AL regimen among pregnant women.
Above all, an interesting pharmacogenetically driven interaction has recently emerged concerning LUM. The geographical distribution overlap between HIV and Malaria in Africa prompts frequent events of co-administration of anti-HIV and antimalarials. The non-nucleoside reverse transcriptase inhibitor (NNRTI) Efavirenz (EFV) is one of the most used anti-HIV drugs in Africa, well documented to be a PXR/NR112 and CAR/NR113 agonist, the former promoting CYP3A and MDR1/ABCB1 transcriptional activation [Citation107,Citation108], the latter, CYP2B6 [Citation109]. This promotes EFV elimination, as this NNRTI is itself extensively metabolized by CYP2B6 [Citation110]. In parallel, EFV treatment interferes with AL pharmacokinetics, leading to significant reductions in Cmax and AUC0-∞ in both LUM and ATM, as well as D7 levels for the former, all interpretable as the result of the aforementioned CYP3A and ABCB1 induction [Citation111–113]. This in turn has consequences in AL efficacy, which plummets below the 90% WHO watermark for ACT performance [Citation114,Citation115]. Interestingly, it has been described that the poor metabolizer CYP2B6*6/*6 genotype carriers, associated with increased EFV plasma levels [Citation116], lead to a significant decreased LUM exposure (D7 blood levels). Consistent with the previous reports, this is likely due to increased nuclear receptor-based activation of CYP3A and ABCB1. Lower D7 levels, in turn, led to a significantly higher rate of recurrent parasitemia [Citation86], showing that the post-treatment protection by the LUM long pharmacokinetic elimination tail was compromised. It has been modeled that an increase from 3-day AL treatment course toward 7 days (akin to the recently trialed long regimen [Citation52]) among CYP2B6*6/*6 patients would offset this effect and restore full AL treatment success [Citation117]. Finally, LUM has been determined in vitro as a CYP2D6 inhibitor. This might also promote interactions with the potential of being exacerbated by specific pharmacogenetic profiles.
A significant involvement of drug transporters in LUM disposition is suggested from the referred Nuclear Receptor driven interactions, as well as the known effect of ketoconazole, a PgP inhibitor [Citation118]. Additionally, one can also argue that as most LUM is eliminated unchanged, functional polymorphism in this ABC transporter is expected to be involved. This is further supported by considering CYP3A participation as part of a proposed ‘battery system’ with MDR1/ABCB1 [Citation119,Citation120], as both the CYPs and this ABC transporter share a range of substrates [Citation121].
MDR1/ABCB1 3435CC allele carriers, a genotype occasionally associated with increased drug exposure, have been reported to be linked with a decreased number of malaria reinfections in an AL efficacy trial in Angola [Citation105]. In other studies, performed in mainland Tanzania and Zanzibar, no trends were found associating ABCB1 C3435T or the A4036G SNPs and pharmacokinetic parameters, including D7 LUM levels [Citation93,Citation106]. Concerning this latter drug-level time point, a significant association was documented between the C1515Y SNP at the MRP2/ABCC2 drug transporter, a key efflux protein present in the hepatic biliary-canalicular membrane. The MRP2/ABCC2 1515YY carriers presented a near 3-fold increase in concentration, when compared with 1515C patients [Citation93], compatible with the in vitro observations of decreased efflux by this variant protein [Citation122].
4.3. Amodiaquine
The Mannich base, 4-aminoquinoline Amodiaquine (AQ) () was synthesized for the first time in the late 1940s [Citation123], and soon recognized as a valid antimalarial [Citation124]. It has been a mainstay in the treatment of uncomplicated malaria in Africa for more than half a Century. Few years after the first inclusion of AQ in the WHO Model List of Essential Drugs in 1979, it was removed due to reports of fatal adverse drug reactions, namely agranulocytosis and/or severe hepatic damage, described among Caucasian travelers under AQ prophylaxis [Citation125,Citation126]. In 1996, an intense review of the available clinical data, including the assessment of adverse events among drug efficacy trials, suggested AQ toxicity as primarily seen in non-Africans [Citation127], specifically under drug exposures associated with prophylaxis regimens, hence supporting its safety and adequate use for the regular treatment of uncomplicated malaria [Citation128]. AQ was subsequently reinstated as a WHO supported option for malaria management [Citation129], leading to its subsequent inclusion in the efforts for developing antimalarial combination therapies, during the early years of the XXI Century. Although AQ remains overall well tolerated, and serious adverse events rather rare, mild ones are very frequent under typical therapeutic doses. This includes abdominal pain, nausea, neutropenia, dizziness, pruritus, and vomiting [Citation130].
AQ is presently mostly used in combination with artesunate (AS) (ASAQ) as a first-generation ACT [Citation131]. ASAQ (weight adjusted 4 mg/kg ATS + 10 mg/kg AQ) is presently a fixed combination originally developed through a collaboration between Sanofi-Aventis and DNDi (Drugs for Neglected Diseases, https://dndi.org) (Winthrop). Additionally, AQ is also used in combination with sulfadoxine-pyrimethamine (SP-AQ: 30 mg AQ base/kg body weight over 3 days, i.e. 10 mg/kg/day) in Seasonal Malaria Chemoprevention (SMC) [Citation132]. This is a key WHO mass drug administration (MDA) strategy based on the monthly distribution of intermittent preventive treatment for young children during disease transmission periods in regions with marked malaria seasonality. This includes most African countries of the Sahel belt sub-region [Citation133]. Finally, AQ is being considered as a partner for novel combinations under development with the old but resurfacing antimalarial methylene blue [Citation134].
AQ is quickly absorbed after oral administration, being primarily metabolized in the liver through N-de-ethylation by the cytochrome P450 2C8 (CYP2C8) toward its main biologically active metabolite desethyl-amodiaquine (DEAQ) [Citation135]. In contrast, with AQ short half-life of 2–12 hours, DEAQ has a longer terminal elimination half-life (4->20 days), commanding a 100 to 240-fold higher internal exposure when compared with AQ [Citation136,Citation137]. Accordingly, AQ has been occasionally even referred to as a pro-drug [Citation138]. Albeit being less active against the parasite [Citation19,Citation139], this large difference in exposure makes DEAQ the main responsible for antimalarial action, including the important temporary post-treatment protective effect during its elimination phase. DEAQ has been detected in plasma and/or blood up to 1 month after drug administration being then further metabolized in vivo, via a still unclear route, into its inactive metabolite, bis-DEAQ [Citation140,Citation141]. A number of minor metabolites from AQ are produced in parallel with DEAQ. This includes the aforementioned bis-DEAQ, now generated directly from AQ [Citation138], hydroxy-DEAQ [Citation142], and the aldehyde compound M2, the latter only detected in vitro. At least M2 is likely a result of CYP1A1 and CYP1B1 action [Citation135] – two mainly extra-hepatic expressed cytochrome P450s – over both AQ and DEAQ [Citation143]. The pharmacological importance of these minor metabolites remains unclear, albeit early research showed hydroxy-DEAQ to be two orders of magnitude less active in vitro then DEAQ against P. falciparum [Citation144], while M2 has been shown in vitro to be further metabolized by CYP1A1 and CYP1B1 toward quinoneimine species [Citation143].
The key importance of CYP2C8 in the metabolism of AQ raises the possibility of its polymorphism to be of pharmacogenetic significance. Albeit less polymorphic than most drug metabolizing CYPs, CYP2C8 carries a range of minor alleles [Citation145]. CYP2C8*2 and CYP2C8*3 are the globally most prevalent [Citation146], both known to code for enzymes with decreased activity in vitro. CYP2C8*2 is common among African populations [Citation147], whilst *3 – common among Europeans – is mainly present in locations in the continent where the admixture between Caucasian and African populations has been historically frequent [Citation148,Citation149]. CYP2C8*2 activity on AQ has been characterized in vitro (recombinant microsomes) with a near 40% reduction in its Vmax, associated with 3-fold increase in its Km [Citation150]. The same authors determined in their experimental setting the *3 variant as having no detectable AQ biotransformation capacity. Additionally, no minor metabolites were detected, this being interpreted as these being mainly produced in vivo in extra-hepatic environments, with no direct role of CYP2C8. These investigations were part of a clinical trial performed in Burkina Faso assessing AQ monotherapy efficacy in uncomplicated malaria patients [Citation151] – no significant differences were reported in treatment success rates between wild-type carriers (*1/*1) and patients harboring the *2 allele. This result has recently been reproduced in a larger analysis of ASAQ trials in Zanzibar [Citation7]. These observations are not surprising, considering that both AQ and DEAQ are active antimalarials. Of note, the Zanzibari studies documented a strikingly similar profile of mild adverse events (mainly abdominal pain), significantly more frequent among carriers of at least one minor allele when compared with the (*1/*1) subjects (ca. 50% vs 30% in Burkina-Faso, 45% vs 28% in Zanzibar) [Citation7]. These observations are of potential importance. Although these adverse events are not on themselves worrying, they have the potential of decreasing compliance in real world, non-supervised treatment courses. This can lead to decreased effectiveness due to incomplete therapies, which additionally can create the conditions for the emergence of drug resistance. Moreover, an altered elimination curve is potentially able to influence the post-treatment protective capacity of DEAQ, which might in turn modulate the risk of selection of less sensitive parasites [Citation152–154]. In this perspective, more intense studies are warranted in the SMC-targeted Sahel regions, where the prevalence of the very low activity CYP2C8*3 is likely to be significant [Citation149].
AQ and DEAQ secondary effects have been associated in animal-based studies and in vitro approaches to a metabolic bio-activation toward quinoneimine (QNM) type of reactive metabolites [Citation155]. AQ is more prone to generate these toxic species (amodiaquine quinoneimine-AQQI) then DEAQ (DEAQ-QI) [Citation156], albeit the orders of magnitude larger exposure to DEAQ largely compensates this difference. The bio-activation of AQ is catalyzed by the cytochrome P450s [Citation157], namely CYP2D6, CYP3A4, and CYP2C8. DEAQ metabolism generating QNMs invoke the same enzymes, plus CYP2C9 [Citation158]. In addition to CYPs, a significant role of leukocyte myeloperoxidases in the production of QNMs has been proposed [Citation159]. All these enzymes harbor polymorphisms leading to significantly altered activities, and hence driving inter-individual differences in risk of AD/DEAQ generated QNM exposure and their linked serious adverse events. In this context, the previously mentioned reports on *2 association with mild adverse can be interpreted as resulting from a higher exposure to AQQIs.
QNMs are detoxified through the glutathione system, likely involving Phase II polymorphic glutathione S-transferases (GSH), producing QNM-GS conjugates [Citation155,Citation160,Citation161]. Specifically, GSTP1, GSTA4, GSTM4, GSTM2, and GSTA2 have been proposed to be involved [Citation162]. Consistent with these observations, high dose or long-term use of AQ potentially leads to depletion of glutathione levels and eventually liver toxicity. The QNM detoxification process also involves the polymorphic defense enzyme NADPH:quinone oxireductase 1 (NQO1) [Citation163], through reduction [Citation162]. This is consistent with the fact that NQO1 is frequently co-induced with the glutathione system as a response battery to environmental stressors [Citation164]. Albeit both GSTs and NQO1 harbor polymorphisms responsible for severe reductions on activity of their coded enzymes [Citation163], no studies have been conducted associating this variation and AQ therapy adverse events. As for QNM-GS, these compounds being a phase II hydrophilic conjugate they are likely to be transported by MRP2/ABCC2, in the liver apical membrane (biliary secretion) as a detoxification route [Citation165].
In terms of possible drug-drug interactions (DDI), it is worth to mention that AQ is a CYP2D6 and CYP2C9 inhibitor in vivo [Citation166], the former cytochrome P450 being involved in the metabolism of Methylene blue, a prospective partner of AQ in antimalarial combination therapy [Citation134]. It is possible that certain pharmacogenetic profiles might exacerbate drug–drug interactions (DDI) which by themselves lead to decreased CYP2C8 activity, e.g. AQ is co-administered with some clinically used drugs capable of CYP2C8 inhibition (ritonavir, ketoconazole, and thrimethoporin) [Citation167]. A CYP2C8*2/*2 carrier, already with lower enzyme activity, would be turned through DDI into a CYP2C8*3 type phenocopy, leading to an even higher risk of AQ associated adverse events.
Finally, one cannot underestimate the diversity Sub-Saharan African populations. This is supported by reports of large inter-individual changes in pharmacokinetic parameters, without the presence of CYP2C8*2 or *3, suggesting that other mutations in the gene might have been involved (e.g. [Citation168]). Such expanded diversity is supported by recent data from studies in African Native populations [Citation46].
4.4. Piperaquine
Piperaquine (PPQ) is a bisquinoline (), with essentially a scaffold involving two chloroquine-like structures linked by a bridge. PPQ was heavily deployed as mass drug administration (MDA) in Southern Asia during the 1980s, with resistance swiftly developing [Citation169,Citation170]. Reintroduced this Century as a combination with DHA (320 mg of PPQ Phosphate +40 mg DHA formulation) in SE Asia, PPQ resistance again rapidly emerged [Citation4]. DHA-PPQ is nevertheless still highly effective in Africa [Citation171], and part of many national malaria control programs [Citation47].
Piperaquine is notorious for its very long elimination half-life (>20 days) [Citation172]. It is bio-transformed toward two main active metabolites, M1 and M2, with ca. 9 and 4 days half-lives. M1 and M2, albeit active, have EC50 values nearly ten times higher than the parent drug. This associated with their shorter T1/2 (especially M2) suggests that faster PPQ metabolizers might confer less protection against post-treatment reinfection events. In vitro data indicates piperaquine as mainly metabolized by CYP3A4, with a possible secondary contribution of CYP2C8 [Citation173]. To our knowledge there are no studies available associating SNPs in these genes and pharmacokinetic/pharmacodynamics outcomes.
4.5. Pyronaridine
Pyronaridine (Malaridine) is a benzonaphthyridine derivative () with strong anti-parasitic action developed in China during the 1980s [Citation174]. In combination with artesunate, it represents the most recent addition to the ACT arsenal, with the 180 mg PYR/60 mg (ATM) combination having been granted European Medicines Agency (EMA) approval in 2015 for medicinal products intended for non-EU markets (Article 58 roadmap). Pyronaridine (PYR) has a long half-life of 12–18 days, and a complex metabolism involving over 10 primary and secondary metabolites, with both hepatic and renal elimination [Citation175]. Unchanged PYR is almost totally eliminated in the liver, representing ~40% of the total recovered drug-related material. Two main metabolites (M1 and M2) dominate >95% of the renal excretion.
Microsome in vitro data during the drug development in China pointed for CYP1A2, CYP2D6, and CYP3A4 as key CYPs in PYR metabolism [Citation176]. CYP2D6 harbors extensive polymorphism with high frequency mutant alleles, raising the possibility of having a significant pharmacogenetic contribution. Trends between CYP2D6 status (probe drug: metoprolol) and pharmacokinetic parameters [Citation177] have not been found, albeit it is to be noted that these relatively limited studies did not fully addressed the complexity of CYP2D6 phenotypes/genotypes. Also, as with LUM, PYR is a documented CYP2D6 inhibitor, which might be of importance in terms of potential drug–drug interactions.
PYR has been determined to be a P-glycoprotein substrate (and inhibitor) [Citation178,Citation179], raising the possibility of a role for MDR1/ABCB1 SNPs in PYR disposition.
5. Transmission blockers: the pharmacogenetics of primaquine and tafenoquine
The drugs commonly used to eradicate blood stages of P. falciparum are not adequately effective against mature gametocytes [Citation180,Citation181]. Since these parasite stages are responsible for transmission to mosquitoes, their persistence jeopardizes transmission-reducing strategies. The World Health Organization (WHO) recommends a single dose of 0.25 mg/kg primaquine (PQ) added to ACT-based P. falciparum treatment in low-transmission areas, or those threatened by emerging or established artemisinin resistance [Citation182]. A systematic review showed recently that this low dose of PQ added to ACT for malaria reduces infectiousness of people to mosquitoes, although it is still not clear whether PQ added to malaria treatment translates into a reduction of community transmission of malaria [Citation183].
Primaquine () is a rapidly eliminated 8-aminoquinoline (elimination half-life, 4–6 hours) developed in 1945 and introduced in clinical use in the early 1950s. PQ is particularly effective against mature gametocytes of P. falciparum and dormant liver stages of Plasmodium vivax and Plasmodium ovale (i.e. hypnozoites) [Citation184]. Although the exact mechanism of action of PQ against the parasite is unknown, a body of evidence points to a similar mechanism of generation of reactive oxygen species (ROS) implicated in PQ efficacy and hemolytic toxicity in patients with G6PD deficiency (G6PDd) () [Citation185,Citation186]. Hydrogen peroxide (H2O2) is the main agent responsible for oxidative damage and hemotoxic effects of PQ [Citation185,Citation187–189].
Figure 3. Mode of action of primaquine and the effects of CYP2D6 polymorphism. Primaquine biotransformation into active metabolites requires the generation of hydrogen peroxide (H2O2) to parasite killing. Reactive oxygen species (ROS) are generated by oxidation and redox cycling of hydroxylated-PQ metabolites (OH-PQm) produced by the complex NADPH cytochrome P450 oxidoreductase (CPR) and the cytochrome P450 2D6 (CYP2D6)[Citation185,Citation186]. Beyond its role as CYP2D6 redox partner required to electron transfer from NADPH, CPR has a direct role in the redox cycling of OH-PQm, leading to generation of H2O2 and parasite killing. PQ gametocytocidal activity is significantly enhanced by the direct reduction of OH-PQm by CPR [Citation185]. Created with BioRender.com.
![Figure 3. Mode of action of primaquine and the effects of CYP2D6 polymorphism. Primaquine biotransformation into active metabolites requires the generation of hydrogen peroxide (H2O2) to parasite killing. Reactive oxygen species (ROS) are generated by oxidation and redox cycling of hydroxylated-PQ metabolites (OH-PQm) produced by the complex NADPH cytochrome P450 oxidoreductase (CPR) and the cytochrome P450 2D6 (CYP2D6)[Citation185,Citation186]. Beyond its role as CYP2D6 redox partner required to electron transfer from NADPH, CPR has a direct role in the redox cycling of OH-PQm, leading to generation of H2O2 and parasite killing. PQ gametocytocidal activity is significantly enhanced by the direct reduction of OH-PQm by CPR [Citation185]. Created with BioRender.com.](/cms/asset/2657a9e4-fc63-41b0-ac03-5ca37fe69e0c/iemt_a_2049235_f0003_oc.jpg)
Host genetic variation can affect PQ metabolism in the liver [Citation187]. Cytochrome P450 2D6 (CYP2D6) is primarily expressed in the liver and is involved in metabolism of 20–25% of all drugs in clinical use, such as antidepressants, antipsychotics, analgesics, beta-blockers, and antiarrhythmics [Citation190–193]. The human CYP2D6 gene is highly polymorphic, with over 140 alleles known to be associated with either complete loss of activity, or decreased, normal or increased function [Citation194,Citation195]. Activity scores (AS) are used by the Clinical Pharmacogenetics Implementation Consortium (CPIC) to infer enzyme activity from genotypes [Citation196]. An AS value ranging between 0 to 1 is assigned for each allele according to the associated enzyme activity and the final AS score corresponds to the sum of AS values of both alleles. AS scores range from 0 (loss of CYP2D6 function) to >2 (ultra-rapid metabolism) [Citation197–199].
Low-activity CYP2D6 polymorphisms were first described to reduce the hypnozoitocidal efficacy of PQ in an experimental P. vivax challenge, with repeated relapses despite standard PQ treatment [Citation200,Citation201]. Several studies have confirmed these findings in different endemic settings [Citation202–204]. CYP2D6 activity is typically predicted from genotype data, but some studies used a probe drug (i.e. dextromethorphan) to directly measure CYP2D6 activity and confirm that individuals with decreased enzyme activity are at elevated risk of P. vivax malaria recurrence [Citation200,Citation203]. Reduced urinary levels of 5,6-ortho-quinone, a surrogate marker of the presumed active metabolite 5-hydroxyprimaquine, are found in individuals with impaired CYP2D6 metabolism, consistent with the key role of CYP2D6-mediated biotransformation for PQ hypnozoitocidal efficacy [Citation205]. Experimental studies using knockout mouse models have also established a link between the anti-relapse activity of PQ and different levels of CYP2D6 enzyme activity [Citation186,Citation206]. Much less is known regarding the impact of CYP2D6 enzyme activity on the gametocytocidal effect of PQ in P. falciparum malaria. A single retrospective study has examined the influence of CYP2D6-mediated PQ metabolism on gametocyte clearance in African populations treated with a single dose of PQ (0.1 to 0.75 mg/kg) in combination with ACT [Citation207]. A higher gametocyte prevalence was found for patients with impaired CYP2D6 activity, compared to normal/ultrarapid metabolizers.
8-aminoquinolines, such as PQ and tafenoquine (TQ), are known to cause hemolytic toxicity in patients with G6PD deficiency. Interestingly, individuals with reduced CYP2D6 activity and G6PDd can also experience hemolysis following PQ treatment [Citation207], suggesting that PQ metabolites causing hemolysis are not necessarily generated by CYP2D6-dependent pathways. G6PDd, the most common inherited disorder of humans, affects over 400 million people worldwide, with a global prevalence of approximately 5%. This X-linked recessive disorder can cause hemolytic anemia and eventually lead to multi-organ failure and mortality, although most individuals with G6PD deficiency are asymptomatic. Multiple allelic variants of the G6PD gene have been identified which affect differently males and females and have a distinct impact on hemolysis and activity of the enzyme [Citation208]. The highest prevalence of G6PDd occurs in sub-Saharan Africa (mainly A−), but locally predominant G6PD alleles in Asian countries are associated with highest risk of severe G6PDd. PQ can be prescribed to prevent P. vivax malaria relapses (adult dose, 0.25–0.5 mg/kg, over 14 days) for individuals with >30% of the normal levels of G6PD enzyme activity. A dose of PQ at 0.75 mg/kg once a week for 8 weeks can be given for individuals with G6PDd. Individuals with G6PDd and those with intermediate (30–80%) levels of activity should be monitored for hemolysis [Citation209]. Also, a single PQ dose (0.25 mg/kg) can be given to G6PDd individuals to rapidly eliminate P. falciparum gametocytes and prevent onwards transmission. Because PQ toxicity is dose-dependent, the currently recommended low gametocytocidal dose is probably safe in individuals with mild-to-moderate G6PDd. The single low dose of PQ was well tolerated and safe even in populations with high prevalence of G6PDd in the North-Western Myanmar-Thailand border where the Mahidol variant associated with a low residual enzymatic activity is predominant [Citation210]. Likewise, African G6PDd males given single low-dose PQ in combination with artemether-lumefantrine or dihydroartemisinin-piperaquine had only mild and transient reductions in hemoglobin in two recent clinical trials, suggesting that this regimen is safe in individuals with the African A-variant [Citation211].
TQ is a slowly eliminated (elimination half-life, 14 days) 8-aminoquinoline () first synthesized in 1979 that advanced to clinical development in 1991 [Citation184]. Only a few clinical studies have assessed the impact of CYP2D6 polymorphisms on the anti-relapse effect of TQ in P. vivax malaria [Citation212,Citation213]. TQ efficacy did not appear to be reduced in intermediate metabolizers of CYP2D6, but TQ efficacy remains to be determined in low metabolizer individuals with AS < 1. Conflicting results have been reported on the effect of TQ biotransformation in CYP2D knock-out against liver-stages of Plasmodium berghei [Citation214–216]. Noticeably, the changes in TQ pharmacokinetic parameters were less pronounced than those reported for PQ in the same mouse model [Citation206,Citation216] TQ is not currently recommended as a gametocytocide for use in P. falciparum infections treated with ACTs and cannot be used without previous G6PDd testing. Moreover, the gametocytocidal effect of TQ may be antagonized by some ACT partner drugs, such as lumefantrine, mefloquine, and amodiaquine [Citation217]. The extent to which human CYP2D6 polymorphism affects TQ effect against gametocytes remains unexplored.
6. Future combinations – carrying over from ACT
The last 15 years have witnessed the establishment of a pipeline for the development of new antimalarials only comparable with the WWII drug discovery effort. Most of these new combinations are based on blending a well-established ACT long half drug with the new compound, in an attempt of replacing the role of the presently used artemisinin derivatives. The information available on the metabolism, and in particular pharmacogenetics of these compounds is still limited. Nevertheless, we herein refer some of the most notable new combinations expected to hit the ground during this decade.
6.1. KAF156 (Ganaplacide)-Lumefantrine
KAF156 is an imidazole-piperazine under development by Novartis, which represents a new type of chemical structure among antimalarial drugs [Citation218] (). This new drug is of interest for this review, as it is planned to be used as a combination with lumefantrine, being to a certain extent a direct heir of the AL ACT.
KAF156 has a very powerful anti-plasmodia effect, with IC50s in the order of the single digit in nano-molarity [Citation219], being reasonably tolerated and with rapid clinical effect [Citation220,Citation221]. The KAF156 combination is presently under phase II trials [Citation25].
According to the available information, Tmax and T1/2 are variable with values of 1–6 h, and 40–70 h, respectively. The inter-individual range in terms of AUC0-∞ is 1.3–2-fold [Citation222]. CYP3A4 has been suggested as involved in KAF156 metabolism, albeit non-P450 phase I enzymes are also expected to have a major role, possibly including flavin monooxygenases. To note that these latter enzymes are generally less inducible then the P450s, which raises the point that any contributions to phenotypic variation will potentially be more related with sequence variation.
6.2. Arterolane (OZ277, RBx11160)-maleate/Piperaquine and artefenomel (OZ439)/Ferroquine (SSR97193)
Since its broader adoption from ca. 2005, the use of ACT has increased almost 40-fold in the following 15 years [Citation8]. Naturally, demand for artemisinin ramp-up in parallel. Most artemisinin is obtained from relatively scarce natural sources, with a recovery amount of 0.1–1% of the Artemisia plants dry weight [Citation223]. This factor alone has been a motivation for the discovery of fully synthetic alternatives. Additionally, Artemisinin and its available derivatives offer a number of limitations, including in some cases (e.g. artesunate) extremely short half-lives (<1 h) and likely limited therapeutic windows.
Recognizing the endoperoxide bridge as the key structure conferring artemisinin anti-Plasmodia effect, several fully synthetic peroxide antimalarials, collectively referred as ozonides have been developed [Citation224].
Arterolane (OZ277, RBx11160) (), represents the first generation of these compounds that reached the market [Citation225]. Not yet approved by WHO, it is nevertheless available in some regions of the developing world in a fixed combination with piperaquine [Citation226,Citation227]. Arterolane has a T1/2 of 2–4 hours, a noticeable but still small improvement over conventional ARTs [Citation228]. Initial in vitro studies using human hepatic microsomes and cryopreserved hepatocytes pointed for the involvement of CYP3A4 and CYP2C9 in Arterolane biotransformation in its two major hydroxylated metabolites [Citation224,Citation229]. To note that arterolane was observed to ‘strongly inhibit’ the CYP2C19 isoform [Citation224], raising the possibility of non-negligible drug–drug interactions.
Artefenomel (OZ439) is a second-generation ozonide (), representing the result of efforts aiming on the development of a more stable chemical scaffold, protecting the peroxide bond pharmacophore [Citation230]. This has resulted in a vastly improved half-life of 46–62 h [Citation231]. No information is readily available concerning factors involved in Artefenomel disposition.
A combination of Artefenomel with the 3rd generation 4-aminoquinoline Ferroquine (SSR97193) () as long elimination partner (>15 days) is presently under phase II/III development. Ferroquine is metabolized by a number of cytochromes P450s – CYP2C9, CYP3A4, CYP2D6 – toward two main active products, N-desmethyl-Ferroquine and N-di-desmethyl-ferroquine [Citation232]. Two- to three-fold variations in Ferroquine exposure has been documented in vivo [Citation233]. No data is available concerning the influence of CYP polymorphism in this inter-individual differences.
6.3. Fosmidomycin-piperaquine
Fosmidomycin () is an antibiotic originally developed during the 1970s for bacterial infection management. Its well-established antimalarial activity [Citation234,Citation235] has motivated the present development of a new combination with the established antimalarial Piperaquine [Citation236]. Fosmidomycin has a short half-life showing a range of inter-individual values (1.5–12 h), and a 3–4-fold varying AUC0-∞ [Citation235,Citation237]. Fosmidomycin seems not to be metabolized, being eliminated unchanged by the kidneys. It is conceivable that the observed range in T1/2 might be in part associated with genetic variation in the large range of ABC and Solute Carrier Family (SLC) transporters present in this organ [Citation238].
6.4. Methylene blue – amodiaquine
The hydrophilic dye methylene blue (MB) () is an historical drug, being actually the first synthetic therapeutic drug, frequently used to treat malaria during the late XIX and early XX Century [Citation239,Citation240]. Recently, there has been a renewed interest in MB, explored as both alone [Citation241], and as a combination partner, in particular, with amodiaquine [Citation242]. MB has a typical T1/2 of 15–24 h. AUC0-∞ values can vary 3–4-fold [Citation243].
Most of MB elimination happens through the renal route, with ca. 40% of MB excreted unchanged. Metabolites include the major metabolite leucomethylene blue, as well as three other minor N-demethylated ones, Azure A, B, and C.
MB is in vitro metabolized predominantly by the phase II glucuronidases UGT1A4 and UGT1A9, with additional contributions of the phase I CYP1A2, CYP2D6, and CYP2C19. Additionally, MB is a wide-spectrum CYP inhibitor, including CYP1A2, CYP2B6, CYP2D6, CYP3A4/5, CYP2C19, CYP2C9, and – of interest for the proposed MB-Amodiaquine combination – CYP2C8 [Citation244]. MB is also a substrate of ABCB1, as well as other ABC-type of transporters [Citation245,Citation246].
Most importantly, its mechanism of action [Citation247] prompts the disturbance of the red blood cell pentose phosphate pathway. As a consequence, MB has the potential to promote hemolysis, especially among G6PDd patients [Citation208]. The actual in vivo penetrance of such effects is still under intense discussion [Citation248–250]. It is possible that specific subpopulations of individuals carrying both G6PD deficiency and an unfavorable ADME set of mutations might be at significantly increased risk.
MB pharmacological characteristics raises a number of concerns of drug–drug interactions, potentially exacerbated by the patient's individual pharmacogene status, namely G6PD. Pharmacogenetic studies are much needed under the perspective of MB-Amodiaquine being a combination presently well established in the MMV pipeline for antimalarial drug discovery and development [Citation134].
7. Conclusions – ten years later, any pharmacogenetic marker in the horizon?
A pharmacogenetic marker, in the sense of full clinical usefulness, is a demanding tool [Citation251]. It involves a number of conditions challenging to fulfill in the context of Developing World health systems. Firstly, it needs to have a high predictive power relative to the clinical/physiological phenotype of interest. This demands high specificity and sensitivity, in order to guarantee an appropriate and useful drug dosing adjustment. The biomarker needs to be simple, preferentially based on only one variation factor, like a SNP or insertion/deletions (indels). If instead, it involves a complex set of genetic markers it will make its application less attractive due to increased costs, more complex technology, more difficult result interpretation, all demanding more specific (and hence less common) training, and increased time of feedback to the health practitioner and the patient. This conjugation of high predictability, assay simplicity, and low cost have rarely been reached.
In the area of malaria, especially in the context of ACT no marker has been found that complies with these demands. The best candidate might not be related to ACT drugs themselves, but instead to primaquine (or tafenoquine), due to the strict importance of CYP2D6 driven metabolism for the generation of the ‘double edge sword’ set of metabolites responsible for both efficacy and adverse events. Anyway, CYP2D6 polymorphism can be intricate and not the straight yes/no bench-side answer that is frequently demanded in clinical settings, especially in low-income regions. Also, and especially concerning the adverse event side of primaquine, it will be important to conjugate this with G6PD pharmacogenetics. The latter is notoriously complex, with the association between gene mutations and final phenotypic outcome being frequently imprecise. An example is the A− mutation, highly frequent among Africans, which is linked with a large range of G6PD decreased activities [Citation252]. Such variation adds doubts as for the availability of an effective precision medicine tool for the optimal personalized use of primaquine.
Population pharmacogenetics have anyway the potential to be useful as public health tools. In particular, when planning large-scale drug administration programs, a knowledge of the pharmacogenetic characteristics of the targeted population will aid decision-making on the type of intended implementation. One clear application relates with SMC, which is based on SP-Amodiaquine MDA. These programs are mainly applied in the Sahel belt and neighboring southern regions, which represent a human genetics interface with the CYP2C8*3 rich Caucasian population and the CYP2C8*2 rich African populations. Consistent with this view, a recent report showed the Eritrean population as harboring intermediate CYP2C8*2 and *3 frequencies between Caucasian and African populations [Citation149]. Increased adverse events associated with being a carrier to these minor alleles, albeit mild, can decrease the compliance of the populations to these important programs.
8. Expert opinion
Since our 2011 review there has been some advances in the understanding of antimalarial pharmacogenetics, with emerging data on other ADME factors, and some information on the possible role of drug transporters. Notably, the first clear data concerning a pharmacogenetics driven interaction between antimalarials and another anti-infective drug, in this case, an anti-HIV non-Nucleoside Reverse Transcriptase inhibitor (NNRTI) have been reported, adding a new dimension on the importance of pharmacogenetics in poly-pharmacy associated with the control of multiple infectious diseases [Citation253]. Still, the body of data continues to be limited, typically scattered in the literature, and inconsistent with the impact and importance of antimalarial therapy.
Also, it is likely that this knowledge gap will widen. A significant number of antimalarial drug therapies are presently being developed, a number already under phase II–III trials, others already in the field waiting for WHO homologation. The information concerning their disposition factors – ADMEs or transporters – is still scarce, curtailing a robust understanding of the potentially involved pharmacogenetics. Nevertheless, genomic analysis tools, namely next-generation sequencing, is becoming less costly at a rapid pace. Most of the information presented in this review cannot yet be considered as an ‘omics’ output, as, with rare exceptions (e.g. [Citation46]), is focused in one or very few genes and SNPs. The arrival of these technologies in the malaria pharmacogenetics field will surely open new views of the expected high diversity of human genomics, particularly in the African Continent. On the other hand, this will demand ever larger, statistically more powerful trials for detecting complex configurations of single low impact genetic polymorphisms associated with relevant pharmacologic phenotypes. These expected increased demands will likely constitute a new bottleneck in the development of the area. Smart new approaches (e.g. adaptive clinical trials [Citation254]) will also need to be considered for the full exploitation of these new possibilities.
In our previous review, we ended with the uplift statement ‘the parallel development of a deeper understanding of the molecular pharmacology of antimalarials should allow in the next 10 years the translation of the first antimalarial pharmacogenetic tools to the endemic areas’ [Citation1]. Reality has proven us too optimistic. As antimalarials continue to constitute one of the most widely prescribed treatments, much more knowledge concerning endemic populations needs to be generated for this hope to materialize [Citation255]. As an example, the understudied pharmacogene diversity in Africa is likely to be larger than presently considered [Citation7], needing focused studies of specific ethnic groups.
Further development of local pharmacogenetic research in malaria affected areas is needed, in particular in scenarios of infectious co-morbidities and associated risk of drug–drug interactions due to polypharmacy, as recently demonstrated between lumefantrine and efavirenz. Particular pharmacogenetic profiles can amplify such interactions resulting on increased risks of adverse events, driving decreases in compliance and treatment success, while potentially influencing the process of resistance development.
Personalized medicine should not be seen as a downstream effect of establishing a successful therapy, but part of the process itself, even when considering that the target populations are amongst the poorest in the world [Citation252]. The review hence does not conclude with the same positive view as its predecessor, but with a more cautious outlook. Albeit we do not dare to predict again that the first individual personalized antimalarial therapy will rise during this decade, the use of population pharmacogenetics might gain a valuable role supporting public health decisions, in particular, with mass drugs administration, aiming for malaria pre-elimination.
Article highlights
Malaria is a vector-borne tropical and subtropical disease affecting over 241 million people just last year. Causing 627,000 deaths in Africa alone mainly driven by Plasmodium falciparum infections.
Artemisinin-based combination therapies (ACTs) are the globally recommended first-line treatments for uncomplicated P. falciparum malaria.
Host genetic variation in genes involved in antimalarial disposition or toxicity constitutes an important determinant of adverse drug reactions and may even prime drug resistance development. The knowledge of its pharmacogenetics continues to largely trail the public health importance of these treatments.
The first clear example of pharmacogenetic exacerbated drug–drug interactions involving antimalarials has been unveiled giving increased urgency for a better understanding of these anti-infection drugs pharmacology.
Albeit individual antimalarial precision medicine is still not achievable with presently available pharmacogenetic knowledge, a better understanding of population-specific pharmacogenetic signature, namely in African populations, can be used for optimizing large-scale drug administration programs.
This box summarizes key points contained in the article.
Declaration of interest
L Pernaute-Lau is a recipient of a fellowship from BioSys PhD program PD65‐2012 (Ref SFRH/BD/142860/2018) from Fundação para a Ciência e Tecnologia (FCT, Portugal). JP Gil, M Camara and U Morris were partially supported by the European Developing Countries Clinical Trial Partnership (EDCTP2) programme supported by the European Union (Grant number RIA2017T-2018 – WANECAM 2). T Nóbrega de Sousa is a recipient of a Senior Research Scholarships from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil). Funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPqants number 404,067/2012-3, 2020/00433-8), and the Fundação de Amparo à Pesquisa do Estado de mInas Gerais (FAPEMIG), grant number CBB-APQ 00952-16). MU Ferreira was funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPqants number 404,067/2012-3, 2020/00433-8), and the Fundação de Amparo à Pesquisa do Estado de mInas Gerais (FAPEMIG), grant number CBB-APQ 00952-16). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Piedade R, Gil JP. The pharmacogenetics of antimalaria artemisinin combination therapy. Expert Opin Drug Metab Toxicol. 2011;7(10):1185–1200. DOI:https://doi.org/10.1517/17425255.2011.608660.
- MMV-supported projects [Internet]. Geneva: Medicines for Malaria Venture; [ cited 2021 Jun 08]. Available from: https://www.mmv.org/research-development/mmv-supported-projects
- Price RN, Uhlemann AC, Brockman A, et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004;364(9432):438–447. DOI:https://doi.org/10.1016/S0140-6736(04)16767-6.
- Amaratunga C, Lim P, Suon S, et al. Dihydroartemisinin–piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis. 2016;16(3):357–365. DOI:https://doi.org/10.1016/S1473-3099(15)00487-9.
- Lawford H, Zurovac D, O’Reilly L, et al. Adherence to prescribed artemisinin-based combination therapy in Garissa and Bunyala districts, Kenya. Malar J. 2011;10(1):281. DOI:https://doi.org/10.1186/1475-2875-10-281.
- Lemma H, Löfgren C, San Sebastian M. Adherence to a six-dose regimen of artemether-lumefantrine among uncomplicated Plasmodium falciparum patients in the Tigray Region, Ethiopia. Malar J. 2011;10(1):349.
- Pernaute-Lau L, Morris U, Msellem M, et al. Influence of cytochrome P450 (CYP) 2C8 polymorphisms on the efficacy and tolerability of artesunate-amodiaquine treatment of uncomplicated Plasmodium falciparum malaria in Zanzibar. Malar J. 2021;20(1):90. DOI:https://doi.org/10.1186/s12936-021-03620-6.
- World Health Organization. World Malaria Report. 2020.
- Macintyre F, Adoke Y, Tiono AB, et al. A randomised, double-blind clinical phase II trial of the efficacy, safety, tolerability and pharmacokinetics of a single dose combination treatment with artefenomel and piperaquine in adults and children with uncomplicated Plasmodium falciparum malaria. BMC Med. 2017;15(1):181. DOI:https://doi.org/10.1186/s12916-017-0940-3.
- Bloland PB, Ettling M, Meek S. Combination therapy for malaria in Africa: hype or hope? Bull World Health Organ. 2000;78(12):1378–1388.
- Chaturvedi R, Chhibber-Goel J, Verma I, et al. Geographical spread and structural basis of sulfadoxine-pyrimethamine drug-resistant malaria parasites. Int J Parasitol. 2021;51(7):505–525. DOI:https://doi.org/10.1016/j.ijpara.2020.12.011.
- Sibley CH, Hyde JE, Sims PF, et al. Pyrimethamine-sulfadoxine resistance in Plasmodium falciparum: what next? Trends Parasitol. 2001;17(12):582–588. DOI:https://doi.org/10.1016/S1471-4922(01)02085-2.
- German PI, Aweeka FT. Clinical pharmacology of artemisinin-based combination therapies. Clin Pharmacokinet. 2008;47(2):91–102.
- Nosten F, Brasseur P. Combination therapy: the way forward? Drugs. 2002;62(9):1315–1329.
- Price RN, Cassar C, Brockman A, et al. The pfmdr1 gene is associated with a multidrug-resistant phenotype in Plasmodium falciparum from the western border of Thailand. Antimicrob Agents Chemother. 1999;43(12):2943–2949. DOI:https://doi.org/10.1128/AAC.43.12.2943.
- Amato R, Lim P, Miotto O, et al. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect Dis. 2017;17(2):164–173. DOI:https://doi.org/10.1016/S1473-3099(16)30409-1.
- Witkowski B, Duru V, Khim N, et al. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype-genotype association study. Lancet Infect Dis. 2017;17(2):174–183. DOI:https://doi.org/10.1016/S1473-3099(16)30415-7.
- Holmgren G, Gil JP, Ferreira PM, et al. Amodiaquine resistant Plasmodium falciparum malaria in vivo is associated with selection of pfcrt 76T and pfmdr1 86Y. Infect Genet Evol. 2006;6(4):309–314. DOI:https://doi.org/10.1016/j.meegid.2005.09.001.
- Echeverry DF, Holmgren G, Murillo C, et al. Short report: polymorphisms in the pfcrt and pfmdr1 genes of Plasmodium falciparum and in vitro susceptibility to amodiaquine and desethylamodiaquine. Am J Trop Med Hyg. 2007;77(6):1034–1038. DOI:https://doi.org/10.4269/ajtmh.2007.77.1034.
- Sá JM, Twu O, Hayton K, et al. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci. 2009;106(45):18883–18889. 10. DOI:https://doi.org/10.1073/pnas.0911317106.
- Plucinski MM, Talundzic E, Morton L, et al. Efficacy of artemether-lumefantrine and dihydroartemisinin-piperaquine for treatment of uncomplicated malaria in children in Zaire and Uíge Provinces, Angola. Antimicrob Agents Chemother. 2015;59(1):437–443. DOI:https://doi.org/10.1128/AAC.04181-14.
- Plucinski MM, Dimbu PR, Macaia AP, et al. Efficacy of artemether-lumefantrine, artesunate-amodiaquine, and dihydroartemisinin-piperaquine for treatment of uncomplicated Plasmodium falciparum malaria in Angola, 2015. Malar J. 2017;16(1):62. DOI:https://doi.org/10.1186/s12936-017-1712-4.
- Dimbu PR, Horth R, Cândido ALM, et al. Continued Low Efficacy of Artemether-Lumefantrine in Angola in 2019. Antimicrob Agents Chemother. 2021;65(2):e01949–20. DOI:https://doi.org/10.1128/AAC.01949-20.
- Marsh K. Malaria disaster in Africa. Lancet. 1998;352(9132):924.
- Koller R, Mombo-Ngoma G, Grobusch MP. The early preclinical and clinical development of ganaplacide (KAF156), a novel antimalarial compound. Expert Opin Invest Drugs. 2018;27(10):803–810.
- Bouwman SA, Zoleko-Manego R, Renner KC, et al. The early preclinical and clinical development of cipargamin (KAE609), a novel antimalarial compound. Travel Med Infect Dis. 2020;36:101765.
- Belete TM. Recent Progress in the Development of New Antimalarial Drugs with Novel Targets. Drug Des Devel Ther. 2020;14:3875–3889.
- Lu G, Nagbanshi M, Goldau N, et al. Efficacy and safety of methylene blue in the treatment of malaria: a systematic review. BMC Med. 2018;16(1):59. DOI:https://doi.org/10.1186/s12916-018-1045-3.
- Bhagavathula AS, Elnour AA, Shehab A. Alternatives to currently used antimalarial drugs: in search of a magic bullet. Infect Dis Poverty. 2016;5(1):103.
- Mischlinger J, Agnandji ST, Ramharter M. Single dose treatment of malaria - current status and perspectives. Expert Rev Anti Infect Ther. 2016;14(7):669–678.
- Wadi I, Nath M, Anvikar AR, et al. Recent advances in transmission-blocking drugs for malaria elimination. Future Med Chem. 2019;11(23):3047–3088. DOI:https://doi.org/10.4155/fmc-2019-0225.
- Sagara I, Beavogui AH, Zongo I, et al. Safety and efficacy of re-treatments with pyronaridine-artesunate in African patients with malaria: a substudy of the WANECAM randomised trial. Lancet Infect Dis. 2016;16(2):189–198. DOI:https://doi.org/10.1016/S1473-3099(15)00318-7.
- Kouakou YI, Tod M, Leboucher G, et al. Systematic review of artesunate pharmacokinetics: implication for treatment of resistant malaria. Int J Infect Dis. 2019;89:30–44.
- Bunnag D, Viravan C, Looareesuwan S, et al. Clinical trial of artesunate and artemether on multidrug resistant falciparum malaria in Thailand. A preliminary report. Southeast Asian J Trop Med Public Health. 1991;22(3):380–385.
- Noedl H, Se Y, Schaecher K, et al. Artemisinin Resistance in Cambodia 1 (ARC1) Study Consortium. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359(24):2619–2620. DOI:https://doi.org/10.1056/NEJMc0805011.
- Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361(5):455–467. DOI:https://doi.org/10.1056/NEJMoa0808859.
- Bethell D, Se Y, Lon C, et al., Dose-dependent risk of neutropenia after 7-day courses of artesunate monotherapy in Cambodian patients with acute Plasmodium falciparum malaria. Clin Infect Dis. 51(12): e105–e114. 2010. . DOI:https://doi.org/10.1086/657402.
- Svensson US, Ashton M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br J Clin Pharmacol. 1999;48(4):528–535.
- Li XQ, Björkman A, Andersson TB, et al. Identification of human cytochrome P(450)s that metabolise anti-parasitic drugs and predictions of in vivo drug hepatic clearance from in vitro data. Eur J Clin Pharmacol. 2003;59(5–6):429–442. DOI:https://doi.org/10.1007/s00228-003-0636-9.
- Yusof W, Hua GS. Gene, ethnic and gender influences predisposition of adverse drug reactions to artesunate among Malaysians. Toxicol Mech Methods. 2012;22(3):184–192.
- Phompradit P, Muhamad P, Cheoymang A, et al. Preliminary investigation of the contribution of CYP2A6, CYP2B6, and UGT1A9 polymorphisms on artesunate-mefloquine treatment response in Burmese patients with Plasmodium falciparum malaria. Am J Trop Med Hyg. 2014;91(2):361–366. DOI:https://doi.org/10.4269/ajtmh.13-0531.
- Ilett KF, Ethell BT, Maggs JL, et al. Glucuronidation of dihydroartemisinin in vivo and by human liver microsomes and expressed UDP-glucuronosyltransferases. Drug Metab Dispos. 2002;30(9):1005–1012. DOI:https://doi.org/10.1124/dmd.30.9.1005.
- Teja-Isavadharm P, Watt G, Eamsila C, et al. Comparative pharmacokinetics and effect kinetics of orally administered artesunate in healthy volunteers and patients with uncomplicated falciparum malaria. Am J Trop Med Hyg. 2001;65(6):717–721. DOI:https://doi.org/10.4269/ajtmh.2001.65.717.
- Evans AM. Membrane transport as a determinant of the hepatic elimination of drugs and metabolites. Clin Exp Pharmacol Physiol. 1996;23(10–11):970–974.
- Jemnitz K, Heredi-Szabo K, Janossy J, et al. ABCC2/Abcc2: a multispecific transporter with dominant excretory functions. Drug Metab Rev. 2010;42(3):402–436. DOI:https://doi.org/10.3109/03602530903491741.
- Pernaute-Lau L, Adegnika AA, Zhou Y, et al. Pharmacogene sequencing of a Gabonese population with severe Plasmodium falciparum malaria reveals multiple novel variants with putative relevance for antimalarial treatment. Antimicrob Agents Chemother. 65(7): e0027521. 2021. . DOI:https://doi.org/10.1128/AAC.00275-21.
- World Health Organization. Drug Policies by Region [Internet]. Geneva: World Health Organization; [cited 2021 May 08]. Available from: https://www.who.int/malaria/am_drug_policies_by_region_afro/en/
- van Agtmael MA, Gupta V, van der Wosten TH, et al. Grapefruit juice increases the bioavailability of artemether. Eur J Clin Pharmacol. 1999;55(5):405–410. DOI:https://doi.org/10.1007/s002280050648.
- Lefevre G, Carpenter P, Souppart C, et al. Pharmacokinetics and electrocardiographic pharmacodynamics of artemether-lumefantrine (Riamet) with concomitant administration of ketoconazole in healthy subjects. Br J Clin Pharmacol. 2002;54(5):485–492. DOI:https://doi.org/10.1046/j.1365-2125.2002.01696.x.
- Staehli Hodel EM, Csajka C, Ariey F, et al. Effect of single nucleotide polymorphisms in cytochrome P450 isoenzyme and N-acetyltransferase 2 genes on the metabolism of artemisinin-based combination therapies in malaria patients from Cambodia and Tanzania. Antimicrob Agents Chemother. 2013;57(2):950–958. DOI:https://doi.org/10.1128/AAC.01700-12.
- van der Pluijm RW, Tripura R, Hoglund RM, et al. Tracking Resistance to Artemisinin Collaboration. Triple artemisinin-based combination therapies versus artemisinin-based combination therapies for uncomplicated Plasmodium falciparum malaria: a multicentre, open-label, randomised clinical trial. Lancet. 2020;395(10233):1345–1360. DOI:https://doi.org/10.1016/S0140-6736(20)30552-3.
- Mhamilawa LE, Ngasala B, Morris U, et al. Parasite clearance, cure rate, post-treatment prophylaxis and safety of standard 3-day versus an extended 6-day treatment of artemether-lumefantrine and a single low-dose primaquine for uncomplicated Plasmodium falciparum malaria in Bagamoyo district, Tanzania: a randomized controlled trial. Malar J. 2020;19(1):216. DOI:https://doi.org/10.1186/s12936-020-03287-5.
- Burk O, Arnold KA, Nussler AK, et al. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol Pharmacol. 2005;67(6):1954–1965. https://doi.org/10.1124/mol.104.009019.
- Zang M, Zhu F, Li X, et al. Auto-induction of phase I and phase II metabolism of artemisinin in healthy Chinese subjects after oral administration of a new artemisinin-piperaquine fixed combination. Malar J. 2014;13:214.
- Lamba J, Lamba V, Strom S, et al. Novel single nucleotide polymorphisms in the promoter and intron 1 of human pregnane X receptor/NR1I2 and their association with CYP3A4 expression. Drug Metab Dispos. 2008;36:169–181.
- Cortes CP, Siccardi M, Chaikan A, et al. Correlates of efavirenz exposure in Chilean patients affected with human immunodeficiency virus reveals a novel association with a polymorphism in the constitutive androstane receptor. Ther Drug Monit. 2013;35:78–83.
- Karbwang J, Looareesuwan S, Phillips RE, et al. Plasma and whole blood mefloquine concentrations during treatment of chloroquine-resistant falciparum malaria with the combination mefloquine-sulphadoxine-pyrimethamine. Br J Clin Pharmacol. 1987;23(4):477–481. DOI:https://doi.org/10.1111/j.1365-2125.1987.tb03079.x.
- Karbwang J, White NJ. Clinical pharmacokinetics of mefloquine. Clin Pharmacokinet. 1990;19(4):264–279.
- Karbwang J, Bangchang KN, Bunnag D, et al. Pharmacokinetics and pharmacodynamics of mefloquine in Thai patients with acute falciparum malaria. Bull World Health Organ. 1991;69(2):207–212.
- Karbwang J, Na Bangchang K, Thanavibul A, et al. Pharmacokinetics of mefloquine alone or in combination with artesunate. Bull World Health Organ. 1994;72(1):83–87.
- Na-Bangchang K, Karbwang J, Palacios PA, et al. Pharmacokinetics and bioequivalence evaluation of three commercial tablet formulations of mefloquine when given in combination with dihydroartemisinin in patients with acute uncomplicated falciparum malaria. Eur J Clin Pharmacol. 2000;55(10):743–748. DOI:https://doi.org/10.1007/s002280050008.
- Ramharter M, Kurth FM, Bélard S, et al. Pharmacokinetics of two paediatric artesunate mefloquine drug formulations in the treatment of uncomplicated falciparum malaria in Gabon. J Antimicrob Chemother. 2007;60(5):1091–1096. DOI:https://doi.org/10.1093/jac/dkm355.
- Gutman J, Green M, Durand S, et al. Mefloquine pharmacokinetics and mefloquine-artesunate effectiveness in Peruvian patients with uncomplicated Plasmodium falciparum malaria. Malar J. 2009;8:58.
- Krudsood S, Looareesuwan S, Tangpukdee N, et al. New fixed-dose artesunate-mefloquine formulation against multidrug-resistant Plasmodium falciparum in adults: a comparative phase IIb safety and pharmacokinetic study with standard-dose nonfixed artesunate plus mefloquine. Antimicrob Agents Chemother. 2010;54(9):3730–3737. DOI:https://doi.org/10.1128/AAC.01187-09.
- Valea I, Tinto H, Traore-Coulibaly M, et al. Pharmacokinetics of co-formulated mefloquine and artesunate in pregnant and non-pregnant women with uncomplicated Plasmodium falciparum infection in Burkina Faso. J Antimicrob Chemother. 2014;69(9):2499–2507. DOI:https://doi.org/10.1093/jac/dku154.
- Ferreira MVD, Vieira JLF, Almeida ED, et al. Pharmacokinetics of mefloquine administered with artesunate in patients with uncomplicated falciparum malaria from the Brazilian Amazon basin. Malar J. 2018;17(1):268. DOI:https://doi.org/10.1186/s12936-018-2416-0.
- Fontaine F, de Sousa G, Burcham PC, et al. Role of cytochrome P450 3A in the metabolism of mefloquine in human and animal hepatocytes. Life Sci. 2000;66(22):2193–2212. DOI:https://doi.org/10.1016/S0024-3205(00)00546-4.
- Ridtitid W, Wongnawa M, Mahatthanatrakul W, et al. Effect of rifampin on plasma concentrations of mefloquine in healthy volunteers. J Pharm Pharmacol. 2000;52(10):1265–1269. DOI:https://doi.org/10.1211/0022357001777243.
- Ridtitid W, Wongnawa M, Mahatthanatrakul W, et al. Ketoconazole increases plasma concentrations of antimalarial mefloquine in healthy human volunteers. J Clin Pharm Ther. 2005;30(3):285–290. DOI:https://doi.org/10.1111/j.1365-2710.2005.00651.x.
- Ozdemir V, Kalow W, Tang BK, et al. Evaluation of the genetic component of variability in CYP3A4 activity: a repeated drug administration method. Pharmacogenetics. 2000;10(5):373–388. DOI:https://doi.org/10.1097/00008571-200007000-00001.
- Kumondai M, Gutiérrez Rico EM, Hishinuma E, et al. Functional Characterization of 40 CYP3A4 Variants by Assessing Midazolam 1’-Hydroxylation and Testosterone 6β-Hydroxylation. Drug Metab Dispos. 2021;49(3):212–220. DOI:https://doi.org/10.1124/dmd.120.000261.
- Apellániz-Ruiz M, Lee MY, Sánchez-Barroso L, et al. Whole-exome sequencing reveals defective CYP3A4 variants predictive of paclitaxel dose-limiting neuropathy. Clin Cancer Res. 2015;21(2):322–328. DOI:https://doi.org/10.1158/1078-0432.CCR-14-1758.
- Westlind-Johnsson A, Hermann R, Huennemeyer A, et al. Identification and characterization of CYP3A4*20, a novel rare CYP3A4 allele without functional activity. Clin Pharmacol Ther. 2006;79(4):339–349. DOI:https://doi.org/10.1016/j.clpt.2005.11.015.
- Rebbeck TR, Jaffe JM, Walker AH, et al. Modification of clinical presentation of prostate tumors by a novel genetic variant in CYP3A4. J Natl Cancer Inst. 1998;90(16):1225–1229. DOI:https://doi.org/10.1093/jnci/90.16.1225.
- Amirimani B, Ning B, Deitz AC, et al. Increased transcriptional activity of the CYP3A4*1B promoter variant. Environ Mol Mutagen. 2003;42(4):299–305. DOI:https://doi.org/10.1002/em.10199.
- Ferreira PE, Veiga MI, Cavaco I, et al. Polymorphism of antimalaria drug metabolizing, nuclear receptor, and drug transport genes among malaria patients in Zanzibar, East Africa. Ther Drug Monit. 2008;30(1):10–15. DOI:https://doi.org/10.1097/FTD.0b013e31815e93c6.
- Cavaco I, Reis R, Gil JP, et al. CYP3A4*1B and NAT2*14 alleles in a native African population. Clin Chem Lab Med. 2003;41(4):606–609. DOI:https://doi.org/10.1515/CCLM.2003.091.
- Kudzi W, Dodoo AN, Mills JJ. Genetic polymorphisms in MDR1, CYP3A4 and CYP3A5 genes in a Ghanaian population: a plausible explanation for altered metabolism of ivermectin in humans? BMC Med Genet. 2010;11:111.
- Lamba JK, Lin YS, Schuetz EG, et al. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002;54:1271–1294.
- Pham YT, Régina A, Farinotti R, et al. Interactions of racemic mefloquine and its enantiomers with P-glycoprotein in an immortalised rat brain capillary endothelial cell line, GPNT. Biochim Biophys Acta. 2000;1524(2–3):212–219. DOI:https://doi.org/10.1016/S0304-4165(00)00160-4.
- Aarnoudse AL, van Schaik RH, Dieleman J, et al. MDR1 gene polymorphisms are associated with neuropsychiatric adverse effects of mefloquine. Clin Pharmacol Ther. 2006;80(4):367–374. DOI:https://doi.org/10.1016/j.clpt.2006.07.003.
- Wu CP, Klokouzas A, Hladky SB, et al. Interactions of mefloquine with ABC proteins, MRP1 (ABCC1) and MRP4 (ABCC4) that are present in human red cell membranes. Biochem Pharmacol. 2005;70(4):500–510. DOI:https://doi.org/10.1016/j.bcp.2005.05.022.
- Ritter CA, Jedlitschky G, Meyer Zu Schwabedissen H, et al. Cellular export of drugs and signaling molecules by the ATP-binding cassette transporters MRP4 (ABCC4) and MRP5 (ABCC5). Drug Metab Rev. 2005;37(1):253–278. DOI:https://doi.org/10.1081/DMR-200047984.
- Piedade R, Traub S, Bitter A, et al. Carboxymefloquine, the major metabolite of the antimalarial drug mefloquine, induces drug-metabolizing enzyme and transporter expression by activation of pregnane X receptor. Antimicrob Agents Chemother. 2015;59(1):96–104. DOI:https://doi.org/10.1128/AAC.04140-14.
- Piedade R, Schaeffeler E, Winter S, et al. PXR variants and artemisinin use in Vietnamese subjects: frequency distribution and impact on the interindividual variability of CYP3A induction by artemisinin. Antimicrob Agents Chemother. 2012;56(4):2153–2157. DOI:https://doi.org/10.1128/AAC.06009-11.
- Maganda BA, Minzi OM, Ngaimisi E, et al. CYP2B6*6 genotype and high efavirenz plasma concentration but not nevirapine are associated with low lumefantrine plasma exposure and poor treatment response in HIV-malaria-coinfected patients. Pharmacogenomics J. 2016;16(1):88–95. https://doi.org/10.1038/tpj.2015.37.
- Hamed K, Grueninger H. Coartem(®): a decade of patient-centric malaria management. Expert Rev Anti Infect Ther. 2012;10(6):645–659.
- Djimdé A, Lefèvre G. Understanding the pharmacokinetics of Coartem. Malar J. 2009;1(Suppl 1):S4. 8 Suppl. DOI:https://doi.org/10.1186/1475-2875-8-S1-S4.
- Borrmann S, Sallas WM, Machevo S, et al. The effect of food consumption on lumefantrine bioavailability in African children receiving artemether-lumefantrine crushed or dispersible tablets (Coartem) for acute uncomplicated Plasmodium falciparum malaria. Trop Med Int Health. 2010;15(4):434–441. DOI:https://doi.org/10.1111/j.1365-3156.2010.02477.x.
- Denis MB, Tsuyuoka R, Lim P, et al. Efficacy of artemether-lumefantrine for the treatment of uncomplicated falciparum malaria in northwest Cambodia. Trop Med Int Health. 2006;11(12):1800–1807. DOI:https://doi.org/10.1111/j.1365-3156.2006.01739.x.
- Rahman MM, Dondorp AM, Day NP, et al. Adherence and efficacy of supervised versus non-supervised treatment with artemether/lumefantrine for the treatment of uncomplicated Plasmodium falciparum malaria in Bangladesh: a randomised controlled trial. Trans R Soc Trop Med Hyg. 2008;102(9):861–867. DOI:https://doi.org/10.1016/j.trstmh.2008.05.022.
- Mutagonda RF, Kamuhabwa AA, Minzi OM, et al. Malaria prevalence, severity and treatment outcome in relation to day 7 lumefantrine plasma concentration in pregnant women. Malar J. 2016;15(1):278. DOI:https://doi.org/10.1186/s12936-016-1327-1.
- Vos K, Sciuto CL, Piedade R, et al. MRP2/ABCC2 C1515Y polymorphism modulates exposure to lumefantrine during artemether-lumefantrine antimalarial therapy. Pharmacogenomics. 2017;18(10):981–985. DOI:https://doi.org/10.2217/pgs-2017-0032.
- Noedl H, Allmendinger T, Prajakwong S, et al. Desbutyl-benflumetol, a novel antimalarial compound: in vitro activity in fresh isolates of Plasmodium falciparum from Thailand. Antimicrob Agents Chemother. 2001;45(7):2106–2109. DOI:https://doi.org/10.1128/AAC.45.7.2106-2109.2001.
- Salman S, Page-Sharp M, Griffin S, et al. Population pharmacokinetics of artemether, lumefantrine, and their respective metabolites in Papua New Guinean children with uncomplicated malaria. Antimicrob Agents Chemother. 2011;55(11):5306–5313. DOI:https://doi.org/10.1128/AAC.05136-11.
- McGready R, Stepniewska K, Lindegardh N, et al. The pharmacokinetics of artemether and lumefantrine in pregnant women with uncomplicated falciparum malaria. Eur J Clin Pharmacol. 2006;62:1021–1031.
- Starzengruber P, Wernsdorfer G, Parizek M, et al. Specific pharmacokinetic interaction between lumefantrine and monodesbutyl-benflumetol in Plasmodium falciparum. Wien Klin Wochenschr. 2007;119(19–20 Suppl 3):60–66. DOI:https://doi.org/10.1007/s00508-007-0861-9.
- Starzengruber P, Kollaritsch H, Sirichaisinthop J, et al. Interaction between lumefantrine and monodesbutyl-benflumetol in Plasmodium falciparum in vitro. Wien. Klin. Wochenschr. 2008;120(19–20 Suppl 4):85–89. DOI:https://doi.org/10.1007/s00508-008-1080-8.
- Wong RP, Salman S, Ilett KF, et al. Desbutyl-lumefantrine is a metabolite of lumefantrine with potent in vitro antimalarial activity that may influence artemether-lumefantrine treatment outcome. Antimicrob Agents Chemother. 2011;55(3):1194–1198.
- Mosha D, Guidi M, Mwingira F, et al. Population pharmacokinetics and clinical response for artemether-lumefantrine in pregnant and nonpregnant women with uncomplicated Plasmodium falciparum malaria in Tanzania. Antimicrob Agents Chemother. 2014;58(8):4583–4592. DOI:https://doi.org/10.1128/AAC.02595-14.
- Hansson HH Human Genetic Variation and their potential role in drug metabolism and susceptibility to malaria [ dissertation]. University of Copenhagen; 2015. https://scholar.google.com/citations?view_op=view_citation&hl=da&user=An7bNBIAAAAJ&citation_for_view=An7bNBIAAAAJ:UeHWp8X0CEIC (accessed, 2021 Nov, 2).
- Okubo M, Murayama N, Shimizu M, et al. CYP3A4 intron 6 C>T polymorphism (CYP3A4*22) is associated with reduced CYP3A4 protein level and function in human liver microsomes. J Toxicol Sci. 2013;38:349–354.
- Wang D, Guo Y, Wrighton SA, et al. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 2011;11:274–286.
- Hodoameda P, Duah-Quashie NO, Hagan CO, et al. Plasmodium falciparum genetic factors rather than host factors are likely to drive resistance to ACT in Ghana. Malar J. 2020;19(1):255. DOI:https://doi.org/10.1186/s12936-020-03320-7.
- Kiaco K, Rodrigues AS, Do Rosário V, et al. The drug transporter ABCB1 c.3435C>T SNP influences artemether-lumefantrine treatment outcome. Malar J. 2017;16(1):383. DOI:https://doi.org/10.1186/s12936-017-2006-6.
- Mutagonda RF, Minzi OMS, Massawe SN, et al. Pregnancy and CYP3A5 Genotype Affect Day 7 Plasma Lumefantrine Concentrations. Drug Metab Dispos. 2019;47(12):1415–1424. DOI:https://doi.org/10.1124/dmd.119.088062.
- Chandler B, Almond L, Ford J, et al. The effects of protease inhibitors and nonnucleoside reverse transcriptase inhibitors on p-glycoprotein expression in peripheral blood mononuclear cells in vitro. J Acquir Immune Defic Syndr. 2003;33(5):551–556. DOI:https://doi.org/10.1097/00126334-200308150-00001.
- Hariparsad N, Nallani SC, Sane RS, et al. Induction of CYP3A4 by efavirenz in primary human hepatocytes: comparison with rifampin and phenobarbital. J Clin Pharmacol. 2004;44:1273–1281.
- Sueyoshi T, Kawamoto T, Zelko I, et al. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J Biol Chem. 1999;274(10):6043–6046. DOI:https://doi.org/10.1074/jbc.274.10.6043.
- Meyer Zu Schwabedissen HE, Oswald S, Bresser C, et al. Compartment-specific gene regulation of the CAR inducer efavirenz in vivo. Clin Pharmacol Ther. 2012;92(1):103–111. DOI:https://doi.org/10.1038/clpt.2012.34.
- Byakika-Kibwika P, Lamorde M, Mayito J, et al. Significant pharmacokinetic interactions between artemether/lumefantrine and efavirenz or nevirapine in HIV-infected Ugandan adults. J Antimicrob Chemother. 2012;67(9):2213–2221. DOI:https://doi.org/10.1093/jac/dks207.
- Huang L, Parikh S, Rosenthal PJ, et al. Concomitant efavirenz reduces pharmacokinetic exposure to the antimalarial drug artemether-lumefantrine in healthy volunteers. J Acquir Immune Defic Syndr. 2012;61(3):310–616. DOI:https://doi.org/10.1097/QAI.0b013e31826ebb5c.
- Maganda BA, Ngaimisi E, Kamuhabwa AA, et al. The influence of nevirapine and efavirenz-based anti-retroviral therapy on the pharmacokinetics of lumefantrine and anti-malarial dose recommendation in HIV-malaria co-treatment. Malar J. 2015;14:179.
- Maganda BA, Minzi OM, Kamuhabwa AA, et al. Outcome of artemether-lumefantrine treatment for uncomplicated malaria in HIV-infected adult patients on anti-retroviral therapy. Malar J. 2014;13:205.
- Parikh S, Kajubi R, Huang L, et al. Antiretroviral Choice for HIV Impacts Antimalarial Exposure and Treatment Outcomes in Ugandan Children. Clin Infect Dis. 2016;63(3):414–422. DOI:https://doi.org/10.1093/cid/ciw291.
- Ngaimisi E, Habtewold A, Minzi O, et al. Importance of ethnicity, CYP2B6 and ABCB1 genotype for efavirenz pharmacokinetics and treatment outcomes: a parallel-group prospective cohort study in two sub-Saharan Africa populations. PLoS One. 2013;8(7):e67946. DOI:https://doi.org/10.1371/journal.pone.0067946.
- Zakaria Z, Badhan RKS. The impact of CYP2B6 polymorphisms on the interactions of efavirenz with lumefantrine: implications for paediatric antimalarial therapy. Eur J Pharm Sci. 2018;119:90–101.
- Huang H, Wang H, Sinz M, et al. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26(2):258–268. DOI:https://doi.org/10.1038/sj.onc.1209788.
- Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of P-glycoprotein and cytochrome P4503A coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49(2):311–318.
- Schuetz EG, Schinkel AH, Relling MV, et al. P-glycoprotein: a major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc Natl Acad Sci. 1996;93(9):4001–4005. DOI:https://doi.org/10.1073/pnas.93.9.4001.
- Dresser GK, SchwarzU.I, Wilkinson G.R, et al. Coordinate induction of both cytochrome P4503A and MDR1 by St John’s wort in healthy subjects. Clin Pharmacol Ther. 2003;73(1):41–50. DOI:https://doi.org/10.1067/mcp.2003.10.
- Elens L, Tyteca D, Panin N, et al. Functional defect caused by the 4544G>A SNP in ABCC2: potential impact for drug cellular disposition. Pharmacogenet Genomics. 2011;21(12):884–893. DOI:https://doi.org/10.1097/FPC.0b013e32834d672b.
- Burckhalter JH, Tendick FH, Jones EM, et al. Aminoalkylphenols as antimalarials (heterocyclicamino)-alpha-amino-o-cresols; the synthesis of camoquin. J Am Chem Soc. 1948;70(4):1363–1373. DOI:https://doi.org/10.1021/ja01184a023.
- Mein RM. Camoquin in the treatment of human malaria. Am J Trop Med Hyg. 1951;31:212–217.
- Hatton CSR, Bunch C, Peto TEA, et al. Frequency of severe neutropenia associated with amodiaquine prophylaxis against malaria. Lancet. 1986;327(8478):411–414. DOI:https://doi.org/10.1016/S0140-6736(86)92371-8.
- Neftel KA, Frick PG, Feh J, et al. Amodiaquine induced agranulocytosis and liver damage. Br Med J (Clin Res Ed). 1986;292(6522):721–723. DOI:https://doi.org/10.1136/bmj.292.6522.721.
- Olliaro P, Nevill C, LeBras J, et al. Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet. 1996;348(9036):1196–1201. DOI:https://doi.org/10.1016/S0140-6736(96)06217-4.
- Phillips-Howard PA, Bjorkman AB. Ascertainment of risk of serious adverse reactions associated with chemoprophylactic antimalarial drugs. Bull World Health Organ. 1990;68(4):493–504.
- World Health Organization. Management of uncomplicated malaria and the use of antimalarial drugs for the protection of travellers: report of an informal consultation. Geneva: World Health Organization; 1995. (no. 18–21).
- Olliaro P, Mussano P. Amodiaquine for treating malaria. Cochrane Database Syst Rev. 2003 2. CD000016. DOI:https://doi.org/10.1002/14651858.CD000016
- Sinclair D, Zani B, Donegan S, et al. Artemisinin-based combination therapy for treating uncomplicated malaria. Cochrane Database Syst Rev. 2009 3:CD007483. DOI:https://doi.org/10.1002/14651858.CD007483.pub2
- Ashley EA, Poespoprodjo JR. Treatment and prevention of malaria in children. Lancet Child Adolesc Health. 2020;4(10):775–789.
- Cairns M, Roca-Feltrer A, Garske T, et al. Estimating the potential public health impact of seasonal malaria chemoprevention in African children. Nat Commun. 2012;3:881.
- Tse EG, Korsik M, Todd MH. The past, present and future of anti-malarial medicines. Malar J. 2019;18(1):93. DOI:https://doi.org/10.1186/s12936-019-2724-z.
- Li XQ, Björkman A, Andersson TB, et al. Amodiaquine clearance and its metabolism to N-desethylamodiaquine is mediated by CYP2C8: a new high affinity and turnover enzyme-specific probe substrate. J Pharmacol Exp Ther. 2002;300(2):399–407. DOI:https://doi.org/10.1124/jpet.300.2.399.
- Laurent F, Saivin S, Chretien P, et al. Pharmacokinetic and pharmacodynamic study of amodiaquine and its two metabolites after a single oral dose in human volunteers. Arzneimittelforschung. 1993;43:612–616.
- Scarsi KK, Fehintola FA, Ma Q, et al. Disposition of amodiaquine and desethylamodiaquine in HIV-infected Nigerian subjects on nevirapine-containing antiretroviral therapy. J Antimicrob Chemother. 2014;69:1370–1376.
- Churchill FC, Patchen LC, Campbell CC, et al. Amodiaquine as a prodrug: importance of metabolite(s) in the antimalarial effect of amodiaquine in humans. Life Sci. 1985;36(1):53–62. DOI:https://doi.org/10.1016/0024-3205(85)90285-1.
- Mariga ST, Gil JP, Sisowath C, et al. Synergism between amodiaquine and its major metabolite, desethylamodiaquine, against Plasmodium falciparum in vitro. Antimicrob Agents Chemother. 2004;48(11):4089–4096. DOI:https://doi.org/10.1128/AAC.48.11.4089-4096.2004.
- Anyorigiya TA, Castel S, Mauff K, et al. Pharmacokinetic profile of amodiaquine and its active metabolite desethylamodiaquine in Ghanaian patients with uncomplicated falciparum malaria. Malar J. 2021;20(1):18. DOI:https://doi.org/10.1186/s12936-020-03553-6.
- Pussard E, Verdier F, Faurisson F, et al. Disposition of monodesethylamodiaquine after a single oral dose of amodiaquine and three regimens for prophylaxis againstPlasmodium falciparum malaria. Eur J Clin Pharmacol. 1987;33(4):409–414. DOI:https://doi.org/10.1007/BF00637639.
- Mount DL, Patchen LC, Nguyen-Dinh P, et al. Sensitive analysis of blood for amodiaquine and three metabolites by high-performance liquid chromatography with electrochemical detection. J Chromatogr. 1986;383(4):409–414. DOI:https://doi.org/10.1016/S0378-4347(00)83483-0.
- Johansson T, Jurva U, Grönberg G, et al. Novel metabolites of amodiaquine formed by CYP1A1 and CYP1B1: structure elucidation using electrochemistry, mass spectrometry, and NMR. Drug Metab Dispos. 2009;37(3):571–579. DOI:https://doi.org/10.1124/dmd.108.025171.
- Churchill FC, Mount DL, Patchen LC, et al. Isolation, characterization and standardization of a major metabolite of amodiaquine by chromatographic and spectroscopic methods. J Chromatogr. 1986;377:307–318.
- PharmVar CYP2C8 [Internet]. Pharmacogene Variation Consortium; [cited 2021 Jun 08]. Available from: https://www.pharmvar.org/gene/CYP2C8
- Lai XS, Yang LP, Li XT, et al. Human CYP2C8: structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr Drug Metab. 2009;10(9):1009–1047. DOI:https://doi.org/10.2174/138920009790711832.
- Baker JL, Shriner D, Bentley AR, et al. Pharmacogenomic implications of the evolutionary history of infectious diseases in Africa. Pharmacogenomics. 2017;17(2):112–120. DOI:https://doi.org/10.1038/tpj.2016.78.
- Cavaco I, Strömberg-Nörklit J, Kaneko A, et al., CYP2C8 polymorphism frequencies among malaria patients in Zanzibar. Eur J Clin Pharmacol. 61(1): 15–18. 2005. . DOI:https://doi.org/10.1007/s00228-004-0871-8.
- Habtemikael L, Russom M, Bahta I, et al. Prevalence of CYP2C8*2 and *3 among eritreans and its potential impact on artesunate/amodiaquine treatment. Pharmgenomics Pers Med. 2020;13:571–575.
- Parikh S, Ouedraogo JB, Goldstein JA, et al., Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther. 82(2): 197–203. 2007. . DOI:https://doi.org/10.1038/sj.clpt.6100122.
- Zongo I, Dorsey G, Rouamba N, et al. Amodiaquine, sulfadoxine-pyrimethamine, and combination therapy for uncomplicated falciparum malaria: a randomized controlled trial from Burkina Faso. Am J Trop Med Hyg. 2005;73(5):826–832. DOI:https://doi.org/10.4269/ajtmh.2005.73.826.
- Paganotti GM, Gallo BC, Verra F, et al. Human genetic variation is associated with Plasmodium falciparum drug resistance. J Infect Dis. 2011;204(11):1772–1778. DOI:https://doi.org/10.1093/infdis/jir629.
- Cavaco I, Mårtensson A, Fröberg G, et al. CYP2C8 status of patients with malaria influences selection of Plasmodium falciparum pfmdr1 alleles after amodiaquine-artesunate treatment. J Infect Dis. 2013;207(4):687–688. DOI:https://doi.org/10.1093/infdis/jis736.
- Paganotti GM. Reply to Cavaco et al. J Infect Dis. 2013;207(4):688–689.
- Harrison AC, Kitteringham NR, Clarke JB, et al. The mechanism of bioactivation and antigen formation of amodiaquine in the rat. Biochem Pharmacol. 1992;43(7):1421–1430. DOI:https://doi.org/10.1016/0006-2952(92)90198-R.
- Tingle MD, Jewell H, Maggs JL, et al. The bioactivation of amodiaquine by human polymorphonuclear leucocytes in vitro: chemical mechanisms and the effects of fluorine substitution. Biochem Pharmacol. 1994;50:1113–1119.
- Jewell H, Maggs JL, Harrison AC, et al. Role of hepatic metabolism in the bioactivation and detoxication of amodiaquine. Xenobiotica. 1995;25(2):199–217. DOI:https://doi.org/10.3109/00498259509061845.
- Zhang Y, Vermeulen NP, Commandeur JN. Characterization of human cytochrome P450 mediated bioactivation of amodiaquine and its major metabolite N-desethylamodiaquine. Br J Clin Pharmacol. 2017;83(3):572–583.
- Uetrecht JP, Rice-Evans C, Halliwell B. Myeloperoxidase as a generator of drug free radicals. Biochem Soc Symp. 1995;61:163–170.
- Shimizu S, Atsumi R, Itokawa K, et al. Metabolism-dependent hepatotoxicity of amodiaquine in glutathione-depleted mice. Arch Toxicol. 2009;83(7):701–707. DOI:https://doi.org/10.1007/s00204-009-0436-9.
- Tafazoli S, O’Brien PJ. Amodiaquine-induced oxidative stress in a hepatocyte inflammation model. Toxicology. 2009;256(1–2):101–109.
- Zhang Y, den Braver-sewradj SP, Vos JC, et al. Human glutathione S-transferases- and NAD(P)H:quinone oxidoreductase 1-catalyzed inactivation of reactive quinoneimines of amodiaquine and N-desethylamodiaquine: possible implications for susceptibility to amodiaquine-induced liver toxicity. Toxicol Lett. 2017;275:83–91.
- Ginsberg G, Guyton K, Johns D, et al. Genetic polymorphism in metabolism and host defense enzymes: implications for human health risk assessment. Crit Rev Toxicol. 2010;40(7):575–619. DOI:https://doi.org/10.3109/10408441003742895.
- Dhakshinamoorthy S, Long DJ 2nd, Jaiswal AK. Antioxidant regulation of genes encoding enzymes that detoxify xenobiotics and carcinogens. Curr Top Cell Regul. 2000;36:201–216.
- Homolya L, Váradi A, Sarkadi B. Multidrug resistance-associated proteins: export pumps for conjugates with glutathione, glucuronate or sulfate. Biofactors. 2003;17(1–4):103–114.
- Wennerholm A, Nordmark A, Pihlsgård M, et al. Amodiaquine, its desethylated metabolite, or both, inhibit the metabolism of debrisoquine (CYP2D6) and losartan (CYP2C9) in vivo. Eur J Clin Pharmacol. 2006;62(7):539–546. DOI:https://doi.org/10.1007/s00228-006-0121-3.
- Walsky RL, Gaman EA, Obach RS. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J Clin Pharmacol. 2005;45(1):68–78.
- Hombhanje FW, Hwaihwanje I, Tsukahara T, et al. The disposition of oral amodiaquine in Papua New Guinean children with falciparum malaria. Br J Clin Pharmacol. 2005;59(3):298–301. DOI:https://doi.org/10.1111/j.1365-2125.2004.02257.x.
- Chen C. Development of antimalarial drugs and their application in China: a historical review. Infect Dis Poverty. 2014;3(1):9.
- Davis TM, Hung TY, Sim IK, et al. Piperaquine: a resurgent antimalarial drug. Drugs. 2005;65(1):75–87. DOI:https://doi.org/10.2165/00003495-200565010-00004.
- Arya A, Kojom Foko LP, Chaudhry S, et al. Artemisinin-based combination therapy (ACT) and drug resistance molecular markers: a systematic review of clinical studies from two malaria endemic regions - India and sub-Saharan Africa. Int J Parasitol Drugs Drug Resist. 2021;15:43–56.
- Tarning J, Lindegårdh N, Annerberg A, et al. Pitfalls in estimating piperaquine elimination. Antimicrob Agents Chemother. 2005;49(12):5127–5128. DOI:https://doi.org/10.1128/AAC.49.12.5127-5128.2005.
- Lee TM, Huang L, Johnson MK, et al. In vitro metabolism of piperaquine is primarily mediated by CYP3A4. Xenobiotica. 2012;42(11):1088–1095. DOI:https://doi.org/10.3109/00498254.2012.693972.
- Bailly C. Pyronaridine: an update of its pharmacological activities and mechanisms of action. Biopolymers. 2021;112(4):e23398.
- Morris CA, Dueker SR, Lohstroh PN, et al. Mass balance and metabolism of the antimalarial pyronaridine in healthy volunteers. Eur J Drug Metab Pharmacokinet. 2015;40(1):75–86. DOI:https://doi.org/10.1007/s13318-014-0182-0.
- Croft SL, Duparc S, Arbe-Barnes SJ, et al. Review of pyronaridine anti-malarial properties and product characteristics. Malar J. 2012;11:270.
- Morris CA, Pokorny R, Lopez-Lazaro L, et al. Pharmacokinetic interaction between pyronaridine-artesunate and metoprolol. Antimicrob Agents Chemother. 2014;58(10):5900–5908. DOI:https://doi.org/10.1128/AAC.02716-14.
- Qi J, Wang S, Liu G, et al. Pyronaridine, a novel modulator of P-glycoprotein-mediated multidrug resistance in tumor cells in vitro and in vivo. Biochem Biophys Res Commun. 2004;319(4):1124–1131. DOI:https://doi.org/10.1016/j.bbrc.2004.05.099.
- Crowe A, Ilett KF, Karunajeewa HA, et al. Role of P glycoprotein in absorption of novel antimalarial drugs. Antimicrob Agents Chemother. 2006;50(10):3504–3506. DOI:https://doi.org/10.1128/AAC.00708-06.
- Targett G, Drakeley C, Jawara M, et al. Artesunate reduces but does not prevent posttreatment transmission of Plasmodium falciparum to Anopheles gambiae. J Infect Dis. 2001;183(8):1254–1259. DOI:https://doi.org/10.1086/319689.
- Bousema JT, Schneider P, Gouagna LC, et al. Moderate effect of artemisinin-based combination therapy on transmission of Plasmodium falciparum. J Infect Dis. 2006;193(8):1151–1159. DOI:https://doi.org/10.1086/503051.
- World Health Organization. Malaria Policy Advisory Committee and Secretariat. Malaria policy advisory committee to the WHO: conclusions and recommendations of September 2012 meeting. Malar J. 2012;11:424.
- Graves PM, Choi L, Gelband H, et al. Primaquine or other 8-aminoquinolines for reducing Plasmodium falciparum transmission. Cochrane Database Syst Rev. 2018;2(2):CD008152. DOI:https://doi.org/10.1002/14651858.CD008152.pub5.
- Baird JK. 8-Aminoquinoline therapy for latent malaria. Clin Microbiol Rev. 2019;32(4):e00011–19.
- Vásquez-Vivar J, Augusto O. Hydroxylated metabolites of the antimalarial drug primaquine. Oxidation and Redox Cycling. J Biol Chem. 1992;267(10):6848–6854.
- Pybus BS, Marcsisin SR, Jin X, et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar J. 2013;12:212.
- Vásquez-Vivar J, Augusto O. Oxidative activity of primaquine metabolites on rat erythrocytes in vitro and in vivo. Biochem Pharmacol. 1994;47(2):309–316.
- Camarda G, Jirawatcharadech P, Priestley RS, et al. Antimalarial activity of primaquine operates via a two-step biochemical relay. Nat Commun. 2019;10(1):3226. DOI:https://doi.org/10.1038/s41467-019-11239-0.
- Pybus BS, Sousa JC, Jin X, et al. CYP450 phenotyping and accurate mass identification of metabolites of the 8-aminoquinoline, anti-malarial drug primaquine. Malar J. 2012;11:259.
- Popovici J, Tebben K, Witkowski B, et al. Primaquine for Plasmodium vivax radical cure: what we do not know and why it matters. Int J Parasitol Drugs Drug Resist. 2021;15:36–42.
- Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005;5(1):6–13.
- Zanger UM, Raimundo S, Eichelbaum M. Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:23–37.
- Zanger UM, Turpeinen M, Klein K, et al. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008;392:1093–1108.
- Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–141.
- PharmVar CYP2D6 [Internet]. Pharmacogene Variation Consortium; [cited 2021 Jun 08]. Available from: https://www.pharmvar.org/gene/CYP2D6
- Gaedigk A, Sangkuhl K, Whirl-Carrillo M, et al. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19:69–76.
- Gaedigk A, Simon SD, Pearce RE, et al. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83:234–242.
- Gaedigk A, Dinh JC, Jeong H, et al. Ten years’ experience with the cyp2d6 activity score: a perspective on future investigations to improve clinical predictions for precision therapeutics. J Pers Med. 2018;8(2):15. DOI:https://doi.org/10.3390/jpm8020015.
- Hicks JK, Swen JJ, Gaedigk A. Challenges in CYP2D6 phenotype assignment from genotype data: a critical assessment and call for standardization. Curr Drug Metab. 2014;15:218–232.
- Bennett JW, Pybus BS, Yadava A, et al. Primaquine failure and cytochrome P-450 2D6 in Plasmodium vivax malaria. N Engl J Med. 2013;369:1381–1382. DOI:https://doi.org/10.1056/NEJMc1301936
- Ferreira MU, Nobrega de Sousa T, Rangel GW, et al. Monitoring Plasmodium vivax resistance to antimalarials: persisting challenges and future directions. Int J Parasitol Drugs Drug Resist. 2021;15:9–24.
- Brasil LW, Rodrigues-Soares F, Santoro AB, et al. CYP2D6 activity and the risk of recurrence of Plasmodium vivax malaria in the Brazilian Amazon: a prospective cohort study. Malar J. 2018;17:57.
- Baird JK, Louisa M, Noviyanti R, et al. Association of impaired cytochrome p450 2d6 activity genotype and phenotype with therapeutic efficacy of primaquine treatment for latent Plasmodium vivax Malaria. JAMA Network Open. 2018;1(4):e181449. DOI:https://doi.org/10.1001/jamanetworkopen.2018.1449.
- Silvino ACR, Kano FS, Costa MA, et al. Novel insights into plasmodium vivax therapeutic failure: cyp2d6 activity and time of exposure to malaria modulate the risk of recurrence. Antimicrob Agents Chemother. 2020;64(5):e02056–19. DOI:https://doi.org/10.1128/AAC.02056-19.
- Spring MD, Sousa JC, Li Q, et al. Determination of Cytochrome P450 isoenzyme 2D6 (CYP2D6) genotypes and pharmacogenomic impact on primaquine metabolism in an active-duty US military population. J Infect Dis. 2019;220:1761–1770.
- Potter BM, Xie LH, Vuong C, et al. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob Agents Chemother. 2015;59(4):2380–2387. DOI:https://doi.org/10.1128/AAC.00015-15.
- Pett H, Bradley J, Okebe J, et al. CYP2D6 polymorphisms and the safety and gametocytocidal activity of single-dose primaquine for Plasmodium falciparum. Antimicrob Agents Chemother. 2019;63(10):e00538–19. DOI:https://doi.org/10.1128/AAC.00538-19.
- Clarke G, Rockett K, Kivinen K, et al. Characterisation of the opposing effects of G6PD deficiency on cerebral malaria and severe malarial anaemia. Elife. 2017;6:e15085.
- World Health Organization. Testing for G6PD deficiency for safe use of primaquine in radical cure of P. vivax and P. ovale malaria. Geneva: World Health Organization; 2016.
- Bancone G, Chowwiwat N, Somsakchaicharoen R, et al. Single low dose primaquine (0.25 mg/kg) does not cause clinically significant haemolysis in g6pd deficient subjects. PLoS One. 2016;11(3):e0151898. DOI:https://doi.org/10.1371/journal.pone.0151898.
- Bastiaens GJH, Tiono AB, Okebe J, et al. Safety of single low-dose primaquine in glucose-6-phosphate dehydrogenase deficient falciparum-infected African males: two open-label, randomized, safety trials. PLoS One. 2018;13(1):e0190272. DOI:https://doi.org/10.1371/journal.pone.0190272.
- St Jean PL, Xue Z, Carter N, et al. Tafenoquine treatment of Plasmodium vivax malaria: suggestive evidence that CYP2D6 reduced metabolism is not associated with relapse in the Phase 2b DETECTIVE trial. Malar J. 2016;15:97.
- Lacerda MVG, Llanos-Cuentas A, Krudsood S, et al. Single-dose tafenoquine to prevent relapse of plasmodium vivax malaria. N Engl J Med. 2019;380(3):215–228. DOI:https://doi.org/10.1056/NEJMoa1710775.
- Vuong C, Xie LH, Potter BM, et al. Differential cytochrome P450 2D metabolism alters tafenoquine pharmacokinetics. Antimicrob Agents Chemother. 2015;59(7):3864–3869.
- Marcsisin SR, Sousa JC, Reichard GA, et al. Tafenoquine and NPC-1161B require CYP 2D metabolism for anti-malarial activity: implications for the 8-aminoquinoline class of anti-malarial compounds. Malar J. 2014;13:2.
- Milner EE, Berman J, Caridha D, et al. Cytochrome P450 2D-mediated metabolism is not necessary for tafenoquine and primaquine to eradicate the erythrocytic stages of Plasmodium berghei. Malar J. 2016;15(1):588. DOI:https://doi.org/10.1186/s12936-016-1632-8.
- Kemirembe K, Cabrera M, Cui L. Interactions between tafenoquine and artemisinin-combination therapy partner drug in asexual and sexual stage Plasmodium falciparum. Int J Parasitol Drugs Drug Resist. 2017;7(2):131–137.
- Wu T, Nagle A, Kuhen K, et al. Imidazolopiperazines: hit to lead optimization of new antimalarial agents. J Med Chem. 2011;54:5116–5130.
- Kuhen KL, Chatterjee AK, Rottmann M, et al. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58(9):5060–5067. DOI:https://doi.org/10.1128/AAC.02727-13.
- White NJ, Duong TT, Uthaisin C, et al. Antimalarial activity of KAF156 in falciparum and vivax malaria. N Engl J Med. 2016;375:1152–1160.
- Kublin JG, Murphy SC, Maenza J, et al. Safety, pharmacokinetics and causal prophylactic efficacy of KAF156 in a Plasmodium falciparum human infection study. Clin Infect Dis. 2021;73(7):e2407–e2414.
- Leong FJ, Jain JP, Feng Y, et al. A phase 1 evaluation of the pharmacokinetic/pharmacodynamic interaction of the anti-malarial agents KAF156 and piperaquine. Malar J. 2018;17(1):7. DOI:https://doi.org/10.1186/s12936-017-2162-8.
- Abdin M, Israr M, Rehman R, et al. Artemisinin, a novel antimalarial drug: biochemical and molecular approaches for enhanced production. Planta Med. 2003;69:289–299.
- Vennerstrom JL, Arbe-Barnes S, Brun R, et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430(7002):900–904. DOI:https://doi.org/10.1038/nature02779.
- Valecha N, Looareesuwan S, Martensson A, et al. Arterolane, a new synthetic tri- oxolane for treatment of uncomplicated Plasmodium falciparum malaria: a phase II, multicenter, randomized, dose-finding clinical trial. Clin Infect Dis. 2010;51:684–691.
- Toure OA, Rulisa S, Anvikar AR, et al. Efficacy and safety of fixed dose com- bination of arterolane maleate and piperaquine phosphate dispersible tablets in paediatric patients with acute uncomplicated Plasmodium falciparum malaria: a phase II, multicentric, open-label study. Malar J. 2015;14:469.
- Toure OA, Valecha N, Tshefu AK, et al. A phase III,double-blind, double dummy,randomized (2:1), multicenter trial comparing the safety and efficacy of FDC tablets of arterolane maleate and piperaquine phosphate (PQP) with Coartem (artemether-lumefantrine tablets) in patients with acute uncomplicated Plasmodium falciparum malaria. Clin Infect Dis. 2016;62:964–971.
- Gautam A, Ahmed T, Sharma P, et al. Pharmacokinetics and pharmacodynamics of arterolane maleate following multiple oral doses in adult patients with P. falciparum malaria. J Clin Pharmacol. 2011;51(11):1519–1528. DOI:https://doi.org/10.1177/0091270010385578.
- Zhou L, Alker A, Ruf A, et al. Characterization of the two major CYP450 metabolites of ozonide (1,2,4-trioxolane) OZ277. Bioorg Med Chem Lett. 2008;18(5):1555–1558. DOI:https://doi.org/10.1016/j.bmcl.2008.01.087.
- Charman SA, Arbe-Barnes S, Bathurst IC, et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci. 2011;108(11):4400–4405. DOI:https://doi.org/10.1073/pnas.1015762108.
- Phyo AP, Jittamala P, Nosten FH, et al. Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: an open-label phase 2 trial. Lancet Infect Dis. 2016;16(1):61–69. DOI:https://doi.org/10.1016/S1473-3099(15)00320-5.
- Daher W, Pelinski L, Klieber S, et al. In vitro metabolism of ferroquine (SSR97193) in animal and human hepatic models and antimalarial activity of major metabolites on Plasmodium falciparum. Drug Metab Dispos. 2006;34(4):667–682. DOI:https://doi.org/10.1124/dmd.104.003202.
- Supan C, Mombo-Ngoma G, Dal-Bianco MP, et al. Pharmacokinetics of ferroquine, a novel 4-aminoquinoline, in asymptomatic carriers of Plasmodium falciparum infections. Antimicrob Agents Chemother. 2012;56(6):3165–3173. DOI:https://doi.org/10.1128/AAC.05359-11.
- Fernandes JF, Lell B, Agnandji ST, et al. Fosmidomycin as an antimalarial drug: a meta-analysis of clinical trials. Future Microbiol. 2015;10(8):1375–1390. DOI:https://doi.org/10.2217/FMB.15.60.
- Na-Bangchang K, Ruengweerayut R, Karbwang J, et al. Pharmacokinetics and pharmacodynamics of fosmidomycin monotherapy and combination therapy with clindamycin in the treatment of multidrug resistant falciparum malaria. Malar J. 2007;6:70.
- Mombo-Ngoma G, Remppis J, Sievers M, et al. Efficacy and safety of fosmidomycin-piperaquine as nonartemisinin-based combination therapy for uncomplicated falciparum malaria: a single-arm, age de-escalation proof-of-concept study in gabon. Clin Infect Dis. 2018;66(12):1823–1830. DOI:https://doi.org/10.1093/cid/cix1122.
- Ruangweerayut R, Looareesuwan S, Hutchinson D, et al. Assessment of the pharmacokinetics and dynamics of two combination regimens of fosmidomycin-clindamycin in patients with acute uncomplicated falciparum malaria. Malar J. 2008;7:225.
- Morrissey KM, Stocker SL, Wittwer MB, et al. Renal transporters in drug development. Annu Rev Pharmacol Toxicol. 2013;53:503–529.
- Guttmann P, Ehrlich P. Ueber die Wirkung des Methylenblau bei Malaria [Effect of methylene blue in malaria]. Vol. 28. German: Berlin Klin Wochenschr; 1891. p. 953–956.
- Couto M. Les injections endo-veineuses du bleu de méthylène dans le paludisme [Endovenous injections of methylene blue in malaria]. Bull Soc Pathol Exot. 1908;1:292–295. French.
- Dicko A, Roh ME, Diawara H, et al. Efficacy and safety of primaquine and methylene blue for prevention of Plasmodium falciparum transmission in Mali: a phase 2, single-blind, randomised controlled trial. Lancet Infect Dis. 2018;18(6):627–639. DOI:https://doi.org/10.1016/S1473-3099(18)30044-6.
- Coulibaly B, Pritsch M, Bountogo M, et al. Efficacy and safety of triple combination therapy with artesunate-amodiaquine-methylene blue for falciparum malaria in children: a randomized controlled trial in Burkina Faso. J Infect Dis. 2015;211(5):689–697. DOI:https://doi.org/10.1093/infdis/jiu540.
- Anh CX, Chavchich M, Birrell GW, et al. Pharmacokinetics and ex vivo antimalarial activity of artesunate-amodiaquine plus methylene blue in healthy volunteers. Antimicrob Agents Chemother. 2020;64(3):e01441–19. DOI:https://doi.org/10.1128/AAC.01441-19.
- Highlights of prescribing information: PROVAYBLUETM [Internet]. Washintong (DC): U.S. Food and Drug Administration; [cited 2021 Jun 08]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/204630s000lbl.pdf
- Bebawy M, Morris MB, Roufogalis BD. A continuous fluorescence assay for the study of P-glycoprotein-mediated drug efflux using inside-out membrane vesicles. Anal Biochem. 1999;268(2):270–277.
- Senarathna SM, Page-Sharp M, Crowe A. The interactions of p-glycoprotein with antimalarial drugs, including substrate affinity, inhibition and regulation. PLoS One. 2016;11(4):e0152677.
- McDonagh EM, Bautista JM, Youngster I, et al. PharmGKB summary: methylene blue pathway. Pharmacogenet Genomics. 2013;23(9):498–508. DOI:https://doi.org/10.1097/FPC.0b013e32836498f4.
- Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf. 2010;33(9):713–726. DOI:https://doi.org/10.2165/11536520-000000000-00000.
- Müller O, Meissner P, Mansmann U. Glucose-6-phosphate dehydrogenase deficiency and safety of methylene blue. Drug Saf. 2012;35(1):85.
- Müller O, Mockenhaupt FP, Marks B, et al. Haemolysis risk in methylene blue treatment of G6PD-sufficient and G6PD-deficient West- African children with uncomplicated falciparum malaria: a synopsis of four RCTs. Pharmacoepidem Dr S. 2013;22(4):376–385. DOI:https://doi.org/10.1002/pds.3370.
- Ingelman-Sundberg M. Pharmacogenomic biomarkers for prediction of severe adverse drug reactions. N Engl J Med. 2008;358(6):637–639.
- Roh ME, Oyet C, Orikiriza P, et al. Screening for glucose-6-phosphate dehydrogenase deficiency using three detection methods: a cross-sectional survey in Southwestern Uganda. Am J Trop Med Hyg. 2016;95(5):1094–1099. DOI:https://doi.org/10.4269/ajtmh.16-0552.
- Pallerla SR, Elion Assiana DO, Linh LTK, et al. Pharmacogenetic considerations in the treatment of co-infections with HIV/AIDS, tuberculosis and malaria in Congolese populations of Central Africa. Int J Infect Dis. 2021;104:207–213.
- Thorlund K, Haggstrom J, Park JJ, et al. Key design considerations for adaptive clinical trials: a primer for clinicians. BMJ. 2018;360:k698.
- Peñas-lledó E, Terán E, Sosa-Macías M, et al. Challenges and opportunities for clinical pharmacogenetic research studies in resource-limited settings: conclusions from the council for international organizations of medical sciences-ibero-American network of pharmacogenetics and pharmacogenomics meeting. Clin Ther. 42(8): 1595–1610.e5. 2020. . DOI:https://doi.org/10.1016/j.clinthera.2020.06.008.