
ABSTRACT
Introduction
The therapeutic scenario of metastatic hormone-sensitive prostate cancer (mHSPC) has dramatically changed in recent years, with the approval of new-generation Androgen Receptor Signaling Inhibitors (ARSIs), in combination with the androgen deprivation therapy (ADT), which was the previous standard of care. Despite showing a similar clinical efficacy, ARSIs, all of which are administered orally, are different in terms of pharmacokinetic and drug-drug interactions (DDIs).
Areas covered
This review covers the main pharmacokinetic characteristics of ARSIs that have been approved for the first-line therapy of mHSPC patients, underlying the differences among these molecules and focusing on the known or possible interactions with other drugs. Full-text articles and abstracts were searched in PubMed.
Expert opinion
Since prostate cancer occurs mainly in older age, comorbidities and the consequent polypharmacy increase the DDI risk in mHSPC patients who are candidates for ARSI. Waiting for new therapeutic options, in the absence of direct comparisons, pharmacokinetic knowledge is essential to guide clinicians in prescribing ARSI in this setting.
1. Introduction
Prostate cancer (PC) is the most frequent type of cancer in the male sex; despite being diagnosed in localized stages in the majority of cases, metastatic disease – de-novo or recurrent – is responsible for a high number of deaths worldwide [Citation1]. Metastatic hormone-sensitive PC (mHSPC) is defined as the presence of metastasis from PC in patients who are naïve to hormonal treatment, except for patients who already received hormonal therapies for localized disease, but whose disease did not recur during the androgen deprivation therapy (ADT) [Citation2].
ADT consists of administering gonadotropin-releasing hormone (GnRH) analogues, suppressing testosterone synthesis, and ultimately inhibiting the testosterone-dependent growth and survival of PC cells [Citation3]. Single-agent GnRH agonists/antagonists had been the standard of care for mHSPC for many years. Despite the initial efficacy of ADT in many patients, the onset of resistance is almost unavoidable, which is why PC progresses and becomes castration-resistant [Citation4].
Androgen receptor (AR) is a ligand-dependent nuclear transcription factor, belonging to the steroid hormone nuclear receptor family [Citation5]; it is activated by dihydrotestosterone (DHT), the active form of testosterone, and in normal prostate gland cells is responsible for the production of secretory proteins such as prostate-specific antigen (PSA) [Citation6]. AR mediates the growth of prostate cancer cells, and its changes – in terms of aberrations or mutations – are frequent during cancer progression, becoming relevant in the castration-resistant phase [Citation7].
To potentiate the effects of ADT and to avoid/delay the onset of resistance, in the last decades, many drugs have been tested – in association with ADT – in the mHSPC setting [Citation2]. Docetaxel, which is a chemotherapy belonging to the taxane family, has demonstrated to improve outcomes in mHSPC patients when added to ADT in 3 randomized phase III trials [Citation8–10]; however, docetaxel seems to be more effective in case of high burden of disease, and its toxicity profile limits the use to younger and fitter patients [Citation2].
Androgen Receptor Signaling Inhibitors (ARSIs) is a term referring to new-generations molecules that block the AR pathway at different levels: through the inhibition of androgen synthesis by blocking cytochrome P450 C17 (CYP17) enzyme – abiraterone acetate (AA) – or through the direct antagonism of AR – apalutamide, enzalutamide, and darolutamide [Citation11].
Both AA and enzalutamide were firstly tested, in addition to ADT, in metastatic castration-resistant PC (mCRPC) patients who had already received chemotherapy: they both demonstrated to prolong survival over placebo in two phase III trials [Citation12,Citation13]. Later, their efficacy was shown also in chemo-naïve mCRPC patients [Citation14,Citation15]. Furthermore, enzalutamide, apalutamide, and darolutamide are able to prolong metastases-free survival (MFS) in patients with non-metastatic castration-resistant PC (nmCPRC) [Citation16].
In this review, the pharmacokinetic of these molecules and their clinical impact in mHSPC patients are discussed. Our goal is to take the stock of the current scenario, schematizing the pharmacological aspects, and focusing on useful notions for prescriber clinicians.
2. Kinetics of ARSIs
2.1. Abiraterone acetate (AA)
AA is a steroidal antiandrogen pro-drug rapidly converted to abiraterone after oral ingestion. AA is a selective inhibitor of the cytochrome P450 17 enzyme (CYP17) and acts as an androgen biosynthesis inhibitor because CYP17 is a crucial enzyme in androgen biosynthesis in testes, adrenals, prostate tissues including prostate tumor cells () [Citation17–19].
Figure 1. Abiraterone acetate (a) and abiraterone (b).
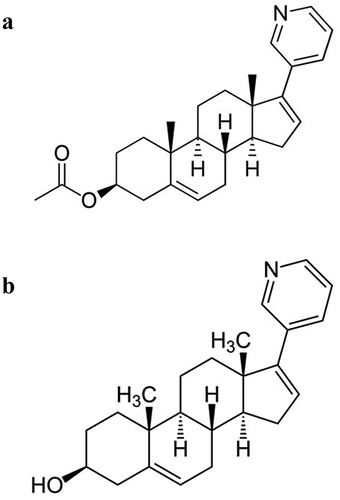
The inhibition of CYP17 pathway decreases the production of endogenous glucocorticoids as cortisol and increases the adrenocorticotropic hormone (ACTH) levels resulting in symptoms of mineralocorticoid excess (i.e. hypokalemia, hypertension, and fluid retention): this is why, in clinical trials, AA was administered in combination with prednisone or prednisolone, countering the potential adverse events (AEs) related to mineralocorticoid excess [Citation20,Citation21].
AA is administered orally at a dose of 1000 mg once daily in a fasting state [Citation22,Citation23]. It is rapidly absorbed and hydrolyzed to abiraterone, achieving maximum plasma concentration within 2 h [Citation24]. Abiraterone has both low solubility and permeability, it reaches its maximum concentrations slower, about 6 h. AA concentration in plasma is 1800-fold lower than the abiraterone concentration; therefore, only abiraterone is responsible for the clinical activity [Citation19,Citation25]. The high lipophilicity of AA causes slow and incomplete dissolution in the gut, resulting in poor absorption and low oral bioavailability; a high lipid content meal can increase bioavailability up to 10-fold because higher bile salt content increases the solubility of both AA and abiraterone [Citation26]. Hydrolysis occurs in the intestinal fluids with esterases and continues in the intestinal and liver microsomes [Citation27,Citation28]. Once AA enters the duodenum, rapid hydrolysis leads to supersaturation and is followed by abiraterone precipitation due to poor solubility in the fasted intestinal contents [Citation29]; this is confirmed by 50% of the dose being recovered as unchanged in feces [Citation30]. In the fed state, the duodenum pH is neutral and esterase activity is reduced, leading to greater absorption of both AA and abiraterone [Citation29,Citation31].
AA has an exposure – response relationship in patients with prostate cancer, with an optimal threshold to serve as a target of 8.4 ng/mL Cmin [Citation32]. The plasma AUC increases 5-10× when administered with food, depending on the fat content of the meal [Citation26]. However, AA has a high interindividual variability in exposure with a coefficient of variation of 46–70% for Cmin and 35–42% of patients do not reach the efficacy threshold of Cmin 8.4 ng/mL [Citation32,Citation33]. Concomitant intake of food and AA can be used in case of low exposure, but this may also lead to higher interindividual variability [Citation23]; this strategy and the existence of an exposure-response relationship provide a strong rationale for therapeutic drug monitoring (TDM) to increase the number of patients with adequate AA exposure [Citation34].
Abiraterone is extensively metabolized through hepatic metabolism by sulfotransferase 2A1 (SULT2A1) to abiraterone sulfate and by SULT2A1 and CYP3A4 to N-oxide abiraterone sulfate [Citation30]. These two major inactive circulating metabolites each account for 40% of the drug present in plasma [Citation30]. About 5% of abiraterone is converted into Δ4-abiraterone (D4A) by 3β-hydroxysteroid dehydrogenase (3βHSD) [Citation35]. This minor metabolite, structurally similar to testosterone, inhibits multiple steroidogenic enzymes and directly binds androgen receptor, blocking its activity [Citation35]. The mean metabolic ratio of D4A and abiraterone is 0.18 ± 0.25 [Citation36]. Conversion of abiraterone by 3βHSD takes place in peripheral tissues resulting in higher on target concentrations despite relatively low plasma concentrations [Citation37].
The mean half-life of abiraterone in plasma is 15 h. A radiolabeled drug study showed that abiraterone and metabolites are mainly excreted in the feces – approximately 88% of the administered dose of AA [Citation30]. In feces, 55% of the dose is unchanged AA and 22% is abiraterone, whilst urine is detectable only 5% of the abiraterone metabolites [Citation30]. In hemodialysis patients, exposure to abiraterone is comparable to that of patients with normal renal function, confirming negligible renal clearance. Healthy volunteers eliminate abiraterone more rapidly than mCRPC subjects, showing an apparent clearance of 2240 and 1505 L/h, respectively [Citation38].
2.2. Apalutamide
Apalutamide is a synthetic biaryl thiohydantoin compound and is a selective androgen receptor (AR) inhibitor [Citation39,Citation40]. It acts by inhibiting the nuclear translocation of the AR and DNA binding to androgen response elements (AREs) and exhibits no significant agonist activity [Citation40]. Apalutamide is a cytochrome P450 substrate and is primarily metabolized to N-desmethyl apalutamide (NDA) that exhibits one third of the activity of apalutamide on the target () [Citation41].
Figure 2. Apalutamide (a) and N-desmethyl apalutamide(b).
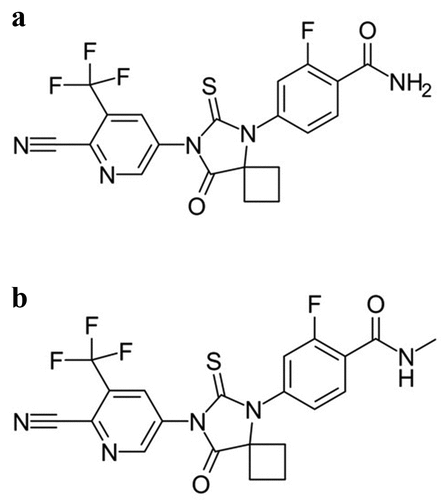
Apalutamide binds weakly to GABAA receptors, comparable to the activity shown by enzalutamide: moreover, it shows greater free fraction compared to enzalutamide but results in four-fold lower steady-state brain-levels, suggesting a lower seizure potential [Citation40].
Apalutamide is rapidly absorbed after oral administration, with measurable plasma concentrations within 30 minutes after administration and peak plasma concentrations (tmax) in 90–180 minutes [Citation42]. Apalutamide concentrations decline slowly, with a half-life at steady-state of approximately 4 days [Citation41]. After oral administration, the pharmacokinetics of apalutamide is linear and dose proportional, the absolute bioavailability of apalutamide is 100% without clinically relevant changes in Cmax and AUC between fasting or after a high-fat meal; however, Tmax achievement was delayed by approximately 2 h with food [Citation42,Citation43]. Patients reach steady-state exposure after 4 weeks of continuous administration [Citation42], with comparable plasma levels of apalutamide and NDA. Steady state Cmax for apalutamide and NDA is similar (~6 µg/m), whilst AUC is 100 and 124 µg h/mL, respectively [Citation42,Citation44]. The interindividual variability on pharmacokinetic parameters of apalutamide and metabolites is low (20%) [Citation45]. Due to its lipophilicity, apalutamide and NDA steady-state volume of distribution are 276 and 238 L, respectively, reflecting a wide distribution to peripheral tissues and cells [Citation45]. Serum albumin is the main binding protein and the unbound fraction of apalutamide and NDA in plasma is 4.2% and 5.1%, respectively [Citation46]. In dogs and mice, apalutamide and NDA show the ability to penetrate the blood – brain barrier although highlighting variety of brain-to-plasma ratio among these species. As for enzalutamide, apalutamide use in humans is associated with a higher incidence of typical central nervous system AEs [Citation47]. The little unchanged drug detected in urine and feces shows that apalutamide is extensively metabolized. Apalutamide is mainly eliminated through CYP450 metabolism to form NDA [Citation41]. CYP2C8 is the main enzyme that contributes for ~60% in the formation of NDA after single dose, followed by CYP3A4 – less than 15% [Citation41,Citation46]. Nevertheless, apalutamide induction effects on CYP3A4 and CYP2C8 influence its own metabolism and the contribution of CYP3A4 to the overall metabolism increases at steady-state, whereas the overall contribution of CYP2C8 decreases [Citation41,Citation44]. Apalutamide and NDA are further metabolized to form the carboxylic acid metabolite by carboxylesterases. Carboxylic acid metabolite is an inactive minor metabolite with minor clinical relevance [Citation41]. Esterases contribution is ~30% after single dose and is not subject to induction or inhibition effects but decreases at steady-state due to CYP autoinduction [Citation46]. The apparent clearance (CL/F) is 1.3 L/h after single dose of apalutamide and increases to 2.0 L/h at steady-state [Citation45]. Apalutamide half-life at steady-state is approximately 3–4 days. Most of apalutamide dose is excreted in urine (65%) and feces (24%). In urine, the carboxylic acid metabolite is the major excreted (31.1%) [Citation41], whilst in feces, unchanged drug, NDA, and carboxylic acid metabolite represent less than 6% of dose; unchanged apalutamide is a minor entity in urine and feces [Citation41].
2.3. Enzalutamide
Enzalutamide is a small molecule with high permeability and limited aqueous solubility having no ionizable groups at physiological pH range [Citation48]. Enzalutamide acts in multiple steps on the androgen pathway: in detail, it inhibits competitively androgen binding to AR and inhibits AR nuclear translocation and interaction with DNA [Citation49,Citation50]. It is mainly metabolized to N‐desmethyl enzalutamide (NDE) and a carboxylic acid metabolite () [Citation48,Citation51].
Figure 3. Enzalutamide (a) and N-desmethyl enzalutamide(b).
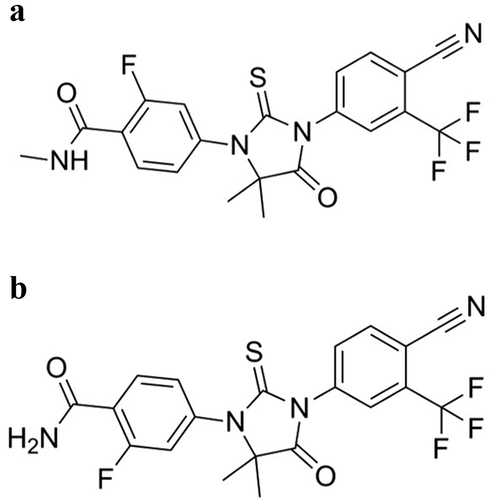
NDE demonstrates primary and secondary pharmacodynamics of similar potency to enzalutamide with plasma concentrations that are similar to enzalutamide. In contrast, the carboxylic acid metabolite is inactive and reaches lower plasma steady state concentrations [Citation48]. When interpreting clinical relevance, conclusions are based on the sum of enzalutamide active moieties, that is enzalutamide plus NDE exposure.
There is no exposure-response relationship for enzalutamide when administered at a single fixed dose of 160 mg daily, probably because it is administered at the higher end of the exposure – response curve [Citation48,Citation52]. After oral administration, enzalutamide is rapidly absorbed [Citation48], being Cmax reached after 1–2 h. NDE and carboxylic acid metabolite are formed slowly and median Cmax is reached after 132 and 96 h, respectively [Citation48]. Cmax is approximately 30% lower and is reached approximately 1 h later when enzalutamide is administered with food, but mean AUC values are similar in fasted and fed conditions establishing negligible absorption change, also in case of a high-fat meal [Citation48]; for this reason, enzalutamide can be taken with or without food. Enzalutamide, NDE, and carboxylic acid metabolite mean terminal half-life is ~6, 8, and 10 days, respectively [Citation48,Citation53]. With daily oral administration, enzalutamide is accumulated 8.3-fold relative to a single dose and steady state is achieved within 28 days [Citation48]. Daily fluctuations in plasma concentrations are low due to the long half-life, with time-linear pharmacokinetics [Citation48,Citation52]. Mean plasma concentrations values for enzalutamide, NDE, and the carboxylic acid metabolite are ~11, 13, and 8 µg/mL, respectively [Citation48]. In patients treated with enzalutamide, the intersubject variability of pharmacokinetic parameters of enzalutamide and NDE is less than 30%, demonstrating low intra-patient variability [Citation48]. Enzalutamide shows linear pharmacokinetics once steady-state is achieved.
The apparent volume of distribution (Vd/F) of enzalutamide is 110 L, indicating that is extensively distributed throughout the body. Enzalutamide and NDE show a high protein bound (approximately 97–98%), mainly to albumin [Citation54].
Enzalutamide is primarily metabolized in the liver [Citation48]. Enzalutamide is extensively metabolized by cytochrome P450. CYP2C8 is primarily responsible for biotransformation of enzalutamide, with CYP3A4/5 playing a minor role, being both responsible for the formation of NDE [Citation48]. On the other hand, NDE is metabolized by carboxylesterase 1 to the carboxyl metabolite [Citation54].
The excretion of enzalutamide through urine and feces concerns 85% of the administered dose. The major route is urine with 71% of dose, but mainly as carboxylic acid metabolite (63% of dose), only a trace amount as unchanged parent enzalutamide and too low to quantitate as NDE [Citation48].
2.4. Darolutamide
Darolutamide is a novel nonsteroidal antagonist of the AR with a flexible pyrazole structure with polar substitutions which is different from enzalutamide and apalutamide [Citation55]. Darolutamide potently inhibits androgen binding to AR and retains antagonistic properties in cells expressing increased AR levels and the oral intake suggests the potential for constant inhibition of the AR signaling pathway and a linear pharmacokinetic after single and multiple dosing [Citation55,Citation56]. Darolutamide comprises a 1:1 mixture of the two active diastereomers, (S, R)-darolutamide and (S, S)-darolutamide, which interconvert through the circulating metabolite ketodarolutamide () [Citation55,Citation57,Citation58].
Figure 4. Darolutamide (a) and Ketodarolutamide (b).
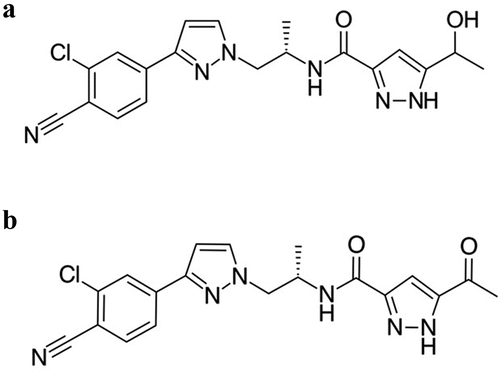
In vitro, the two diastereomers and ketodarolutamide show similar binding affinity for the AR and no major differences in pharmacological activity [Citation55,Citation57,Citation59]. Darolutamide exposure is influenced by food. Oral tablet bioavailability is about 30% in fasted conditions, indicating low solubility (logPdarolutamide = 1.9) and incomplete absorption [Citation60], whilst it reaches 60–75% when given with food, regardless of meal type and calorie [Citation61,Citation62]. The fat content of the meal does not play a determining role and does not affect absorption, so the solubility of darolutamide is probably related to pancreatic enzymes or bile salts rather than lipophilicity [Citation63]. For all these reasons, it is recommended to take darolutamide with a meal. Darolutamide is gradually absorbed after oral dosing, with a median time to peak concentration of 4–6 h (Cmax). Plasma concentrations declined in a biphasic manner, with a half-life of 18–20 h [Citation60]. The major metabolite, ketodarolutamide, is formed rapidly, and the plasma exposure of ketodarolutamide is higher than the darolutamide one [Citation60,Citation62]. Darolutamide steady state is reached after 2–5 days of twice-daily dosing and exposure increased in a linear, dose-related manner up to 1400 mg/day. At steady state, peak plasma concentrations of darolutamide are approximately 4 mg/L [Citation64].
The apparent volume of distribution (Vd/F) of darolutamide is 119 L, indicating that is widely distributed throughout the body [Citation63]. The unbound fraction of darolutamide in plasma is ~8%. In vitro data show no difference between the two diastereomers, moderately bound to plasma proteins (92%). The major metabolite ketodarolutamide is highly bound and the unbound fraction of ketodarolutamide in human plasma is only 0.1%. For this reason, Ketodarolutamide suggests no relevance in terms of mediating efficacy and potential drug interactions, and pharmacologic activity is considered to be driven by the diastereomers [Citation60]. Darolutamide shows low penetration of the blood-brain barrier, resulting only 4.5% of the plasma exposure after a single dose of darolutamide in rats and less than 4% in mice after multiple doses. The lower penetration it is also confirmed in humans, in fact Darolutamide records a lower incidence of seizure and CNS effects compared to the other ARSIs [Citation55,Citation65]. Furthermore, the negligible blood – brain barrier penetration, lack of stimulation of the androgen feedback loop at the hypothalamic – pituitary – gonadal axis, justifies the lack of serum testosterone elevations, and reduced activation of the AR pathway in PC cells, in contrast to the other ARSIs [Citation55,Citation66].
Darolutamide is mainly metabolized by oxidation and glucuronidation. In vitro data show that CYP3A4 mediates darolutamide oxidation and DDI studies with itraconazole and rifampicin confirmed it [Citation67]. Ketodarolutamide is also a substrate of CYP3A4. UGT1A1/9/3 and UGT2B10 catalyze further O- and N-glucuronidation of both darolutamide and ketodarolutamide [Citation68]. CYP3A4 – and lesser CYP1A1 – oxidates darolutamide diastereomers leading to ketodarolutamide formation.
The excretion of darolutamide and its metabolites is rapid and complete [Citation68]. Darolutamide is excreted 63% in the urine, but only 7% as the two diastereomers unchanged fraction consisting of 1.5% (S, R)-darolutamide and 5.7% (S, S)-darolutamide; O-glucuronides are the most common metabolites, while no relevant amount of keto-darolutamide was detected [Citation68]. Although the most darolutamide is excreted renally, the high concentrations and number of metabolites excreted in the urine suggests that hepatic metabolism contributes to darolutamide clearance more than renal elimination [Citation68]. Darolutamide is excreted predominantly via glomerular filtration, but transporter-mediated secretion of (S, R)-darolutamide could not be excluded [Citation68]. Thirty-two percent of darolutamide is excreted in feces: the fecal excretion pattern is different than what is observed in urine, being the 25–40% unabsorbed drug fraction with oral tablets probably responsible for it [Citation68].
3. Special populations and drug interactions
3.1. Renal impairment
In patients with end-stage renal disease on hemodialysis, Cmax and AUC of abiraterone are similar to those of healthy controls, consistent with the findings that renal excretion is not an important route of elimination [Citation30,Citation38]. Therefore, AA does not require dose adjustment in patients with renal impairment.
No clinical differences in apalutamide pharmacokinetics are observed in patients with mild to moderate renal impairment, but the use is not recommended due to lack of data especially in case of severe impairment [Citation69].
Concerning enzalutamide, no dose adjustments are required for patients with creatinine clearance (CrCL) ≥30 mL/min, whilst it has not been evaluated in case of severe renal impairment (CrCL <30 mL/min) apart from case reports [Citation70].
Darolutamide summary of product characteristics recommended reduction of the starting dose to 300 mg BID in patients with severe renal impairment [Citation60]. A clinical study shows that the increase in darolutamide exposure in patients with mild or moderate renal impairment is negligible and does not require dose adjustment [Citation63].
3.2. Hepatic impairment
As hepatic metabolism plays a critical role, an effect of hepatic impairment on the pharmacokinetics of AA is expected. The unbound fraction of abiraterone is 78% higher in patients with severe hepatic impairment compared to healthy controls, on the other hand it is relatively insensitive to mild or moderate hepatic impairment [Citation24]. AA should be used with caution in patients with moderate hepatic impairment, possibly considering a reduced initial dose, while it is contraindicated in subjects with severe hepatic impairment [Citation24,Citation54].
As for renal impairment, the use of apalutamide is not recommended in case of severe hepatic impairment due to lack of data [Citation63].
Enzalutamide mean Cmax is 24% higher in patients with mild hepatic impairment compared with healthy controls, while it is 11% and 41% lower in patients with moderate and severe hepatic impairment; the apparent clearance (CL/F) showed minimal difference, otherwise the apparent volume of distribution (Vd/F) is higher in patients with severe hepatic impairment because albumin concentration tends to decline if hepatic impairment is present and baseline albumin concentration is lower [Citation71]. Exposure to enzalutamide active moieties (AUC enzalutamide plus NDE) is comparable in subjects with severe hepatic impairment and healthy controls and is slightly higher in subjects with mild or moderate hepatic impairment than in control subjects but is not clinically meaningful [Citation71]. Despite it all, the metabolism of enzalutamide is relatively insensitive to hepatic impairment, and no initial dose adjustment is necessary [Citation48,Citation71]. Overall, enzalutamide does not require dose reduction in patients with impaired hepatic function.
As for renal impairment, in patients with moderate hepatic impairment the starting dose of darolutamide should be 300 mg BID [Citation63], whilst the increase in darolutamide exposure is negligible in case of mild hepatic impairment, not requiring dose adjustment [Citation60].
3.3. Drug-drug interactions (DDI)
Abiraterone is extensively metabolized by SULT2A1 and CYP3A4. Concomitant administration with a strong inducer of CYP3A4 enzyme decreases the exposure to abiraterone by 55%, while strong CYP3A4 inhibitors increase exposure to abiraterone by only 15%, which is not considered to be clinically relevant [Citation72]. Patients treated with abiraterone acetate had a 2.9-fold increase in exposure to the CYP2D6 substrate dextromethorphan, while coadministration of the CYP1A2 substrate theophylline does not alter theophylline exposure [Citation73]. Coadministration of abiraterone acetate with the CYP2C8 substrate pioglitazone increased pioglitazone exposure by 46%, and this is why abiraterone is considered a CYP2C8 inhibitor [Citation74]. Differently from AA, abiraterone is not a substrate for P-gp and has a no clinically relevant inhibitory effect on P-gp [Citation75]. Based on these data, the administration of AA together with strong CYP3A4 inducers should be avoided, while caution is recommended in case of concomitant administration of AA and substrates of CYP2D6 or CYP2C8 – especially for drugs with a narrow therapeutic index – possibly considering dose reductions.
Apalutamide is mainly metabolized by CYP2C8 and CYP3A4, but no dose adjustment is necessary when apalutamide is co-administered with inhibitors or inducers of these enzymes; this is due to the metabolic clearance of apalutamide which is shared equally by CYP2C8 and CYP3A4 at steady-state and mitigates, with a compensatory mechanism, the drug interaction if either pathway is inhibited [Citation46]. Furthermore, the steady-state autoinduction activity of apalutamide limits the effect of a CYP inducer [Citation46]. Apalutamide is considered a strong inducer of CYP3A4, CYP2C19 and a weak inducer of CYP2C9 and it is recommended to substitute concomitant medications if substrates of these enzymes or to monitor their activity if continued [Citation44]. Being apalutamide considered a potential inducer of UGT and a weak inducer of BCRP, P-gp and OATP1B1, it may reduce exposures of drugs if substrates of these enzymes and should be used with caution, evaluating a possible dose adjustment [Citation44]. Apalutamide inhibits CYP2C8 at steady-state Cmax and is considered a moderate inhibitor of CYP2C8. Apalutamide and NDA inhibit CYP1A2, CYP2B6, CYP2C9/19, CYP2D6, and CYP3A4, but at concentrations much greater than apalutamide Cmax and this is why they are considered weak inhibitors. Apalutamide and NDA are also weak inhibitors of OATP1B3 [Citation44].
Enzalutamide is mainly metabolized by CYP2C8 and minorly by CYP3A and has a considerable potential for DDIs due to induction activity on several metabolic enzymes [Citation54]. In order to mitigate the risks of seizure to patients, it is recommended to reduce the dose of enzalutamide to 80 mg once daily during concomitant use with a strong CYP2C8 inhibitor [Citation48]. Coadministration of itraconazole, a strong CYP3A4 inhibitor, has a small impact on enzalutamide active moieties, so that a dose adjustment is not recommended [Citation48,Citation76]. Enzalutamide influences the metabolism of CYP2C9/19 and CYP3A4 substrates, being considered a moderate inducer of CYP2C9/19 and a strong inducer of CYP3A4 [Citation77–79]. It is recommended to avoid concomitant use of enzalutamide with administered narrow therapeutic index drugs substrates of CYP2C9/19 or CYP3A4 [Citation48]. If co-administered, the maximum induction potential occurs when steady-state plasma concentrations of enzalutamide are reached, approximately after 1 month of treatment, and due to long enzalutamide half-life inducer effects may persist for 1 month or longer after stopping enzalutamide; therefore, a gradual dose reduction of the concomitant drug should be considered when stopping enzalutamide administration [Citation48]. Enzalutamide is not a substrate of OATP1B1/3, P-gp, or BCRP [Citation54].
Darolutamide is mainly metabolized by CYP3A4 and is identified as a substrate for P-gp and BCRP, the remaining enzymes involved have a minor contribution in the metabolism of darolutamide such that they would not be involved in clinically relevant DDIs [Citation55]. Neither P-gp nor BCRP inhibitors have significant impact on the pharmacokinetics of darolutamide [Citation80]. Darolutamide exposure decreased by 72% when co-administered with rifampicin (UGT1A9 inducer), but probably due to the induction of CYP3A4 and P-glycoprotein since the exposure of dapagliflozin (sensitive UGT1A9 substrate) is reduced only for 22% by rifampicin [Citation67,Citation81]. Darolutamide showed no evidence of CYP3A4 inhibition and is unlikely to inhibit other CYP enzymes, suggesting a low potential for CYP-mediated DDIs [Citation55,Citation67]. In patients treated with darolutamide, the concomitant use of moderate and strong CYP3A4 inducers and P-gp inducers is not recommended. Patients treated with strong CYP3A and P-gp transport combined inhibitors should be monitored more closely for the development of AEs [Citation82]. Use of darolutamide with BCRP substrates should be avoided, while patients taking concomitant drugs that are OATP1B1/3 substrates should be monitored [Citation67]. Darolutamide has a low potential for clinically relevant DDIs with drugs that are substrates for CYP or P-gp; increased exposure of BCRP and probably OATP substrates was the main interaction of note [Citation67]. Moreover, darolutamide does not affect OATP1B3-mediated transport of docetaxel as assessed by Buck et al. in both in vitro and in vivo models, thus supporting the combined treatment with docetaxel and darolutamide [Citation83].
In are resumed the main DDI for each drug.
Table 1. Main drug-drug interactions (DDI) of ARSI administered in mHSPC patients.
4. Clinical impact of ARSIs in mHSPC
In the latest years, the therapeutic scenario of mHSPC has rapidly evolved thanks to the approval of ARSIs in association with ADT as first-line treatment.
AAP was studied in two different trials. In the phase III LATITUDE trial, 1199 patients were randomized to receive either ADT plus AAP or ADT plus dual placebos; the first interim analysis showed a substantial benefit from the addition of AAP to ADT, with a statistically significant risk reduction for death and for radiographic progression (38% and 53%, respectively), allowing crossover for patients in the placebo group to receive AAP [Citation84]. The final OS analysis from the same trial confirmed the OS benefit (53.3 vs 36.5 months, HR: 0.66, p < 0.0001) [Citation85]. In the multistage STAMPEDE trial, 1917 patients were randomized to receive either ADT plus AAP or ADT alone; risk reduction for death was similar to what observed in the LATITUDE trial (37%) [Citation86]. Among AEs, the most frequent grade 3 AEs were hypertension and hypokalemia. It must be noted that the dose of prednisone in mHSPC trials was 5 mg once daily, differently from the dose adopted in mCRPC trial (5 mg twice daily), and this lower dose did not result in an increase rate of pharmacologically relevant severe AEs [Citation75]. In the phase III PEACE-1 clinical trial, among 582 mHSPC patients assigned to AAP + standard of care (± radiotherapy), 355 of them received concurrent docetaxel; this trial showed that the addition of AAP to docetaxel can improve OS without increasing the rate of docetaxel-related AEs [Citation87].
The role of apalutamide in mHSPC was evaluated in the phase III TITAN trial; in detail, 1042 patients were randomized 1:1 between apalutamide + ADT and placebo + ADT [Citation88]. The first interim analysis showed a statistically significant improvement in radiographic PFS at 24 months (HR: 0.48; p < 0.001) and OS at 24 months (HR:0.67; p = 0.005) [Citation88]. Final results from this trial confirmed the risk reduction for death despite crossover; moreover, apalutamide delayed second PFS and castration resistance (p < .0001 for both), maintaining health-related quality of life [Citation89]. Among AEs, rash was more common in the apalutamide group. It is noteworthy that the subgroup analysis of the TITAN trial showed that adding apalutamide to ADT and docetaxel – whose administration was permitted by study protocol before starting apalutamide – failed to improve OS compared to ADT and docetaxel alone [Citation89].
Enzalutamide, in addition to ADT, was compared to standard nonsteroidal antiandrogen therapy + ADT (standard-care group) in the phase III ENZAMET trial. OS, which is the primary endpoint of the trial, was improved by enzalutamide (HR: 0.67, p = 0.002) [Citation90]; secondary endpoints included PSA PFS and in clinical PFS (HR 0.39 and HR: 0.40, respectively, both p < 0.001). Among AEs, fatigue was more common in the enzalutamide group. In the ARCHES trial 1,150 mHSPC patients were randomized between enzalutamide and placebo, plus ADT [Citation91]; rPFS was the primary endpoint of this trial and it was significantly prolonged by enzalutamide (HR: 0.39; p < 0.001), regardless of volume disease and prior docetaxel therapy. Enzalutamide was tested together with docetaxel in mCRPC patients in a phase Ib trial [Citation92] showing feasibility even if at the costs of higher docetaxel-related toxicities. Similarly to apalutamide, the subgroup analysis of the ENZAMET trial showed that adding enzalutamide to ADT and docetaxel did not translate into OS improvement, as compared to ADT and docetaxel alone [Citation90].
Darolutamide, differently from the other ARSIs, has been tested in association with docetaxel in the mHSPC setting, given its manageable tolerability profile: in the phase III ARASENS trial, 1306 patients were randomized to receive darolutamide or placebo, both in combination with ADT and docetaxel [Citation93]. The primary endpoint of the trial, OS, was significantly improved by the addition of darolutamide (HR: 0.68, p < 0.001); among grade 3–4 AEs, neutropenia was the most common in both treatment arms.
In summary, new-generation ARSIs have a similar clinical benefit in terms of PFS and OS (). It must be noted that no direct comparison between ARSIs in this setting has been made to date: the choice between these molecules in the clinical practice should be guided by comorbidities and patients’ preference, with the exception of darolutamide which has been approved only in association with docetaxel.
Table 2. Clinical trials with ARSI in mHSPC patients.
5. Expert opinion
The advent of new drugs in the mHSPC setting is , certainly, an important innovation given the survival advantages shown by the aforementioned clinical trials, but several questions remain unanswered. Despite the absence of direct comparison, assuming a similar efficacy of these molecules, the choice between them is mainly driven by patients’ comorbidities, safety profile, and possible DDIs.
Comorbidities are , certainly, an important factor when deciding among ARSIs, for example, uncontrolled blood hypertension is a contraindication for AA, whilst a history of epilepsy is a contraindication for enzalutamide [Citation94]; however, darolutamide, which does not require concomitant steroid and displays a lower blood-brain barrier penetration, could be administered also in patients with multiple comorbidities [Citation80].
The phase III clinical trials described above, which led to the approval of these molecules in the mHSPC setting, adopted placebo as comparator in most cases, showing homogeneous efficacy according to primary endpoints but not in terms of side effects, which are specific for each drug. It must be noted that side effects are easily manageable in most cases, generally not requiring drug interruption.
It is acknowledged that older cancer patients are more vulnerable to DDI given the age-related polypharmacy [Citation95], and prostate cancer incidence is higher in men older than 65 years: this means that mHSPC patients eligible for an ARSI treatment have de facto a high DDI risk. As shown before, among ARSIs, the DDI risk is different according to specific drugs but none of these molecules is free from it. Clinicians should be persuaded to deprescribe inappropriate or unnecessary medications, reducing possible DDIs but also improving patients’ quality of life [Citation96]; moreover, in case of drug assumption for serious concomitant illness, shifting to drugs with similar efficacy but less interactions, if possible, should be always considered. All prescribers should verify potential DDIs before starting an ARSI in prostate cancer patients; different tools to check DDIs are available on the Internet or on dedicated software [Citation97].
Research in this field is evaluating new strategies to block the androgen receptor [Citation98]. It is clear that in the future new molecules will be available for mHSPC, but safety and DDIs must remain a priority when evaluating them for subsequent introduction in the clinical practice. The use of predictive biomarkers will certainly help in identifying patients that will – or that will not – benefit from ARSI in the first line setting; on the other hand, escalating treatment by combining other drugs – either targeting the AR pathway or targeting other mechanism – seems to be a less feasible option for all-comers given the increased risk of toxicities, even if some specific populations (i.e. patients with homologous recombination deficiency, HRD) would benefit from tailored therapies [Citation99,Citation100].
Artificial intelligence (AI) is gaining ground in the current precision oncology era [Citation101]. In this field, AI could be useful for clinicians to identify patients at higher risk of DDI: implementing this tool will be important to optimize therapeutic choices, limiting toxicities and avoidable interactions.
It must also be noted that the use of PSMA PET in the staging of treatment-naïve prostate cancer patients may increase the number of patients eligible for ARSI, given its higher diagnostic sensitivity as compared to ‘conventional imaging’ [Citation102,Citation103]; on the other hand, PSMA PET use may also increase the proportion of patients with oligometastatic disease amenable for localized therapies (i.e. radiotherapy) [Citation104]. Clinicians should be also aware that discordant cases (negative conventional imaging and positive PSMA PET imaging) may benefit from the early use of ARSI in a similar way to those that are negative to both (non-metastatic hormone-sensitive prostate cancer, nmHSPC) [Citation105,Citation106]; the early use of ARSI will probably be the therapeutic standard in high-risk nmHSPC patients in the next future [Citation107].
In conclusion, waiting for new data, the optimization of ARSI use in first-line mHSPC therapy cannot prescind from the thorough knowledge of pharmacokinetics and DDI of these molecules.
Article highlights
Androgen Receptor Signaling Inhibitors (ARSIs) are now adopted in metastatic hormone-sensitive prostate cancer (mHSPC) patients in addition to androgen deprivation therapy (ADT).
Despite a similar meaningful improvement in survival, they show different pharmacokinetic profiles and a high potential for drug-drug interactions (DDIs).
Given comorbidities and the consequent polypharmacy, older mHSPC patients have an increased risk of DDI when receiving ARSI.
Deep pharmacokinetic knowledge and future research will be able to overcome the problem of DDI and optimize the use of ARSIs in this therapeutic setting.
Declaration of interest
E. Francesco Giunta reports travel support from Janssen-Cilag and Bayer. UDG reports honoraria for advisory boards or speaker fees for Pfizer, BMS, MSD, PharmaMar, Astellas, Bayer, Ipsen, Roche, Novartis, Clovis, GSK, AstraZeneca, institutional research grants from AstraZeneca, Sanofi and Roche. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgment
This work was partly supported thanks to the contribution of R Corrente by the Italian Ministry of Health
Additional information
Funding
References
- Wang L, Lu B, He M, et al. Prostate cancer incidence and mortality: global status and temporal trends in 89 countries from 2000 to 2019. Front Public Health. [2022 Feb 16];10:811044. doi: 10.3389/fpubh.2022.811044
- Ng K, Smith S, Shamash J. Metastatic hormone-sensitive prostate cancer (mHSPC): advances and treatment strategies in the first-line setting. Oncol Ther. 2020 Dec;8(2):209–230. doi: 10.1007/s40487-020-00119-z
- Choi E, Buie J, Camacho J, et al. Evolution of androgen deprivation therapy (ADT) and its new emerging modalities in prostate cancer: an update for practicing urologists, Clinicians and medical providers. Res Rep Urol. [2022 Mar 30];14:87–108. doi: 10.2147/RRU.S303215
- Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. [2013 Dec 5];32(49):5501–5511. doi: 10.1038/onc.2013.206
- Chang C, Saltzman A, Yeh S, et al. Androgen receptor: an overview. Crit Rev Eukaryot Gene Expr. 1995;5(2):97–125. doi: 10.1615/CritRevEukarGeneExpr.v5.i2.10
- Fujita K, Nonomura N. Role of androgen receptor in prostate cancer: a review. World J Mens Health. 2019 Sep;37(3):288–295. doi: 10.5534/wjmh.180040
- Dai C, Dehm SM, Sharifi N. Targeting the androgen signaling axis in prostate cancer. J Clin Oncol. [2023 Sep 10];41(26):4267–4278. doi: 10.1200/JCO.23.00433
- Kyriakopoulos CE, Chen YH, Carducci MA, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer: long-term survival analysis of the randomized phase III E3805 CHAARTED trial. J Clin Oncol. [2018 Apr 10];36(11):1080–1087. doi: 10.1200/JCO.2017.75.3657
- James ND, Sydes MR, Clarke NW, et al. STAMPEDE investigators. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. [2016 Mar 19];387(10024):1163–1177. doi: 10.1016/S0140-6736(15)01037-5
- Gravis G, Fizazi K, Joly F, et al. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013 Feb;14(2):149–158. doi: 10.1016/S1470-2045(12)70560-0
- Jacob A, Raj R, Allison DB, et al. Androgen receptor signaling in prostate cancer and therapeutic strategies. Cancers (Basel). [2021 Oct 28];13(21):5417. doi: 10.3390/cancers13215417
- de Bono JS, Logothetis CJ, Molina A, et al. COU-AA-301 investigators. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. [2011 May 26];364(21):1995–2005. doi: 10.1056/NEJMoa1014618.
- Scher HI, Fizazi K, Saad F, et al. AFFIRM investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. [2012 Sep 27];367(13):1187–1197. doi: 10.1056/NEJMoa1207506.
- Ryan CJ, Smith MR, Fizazi K, et al. COU-AA-302 investigators. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015 Feb;16(2):152–160. doi: 10.1016/S1470-2045(14)71205-7
- Beer TM, Armstrong AJ, Rathkopf DE, et al. PREVAIL investigators. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. [2014 Jul 31];371(5):424–433. doi: 10.1056/NEJMoa1405095
- Berruti A, Bracarda S, Caffo O, et al. nmCRPC, a look in the continuous care of prostate cancer patients: state of art and future perspectives. Cancer Treat Rev. 2023 Apr;115:102525. doi: 10.1016/j.ctrv.2023.102525
- Jarman M, Barrie SE, Llera JM. The 16,17-double bond is needed for irreversible inhibition of human cytochrome p45017alpha by abiraterone (17-(3-pyridyl)androsta-5, 16-dien-3beta-ol) and related steroidal inhibitors. J Med Chem. [1998 Dec 31];41(27):5375–5381. doi: 10.1021/jm981017j
- O’Donnell A, Judson I, Dowsett M, et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. [2004 Jun 14];90(12):2317–2325. doi: 10.1038/sj.bjc.6601879.
- Ryan CJ, Smith MR, Fong L, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. [2010 Mar 20];28(9):1481–1488. doi: 10.1200/JCO.2009.24.1281
- Vasaitis TS, Bruno RD, Njar VC. CYP17 inhibitors for prostate cancer therapy. J Steroid Biochem Mol Biol. 2011 May;125(1–2):23–31. doi: 10.1016/j.jsbmb.2010.11.005
- Auchus RJ, Yu MK, Nguyen S, et al. Use of prednisone with abiraterone acetate in metastatic castration-resistant prostate cancer. Oncology. 2014 Dec;19(12):1231–1240. doi: 10.1634/theoncologist.2014-0167
- Groenland SL, van Nuland M, Bergman AM, et al; Dutch Pharmacology Oncology Group (DPOG). Concomitant intake of abiraterone acetate and food to increase pharmacokinetic exposure: real life data from a therapeutic drug monitoring programme. Eur J Cancer. 2020 May;130:32–38. doi: 10.1016/j.ejca.2020.02.012
- Danielak D, Krejčí T, Beránek J. Increasing the efficacy of abiraterone - from pharmacokinetics, through therapeutic drug monitoring to overcoming food effects with innovative pharmaceutical products. Eur J Pharm Sci. [2022 Sep 1];176:106254. doi: 10.1016/j.ejps.2022.106254
- Marbury T, Lawitz E, Stonerock R, et al. Single-dose pharmacokinetic studies of abiraterone acetate in men with hepatic or renal impairment. J Clin Pharmacol. 2014 Jul;54(7):732–741. doi: 10.1002/jcph.253
- Bouhajib M, Tayab Z. Evaluation of the pharmacokinetics of abiraterone acetate and abiraterone following single-dose administration of abiraterone acetate to healthy subjects. Clin Drug Investig. 2019 Mar;39(3):309–317. doi: 10.1007/s40261-019-00752-1
- Chi KN, Spratlin J, Kollmannsberger C, et al. Food effects on abiraterone pharmacokinetics in healthy subjects and patients with metastatic castration-resistant prostate cancer. J Clin Pharmacol. 2015 Dec;55(12):1406–1414. doi: 10.1002/jcph.564
- Basa-Dénes O, Solymosi T, Ötvös Z, et al. Investigations of the mechanism behind the rapid absorption of nano-amorphous abiraterone acetate. Eur J Pharm Sci. [2019 Mar 1];129:79–86. doi: 10.1016/j.ejps.2019.01.001
- Sakai Y, Fukami T, Nagaoka M, et al. Arylacetamide deacetylase as a determinant of the hydrolysis and activation of abiraterone acetate in mice and humans. Life Sci. [2021 Nov 1];284:119896. doi: 10.1016/j.lfs.2021.119896
- Geboers S, Stappaerts J, Mols R, et al. The effect of food on the intraluminal behavior of abiraterone acetate in man. J Pharm Sci. 2016 Sep;105(9):2974–2981. doi: 10.1016/j.xphs.2016.03.008
- Acharya M, Gonzalez M, Mannens G, et al. A phase I, open-label, single-dose, mass balance study of 14C-labeled abiraterone acetate in healthy male subjects. Xenobiotica. 2013 Apr;43(4):379–389. doi: 10.3109/00498254.2012.721022
- Van Speybroeck M, Mellaerts R, Mols R, et al. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur J Pharm Sci. [2010 Dec 23];41(5):623–630. doi: 10.1016/j.ejps.2010.09.002
- Carton E, Noe G, Huillard O, et al. Relation between plasma trough concentration of abiraterone and prostate-specific antigen response in metastatic castration-resistant prostate cancer patients. Eur J Cancer. 2017 Feb;72:54–61. doi: 10.1016/j.ejca.2016.11.027
- van Nuland M, Groenland SL, Bergman AM, et al. Exposure-response analyses of abiraterone and its metabolites in real-world patients with metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2020 Jun;23(2):244–251. doi: 10.1038/s41391-019-0179-5
- Mueller-Schoell A, Groenland SL, Scherf-Clavel O, et al. Therapeutic drug monitoring of oral targeted antineoplastic drugs. Eur J Clin Pharmacol. 2021 Apr;77(4):441–464. doi: 10.1007/s00228-020-03014-8
- Li Z, Bishop AC, Alyamani M, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. [2015 Jul 16];523(7560):347–351. doi: 10.1038/nature14406
- Blanchet B, Carton E, Alyamani M, et al. A PK/PD study of delta-4 abiraterone metabolite in metastatic castration-resistant prostate cancer patients. Pharmacol Res. 2018 Oct;136:56–61. doi: 10.1016/j.phrs.2018.08.016
- Crombag MBS, van Nuland M, Bergman AM, et al. Impact of age on exposure to oral antiandrogen therapies in clinical practice. Prostate Cancer Prostatic Dis. 2019 Mar;22(1):168–175. doi: 10.1038/s41391-018-0096-z
- Stuyckens K, Saad F, Xu XS, et al. Population pharmacokinetic analysis of abiraterone in chemotherapy-naïve and docetaxel-treated patients with metastatic castration-resistant prostate cancer. Clin Pharmacokinet. 2014 Dec;53(12):1149–1160. doi: 10.1007/s40262-014-0178-6
- Jung ME, Ouk S, Yoo D, et al. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J Med Chem. [2010 Apr 8];53(7):2779–2796. doi: 10.1021/jm901488g
- Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. [2012 Mar 15];72(6):1494–1503. doi: 10.1158/0008-5472.CAN-11-3948
- de Vries R, Jacobs F, Mannens G, et al. Apalutamide absorption, metabolism, And excretion in healthy men, and enzyme reaction in human hepatocytes. Drug Metab Dispos. 2019 May;47(5):453–464. doi: 10.1124/dmd.118.084517
- Rathkopf DE, Morris MJ, Fox JJ, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. [2013 Oct 1];31(28):3525–3530. doi: 10.1200/JCO.2013.50.1684
- Yu A, Erba M, Hazra A. Pharmacokinetics and use-testing of apalutamide prepared in aqueous food vehicles for alternative administration. Clin Pharmacol Drug Dev. 2021 Nov;10(11):1375–1384. doi: 10.1002/cpdd.1001
- Duran I, Carles J, Bulat I, et al. Pharmacokinetic drug-drug interaction of apalutamide, part 1: clinical studies in healthy men and patients with castration-resistant prostate cancer. Clin Pharmacokinet. 2020 Sep;59(9):1135–1148. doi: 10.1007/s40262-020-00882-2
- Pérez-Ruixo C, Pérez-Blanco JS, Chien C, et al. Population pharmacokinetics of apalutamide and its active metabolite N-Desmethyl-apalutamide in healthy and castration-resistant prostate cancer subjects. Clin Pharmacokinet. 2020 Feb;59(2):229–244. doi: 10.1007/s40262-019-00808-7
- Van den Bergh A, Snoeys J, De Zwart L, et al. Pharmacokinetic drug-drug interaction of apalutamide, part 2: investigating interaction potential using a physiologically based pharmacokinetic model. Clin Pharmacokinet. 2020 Sep;59(9):1149–1160. doi: 10.1007/s40262-020-00881-3.
- Smith MR, Saad F, Chowdhury S, et al. SPARTAN investigators. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med. [2018 Apr 12];378(15):1408–1418. doi: 10.1056/NEJMoa1715546
- Gibbons JA, Ouatas T, Krauwinkel W, et al. Clinical pharmacokinetic studies of enzalutamide. Clin Pharmacokinet. 2015 Oct;54(10):1043–1055. doi: 10.1007/s40262-015-0271-5.
- Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. [2009 May 8];324(5928):787–790. doi: 10.1126/science.1168175
- Armstrong CM, Gao AC. Drug resistance in castration resistant prostate cancer: resistance mechanisms and emerging treatment strategies. Am J Clin Exp Urol. [2015 Aug 8];3(2):64–76.
- Bennett D, Gibbons JA, Mol R, et al. Validation of a method for quantifying enzalutamide and its major metabolites in human plasma by LC-MS/MS. Bioanalysis. 2014 Mar;6(6):737–744. doi: 10.4155/bio.13.325
- van Nuland M, Bergman AM, Rosing H, et al. Exposure-response assessment of enzalutamide and its major metabolites in a real-world cohort of patients with metastatic castration-resistant prostate cancer. Pharmacotherapy. 2019 Dec;39(12):1137–1145. doi: 10.1002/phar.2339
- Joulia ML, Carton E, Jouinot A, et al. Pharmacokinetic/Pharmacodynamic relationship of enzalutamide and its active metabolite N-Desmethyl enzalutamide in metastatic castration-resistant prostate cancer patients. Clin Genitourin Cancer. 2020 Apr;18(2):155–160. doi: 10.1016/j.clgc.2019.05.020
- Benoist GE, Hendriks RJ, Mulders PF, et al. Pharmacokinetic aspects of the two novel oral drugs used for metastatic castration-resistant prostate cancer: abiraterone acetate and enzalutamide. Clin Pharmacokinet. 2016 Nov;55(11):1369–1380. doi: 10.1007/s40262-016-0403-6
- Moilanen AM, Riikonen R, Oksala R, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. [2015 Jul 3];5(1):12007. doi: 10.1038/srep12007
- Fizazi K, Massard C, Bono P, et al. ARADES study group. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. 2014 Aug;15(9):975–985. doi: 10.1016/S1470-2045(14)70240-2
- Taavitsainen P, Gieschen H, Korjamo T, et al. Absorption, distribution, metabolism and excretion of darolutamide (a novel non-steroidal androgen receptor antagonist) in rats. Xenobiotica. 2020 Aug;50(8):967–979. doi: 10.1080/00498254.2020.1723038
- Nykänen P, Korjamo T, Gieschen H, et al. Pharmacokinetics of darolutamide, its diastereomers and active metabolite in the mouse: response to Saini NK et al. Drug Metab Lett. 2021;14(1):9–16. doi: 10.2174/1872312814666201112121129
- Sugawara T, Baumgart SJ, Nevedomskaya E, et al. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int J Cancer. [2019 Sep 1];145(5):1382–1394. doi: 10.1002/ijc.32242
- Zurth C, Nykänen P, Wilkinson G, et al. Clinical pharmacokinetics of the androgen receptor inhibitor darolutamide in healthy subjects and patients with hepatic or renal impairment. Clin Pharmacokinet. 2022 Apr;61(4):565–575. doi: 10.1007/s40262-021-01078-y
- Massard C, Penttinen HM, Vjaters E, et al. Pharmacokinetics, antitumor activity, and safety of ODM-201 in patients with chemotherapy-naive metastatic castration-resistant prostate cancer: an open-label phase 1 study. Eur Urol. 2016 May;69(5):834–840. doi: 10.1016/j.eururo.2015.09.046
- Matsubara N, Mukai H, Hosono A, et al. Phase 1 study of darolutamide (ODM-201): a new-generation androgen receptor antagonist, in Japanese patients with metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2017;80(6):1063–1072. doi: 10.1007/s00280-017-3417-3
- Podgoršek E, Mehra N, van Oort IM, et al. Clinical pharmacokinetics and pharmacodynamics of the next generation androgen receptor inhibitor-darolutamide. Clin Pharmacokinet. 2023 Aug;62(8):1049–1061. doi: 10.1007/s40262-023-01268-w
- Shore ND. Darolutamide (ODM-201) for the treatment of prostate cancer. Expert Opin Pharmacother. 2017 Jun;18(9):945–952. doi: 10.1080/14656566.2017.1329820
- Zurth C, Sandmann S, Trummel D, et al. Blood-brain barrier penetration of [14C]darolutamide compared with [14C]enzalutamide in rats using whole body autoradiography. J Clin Oncol. 2018;36(6_suppl):345. doi: 10.1200/JCO.2018.36.6_suppl.345
- Fizazi K, Albiges L, Loriot Y, et al. ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2015;15(9):1007–1017. doi: 10.1586/14737140.2015.1081566
- Zurth C, Koskinen M, Fricke R, et al. Drug-drug interaction potential of Darolutamide: In Vitro and clinical studies. Eur J Drug Metab Pharmacokinet. 2019 Dec;44(6):747–759. doi: 10.1007/s13318-019-00577-5
- Taavitsainen P, Prien O, Kähkönen M, et al. Metabolism and Mass balance of the novel nonsteroidal androgen receptor inhibitor darolutamide in humans. Drug Metab Dispos. 2021 Jun;49(6):420–433. doi: 10.1124/dmd.120.000309
- Bögemann M, Facchini G, Bauernhofer T, et al. Role of apalutamide in the treatment landscape for patients with advanced prostate cancer: an expert opinion statement of European clinical practice. Ir J Med Sci. 2023 Dec;192(6):2643–2651. doi: 10.1007/s11845-023-03303-y
- Tsang ES, de Haan M, Eigl BJ. A case report of enzalutamide administration in a dialysis-dependent patient with castration-resistant prostate cancer. J Oncol Pharm Pract. 2018 Mar;24(2):143–145. doi: 10.1177/1078155216689381
- Krauwinkel W, Noukens J, van Dijk J, et al. Comparison of the pharmacokinetics and safety of enzalutamide in subjects with hepatic impairment and matched healthy subjects. J Clin Pharm Ther. 2017 Jun;42(3):268–275. doi: 10.1111/jcpt.12503
- Bernard A, Vaccaro N, Acharya M, et al. Impact on abiraterone pharmacokinetics and safety: open-label drug-drug interaction studies with ketoconazole and rifampicin. Clin Pharmacol Drug Dev. 2015 Jan;4(1):63–73. doi: 10.1002/cpdd.132
- Chi KN, Tolcher A, Lee P, et al. Effect of abiraterone acetate plus prednisone on the pharmacokinetics of dextromethorphan and theophylline in patients with metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol. 2013 Jan;71(1):237–244. doi: 10.1007/s00280-012-2001-0
- Monbaliu J, Gonzalez M, Bernard A, et al. In vitro and in vivo drug-drug interaction studies to assess the effect of abiraterone acetate, abiraterone, and metabolites of abiraterone on CYP2C8 activity. Drug Metab Dispos. 2016 Oct;44(10):1682–1691. doi: 10.1124/dmd.116.070672
- Boujonnier F, Lemaitre F, Scailteux LM. Pharmacokinetic interactions between abiraterone, apalutamide, darolutamide or enzalutamide and antithrombotic drugs: prediction of clinical events and review of pharmacological information. Cardiovasc Drugs Ther. 2023 May 1. doi: 10.1007/s10557-023-07453-0
- Lehr T, Staab A, Trommeshauser D, et al. Semi-mechanistic population pharmacokinetic drug-drug interaction modelling of a long half-life substrate and itraconazole. Clin Pharmacokinet. 2010;49(1):53–66. doi: 10.2165/11317210-000000000-00000
- Seo KA, Bae SK, Choi YK, et al. Metabolism of 1’- and 4-hydroxymidazolam by glucuronide conjugation is largely mediated by UDP-glucuronosyltransferases 1A4, 2B4, and 2B7. Drug Metab Dispos. 2010 Nov;38(11):2007–2013. doi: 10.1124/dmd.110.035295
- Jones DR, Moran JH, Miller GP. Warfarin and UDP-glucuronosyltransferases: writing a new chapter of metabolism. Drug Metab Rev. 2010 Feb;42(1):55–61. doi: 10.3109/03602530903209395
- Bellesoeur A, Thomas-Schoemann A, Allard M, et al. Pharmacokinetic variability of anticoagulants in patients with cancer-associated thrombosis: clinical consequences. Crit Rev Oncol Hematol. 2018 Sep;129:102–112. doi: 10.1016/j.critrevonc.2018.06.015
- Shore N, Zurth C, Fricke R, et al. Evaluation of clinically relevant drug-drug interactions and population pharmacokinetics of darolutamide in patients with nonmetastatic castration-resistant prostate cancer: results of pre-specified and post hoc analyses of the phase III ARAMIS Trial. Target Oncol. 2019 Oct;14(5):527–539. doi: 10.1007/s11523-019-00674-0
- Kasichayanula S, Liu X, Griffen SC, et al. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diab Obes Metab. 2013 Mar;15(3):280–283. doi: 10.1111/dom.12024
- Konig J, Muller F, Fromm MF. Transporters and drug-drug interactions: important determinants of drug disposition and efects. Pharmacol Rev. 2013;65(3):944–966. doi: 10.1124/pr.113.007518
- Buck SAJ, Talebi Z, Drabison T, et al. Darolutamide does not interfere with OATP-mediated uptake of docetaxel. Int J Cancer. [2024 Mar 16]. doi: 10.1002/ijc.34922
- Fizazi K, Tran N, Fein L, et al. LATITUDE investigators. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med. [2017 Jul 27];377(4):352–360. doi: 10.1056/NEJMoa1704174
- Fizazi K, Tran N, Fein L, et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019 May;20(5):686–700. doi: 10.1016/S1470-2045(19)30082-8
- James ND, de Bono JS, Spears MR, et al. STAMPEDE investigators. Abiraterone for prostate cancer not Previously treated with hormone therapy. N Engl J Med. [2017 Jul 27];377(4):338–351. doi: 10.1056/NEJMoa1702900
- Fizazi K, Foulon S, Carles J, et al. PEACE-1 investigators. Abiraterone plus prednisone added to androgen deprivation therapy and docetaxel in de novo metastatic castration-sensitive prostate cancer (PEACE-1): a multicentre, open-label, randomised, phase 3 study with a 2 × 2 factorial design. Lancet. [2022 Apr 30];399(10336):1695–1707. doi: 10.1016/S0140-6736(22)00367-1
- Chi KN, Agarwal N, Bjartell A, et al; TITAN Investigators. Apalutamide for metastatic, castration-sensitive prostate cancer. N Engl J Med. [2019 Jul 4];381(1):13–24. doi: 10.1056/NEJMoa1903307
- Chi KN, Chowdhury S, Bjartell A, et al. Apalutamide in Patients with metastatic castration-sensitive prostate cancer: final survival analysis of the randomized, double-blind, phase III TITAN study. J Clin Oncol. [2021 Jul 10];39(20):2294–2303. doi: 10.1200/JCO.20.03488
- Davis ID, Martin AJ, Stockler MR, et al; ENZAMET Trial Investigators and the Australian and New Zealand Urogenital and Prostate Cancer Trials Group. Enzalutamide with standard first-line therapy in metastatic prostate cancer. N Engl J Med. [2019 Jul 11];381(2):121–131. doi: 10.1056/NEJMoa1903835
- Armstrong AJ, Szmulewitz RZ, Petrylak DP, et al. ARCHES: a randomized, phase III study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J Clin Oncol. [2019 Nov 10];37(32):2974–2986. doi: 10.1200/JCO.19.00799
- Morris MJ, Rathkopf DE, Novotny W, et al. Phase ib study of enzalutamide in combination with docetaxel in men with metastatic castration-resistant prostate cancer. Clin Cancer Res. [2016 Aug 1];22(15):3774–3781. doi: 10.1158/1078-0432.CCR-15-2638
- Smith MR, Hussain M, Saad F, et al. ARASENS trial investigators. Darolutamide and survival in metastatic, hormone-sensitive prostate cancer. N Engl J Med. [2022 Mar 24];386(12):1132–1142. doi: 10.1056/NEJMoa2119115
- Mir N, Burke O, Yates S, et al. Androgen receptor pathway inhibitors, prostate cancer, and older adults: a global young international society of geriatric oncology drug review. Ther Adv Med Oncol. [2023 Jan 28];15:17588359221149887. doi: 10.1177/17588359221149887
- Ramsdale E, Mohamed M, Yu V, et al. Polypharmacy, potentially inappropriate medications, and drug-drug interactions in vulnerable older adults with advanced cancer initiating cancer treatment. Oncology. [2022 Jul 5];27(7):e580–e588. doi: 10.1093/oncolo/oyac053
- Turner JP, Shakib S, Bell JS. Is my older cancer patient on too many medications? J Geriatr Oncol. 2017 Mar;8(2):77–81. doi: 10.1016/j.jgo.2016.10.003
- Marcath LA, Xi J, Hoylman EK, et al. Comparison of nine tools for screening drug-drug interactions of oral oncolytics. J Oncol Pract. 2018 Jun;14(6):e368–e374. doi: 10.1200/JOP.18.00086
- Henry MC, Riley CM, Hunter I, et al. Synthesis and evaluation of small molecule inhibitors of the androgen receptor N-Terminal domain. ACS Med Chem Lett. [2023 Nov 17];14(12):1800–1806. doi: 10.1021/acsmedchemlett.3c00426
- De Giorgi U, Giunta EF, Verzoni E, et al. Adding PARP inhibitor to an androgen-receptor signaling inhibitor in metastatic prostate cancer: what are we missing? Ann Oncol. 2023 Sep;34(9):729–731. doi: 10.1016/j.annonc.2023.06.008
- Bourlon MT, Valdez P, Castro E. Development of PARP inhibitors in advanced prostate cancer. Ther Adv Med Oncol. [2024 Jan 9];16:17588359231221337. doi: 10.1177/17588359231221337
- Shreve JT, Khanani SA, Haddad TC. Artificial intelligence in oncology: Current capabilities, future opportunities, and ethical considerations. Am Soc Clin Oncol Educ Book. 2022 Apr;42(42):1–10. doi: 10.1200/EDBK_350652
- Caroli P, Sandler I, Matteucci F, et al. 68Ga-PSMA PET/CT in patients with recurrent prostate cancer after radical treatment: prospective results in 314 patients. Eur J Nucl Med Mol Imaging. 2018 Nov;45(12):2035–2044. doi: 10.1007/s00259-018-4067-3
- Hofman MS, Lawrentschuk N, Francis RJ, et al. proPSMA Study Group Collaborators. Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): a prospective, randomised, multicentre study. Lancet. [2020 Apr 11];395(10231):1208–1216. doi: 10.1016/S0140-6736(20)30314-7
- Ayati N, Herrmann K, Fanti S, et al. More accurate imaging is not stage migration: time to move from “hubble” to “webb” in hormone-sensitive prostate cancer. Eur Urol. 2023 Jan;83(1):6–9. doi: 10.1016/j.eururo.2022.10.005
- Freedland SJ, de Almeida Luz M, De Giorgi U, et al. Improved outcomes with enzalutamide in biochemically recurrent prostate cancer. N Engl J Med. [2023 Oct 19];389(16):1453–1465. doi: 10.1056/NEJMoa2303974
- Armstrong WR, Clark KJ, Smith CP, et al. PSMA PET findings in an “EMBARK-like” cohort of patients with high-risk non-metastatic hormone-sensitive prostate cancer: a single center post-hoc retrospective analysis. J Clin Oncol. 2023;41(16_suppl):5091–5091. doi: 10.1200/JCO.2023.41.16_suppl.5091
- Giunta EF, Gasperoni L, Giorgi U. Enzalutamide and leuprolide acetate in non-metastatic hormone-sensitive prostate cancer: the sooner the better? Future Oncol. 2024 Feb;20(4):163–166. doi: 10.2217/fon-2023-1019