
ABSTRACT
Introduction
Aspirin is known for its therapeutic benefits in preventing strokes and relieving pain. However, it is toxic to some individuals, and the biological mechanisms causing toxicity are unknown. Limited literature is available on the role of glycine conjugation as the principal pathway in aspirin detoxification. Previous studies have quantified this two-step enzyme reaction as a singular enzymatic process. Consequently, the individual contributions of these enzymes to the kinetics remain unclear.
Areas covered
This review summarized the available information on the pharmacokinetics and detoxification of aspirin by the glycine conjugation pathway. Literature searches were conducted using Google Scholar and the academic journal databases accessible through the North-West University Library. Furthermore, the factors affecting interindividual variation in aspirin metabolism and what is known regarding aspirin toxicity were discussed.
Expert opinion
The greatest drawback in understanding the pharmacokinetics of aspirin is the limited information available on the substrate preference of the xenobiotic ligase (ACSM) responsible for activating salicylate to salicyl-CoA. Furthermore, previous pharmacokinetic studies did not consider the contribution of other substrates from the diet or genetic variants, to the detoxification rate of glycine conjugation. Impaired glycine conjugation might contribute to adverse health effects seen in Reye’s syndrome and cancer.
1. Introduction
Aspirin was discovered when it was realized that willow extract could be used for the treatment of pain and inflammation [Citation1–6]. Further investigations in the early 1800s identified the active components as salicin, salicylic acid, and acetylsalicylic acid. Henri Leroux isolated salicin in 1829 [Citation7,Citation8], and the Bayer Company subsequently registered the compound under the trade name Aspirin in 1899 [Citation9]. The pharmacology of aspirin was evaluated using clinical trials, and it was shown in 1971 that aspirin interferes with the synthesis of prostaglandins (PGs) [Citation10].
Various epidemiological studies since the 1980s have shown that aspirin prevents recurrent transient ischemic attacks or mini-strokes in men [Citation11], is a potential anticarcinogenic drug [Citation11–14], possibly prevents pre-eclampsia [Citation15], lowers blood glucose in patients with type 2 diabetes (T2D) [Citation16] and at relatively high doses (5–20 mM), it has been shown to have cell proliferation inhibitory potential in various cancer cell populations [Citation17–23].
Aspirin, however, has also been linked to deleterious effects such as hearing loss, tinnitus [Citation24–28], aspirin hypersensitivity causing ailments of the skin (urticaria, angioedema), respiratory tract (rhinitis, asthma) [Citation29], gastrointestinal symptoms (stomach bleeding, heart burn, nausea and vomiting) [Citation30,Citation31], and Reye’s syndrome [Citation32]. The biological mechanisms causing these disorders still need to be investigated.
Although there are derivatives and similar common non-steroidal anti-inflammatory drugs (NSAIDs) such as Ibuprofen, Ketoprofen, Diclofenac, Naproxen, Meloxicam, and Co-codamol, this review will discuss the pharmacokinetics of aspirin as well as the current limitations of the studies that were performed. We will also highlight important future directives that can aid in understanding the anticarcinogenic and toxicity mechanisms of aspirin.
To achieve the goal and objectives of this review, the literature search was conducted using two primary resources: Google Scholar and the academic journal databases accessible through the North-West University (NWU) Library. These databases were selected due to their comprehensive coverage of academic and scientific publications relevant to the pharmacokinetics of aspirin. A broad approach was used at the initial stages of the search, utilizing key terms related to the pharmacokinetics of aspirin. At this stage, no date restrictions were applied, allowing the identification of foundational studies and historical perspectives on the topic. After the initial search, date restrictions were applied, starting with the earliest studies and progressing to the most recent. This approach helped trace the evolution of research and understand the progression of knowledge on the subject. As gaps in the literature were identified, targeted searches were conducted using specific phrases and questions related to these gaps. This helped focus on studies that addressed aspects of aspirin pharmacokinetics and toxicity.
To ensure relevance, the search results were filtered to include articles specifically related to the mechanisms of action of aspirin and its detoxification process. Emphasis was placed on identifying original publications that unraveled the role of glycine conjugation in aspirin metabolism. Studies not directly related to the pharmacokinetics or mechanisms of aspirin were excluded.
2. Pharmacokinetics of aspirin
Aspirin is widely used as a drug to treat various health conditions [Citation33,Citation34]. Salicylates are also found in many common food items, e.g. vegetables, fruits, herbs/spices, and beverages [Citation35,Citation36]. Significantly higher levels of salicylic acid were found in the serum of vegetarians not taking aspirin, with the reported levels comparable to that of patients taking 75 mg of aspirin [Citation37].
2.1. Absorption of aspirin after ingestion
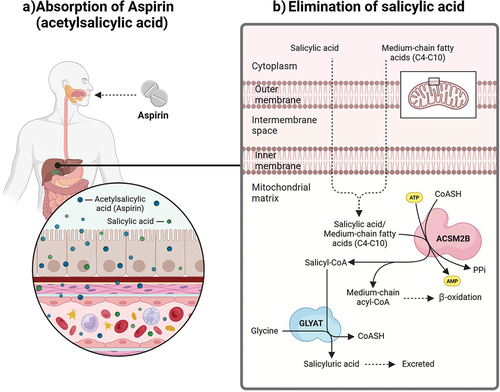
Aspirin is usually administered in one of four ways: orally, rectally, intravenously, or topical. The method of administration depends on the relief desired [Citation38]. Following ingestion, aspirin is rapidly deacetylated to form salicylic acid. The absorption of both aspirin and salicylates is rapid, and the serum half-life of salicylate ranges from an hour (low doses) to 30 hours (overdose) [Citation39,Citation40]. Gastrointestinal absorption of aspirin has been observed to occur in the stomach [Citation41,Citation42], small [Citation43,Citation44], and large intestine [Citation45] by passive diffusion ().
Figure 1. The absorption of aspirin and the excretion of salicylic acid.
2.2. Mechanism of action of aspirin
Aspirin remains the only NSAID to irreversibly acetylate a serine residue in the cyclooxygenase active site of the bifunctional cyclooxygenase (COX-1 and COX-2) enzymes [Citation46,Citation47]. While aspirin-mediated acetylation of COX-1 effectively inhibits the oxygenation of arachidonic acid to the prostaglandin G2 (PGG2) intermediate of PGs and thromboxanes [Citation48,Citation49], the acetylation of COX-2 leaves the cyclooxygenase with an altered lipoxygenase activity mediating the generation of 15(R)-hydroxyeicosatetraenoic acid (15 R-HETE) from arachidonic acid [Citation46,Citation50]. The constitutive expression of COX-1 is miscellaneous, yet its most important contribution is localized throughout the circulatory system, recurringly present in platelets, endothelial, and smooth muscle cells across the body [Citation51]. Therefore, the inhibition of COX-1 in an anucleate cell i.e. platelet terminate platelet function for the remainder of the cell’s life span (7–10 days) as no “new” enzyme can be transcribed [Citation52,Citation53]. Endothelial cells regain the ability to produce prostaglandins, primarily the vasodilator and platelet aggregation inhibitor, prostacyclin (PGI2), after de novo synthesis of COX-1 [Citation54]. Therefore, the level of prostacyclin in the blood would be greater than the platelet activator and vasoconstrictor, thromboxane A2 (TxA2), and would mediate a blood thinning effect. It has also been noted that aspirin cannot block adenosine diphosphate (ADP)-mediated α-granule release of the binding glycoproteins, fibrinogen, and von Willebrand Factor [Citation55]. This is suggestive that aspirin does not completely inactivate platelets but rather inhibits the synthesis of the most potent platelet activator and vasoconstrictor.
Endothelial cells also express the inducible cyclooxygenase, COX-2 [Citation51], which when acetylated by aspirin mediates a lipoxygenase reaction to convert arachidonic acid to 15 R-HETE. However, it has been seen that aspirin inhibits the expression of COX-2 [Citation56]. The availability of 15 R-HETE triggers the transcellular synthesis of 15-epi-lipoxin A4, a more potent 15 R-epimer of lipoxin A4, within leukocytes by 5-Lipoxygenase [Citation57,Citation58]. The 15-epi-lipoxin A4 mediates an anti-inflammatory response by abolishing the endothelial cell-leukocyte interaction, and it also has an antiplatelet effect by inhibiting thromboxane [Citation59]. Additionally, the nitric oxide-producing enzyme, Endothelial nitric oxide synthase (eNOS), found in endothelial cells and platelets, is acetylated by aspirin, resulting in an increase in enzyme activity [Citation60]. The available nitric oxide diffuses to the vascular smooth muscle and aids in the dilation of the vessel in a soluble Guanylate cyclase-cyclic guanosine monophosphate (cGMP) activation of Protein kinase G [Citation61]. Furthermore, nitric oxide has been found to attenuate the adherence and invasion of leukocytes to the endothelium [Citation62]. The increase in nitric oxide, 15-epi-lipoxin A4, and prostacyclin (PGI2), along with the decrease in thromboxane A2 and prostaglandins, mediate the antipyretic, analgesic, and anti-inflammatory effects of aspirin.
2.3. Detoxification of salicylic acid by the glycine conjugation pathway
Although approximately 10% of salicylates are excreted in the urine unchanged [Citation63], three different processes are involved in detoxification. The primary route is the conjugation with glycine to produce salicyluric acid, which accounts for 75 to 84% of the metabolites [Citation64,Citation65]. Additionally, salicylic acid can be conjugated to glucuronides through glucuronidation, forming ester and ether glucuronides. These metabolic pathways involve a range of uridine 5’-diphosphoglucuronosyltransferases (UGTs) enzymes. Oxidation by cytochrome P450 enzymes and non-enzymatic reactions to minor metabolites of salicylic acids, such as 2,3-dihydroxybenzoic acid and 2,5-dihydroxybenzoic acid, constitute the third process [Citation66,Citation67].
The excretion of salicyluric acid reaches a maximum and then stays constant until the entire dose has been excreted. Therefore, when a specific concentration of salicylate is exceeded, the amount of salicyluric acid excreted will not increase, i.e. the half-life of salicylate will rather increase. The bottleneck in the rate of excretion seems to lie at the rate of formation of salicyl-CoA rather than the rate of absorption [Citation65,Citation68]. This limit of excretion is reached at therapeutic doses of aspirin (360 mg) and contributes to the concentration-dependent nonlinear pharmacokinetics observed [Citation69].
Even though aspirin is one of the most widely used drugs, the literature on the detoxification of salicylic acid by the major glycine conjugation route is severely limited. Salicylic acid is detoxified via a two-step enzymatic process in the mitochondria of the liver (). The xenobiotic/medium chain fatty acid: CoA ligase (ACSM2B, EC 6.2.1.2) uses ATP to activate salicylic acid to salicyl-CoA releasing AMP and pyrophosphate [Citation70]. Glycine N-acyltransferase (GLYAT, EC 2.3.1.13) forms salicyluric acid and releases CoA by conjugating salicyl-CoA to glycine [Citation71].
There are conflicting reports on whether ACSM2B can effectively activate salicylic acid [Citation72–75]. This is because it is very difficult to purify a single ACSM enzyme from tissue and subcellular preparations [Citation76]. Previous studies used salicylic acid concentrations in the range (0.5 mM or higher) that would inhibit ACSM2B [Citation73]. Various NSAIDs have also been shown to inhibit the mouse and bovine ACSM2 [Citation77]. From the pharmacokinetic studies it seems that human ACSM2B is not inhibited by salicylate as hippurate is still formed in the presence of salicylate [Citation69,Citation78]. However, a subsequent study using adult livers showed that salicylic acid and diflunisal strongly inhibit the formation of hippuric acid, while ibuprofen was shown to be a weak inhibitor [Citation79]. To date, no ACSM has been identified that can preferentially activate salicylic acid to salicyl-CoA. GLYAT preferentially conjugates benzoyl-CoA to glycine [Citation71], and benzoate consumption can inhibit the excretion of salicyluric acid [Citation69]. This indicates that salicyl-CoA is not a suitable substrate for GLYAT.
2.4. Interindividual variation in aspirin metabolism
There is significant interindividual variation in the rate of glycine conjugation of aspirin [Citation80–86] with gender related differences (61% higher in males) [Citation81,Citation87,Citation88] and inter-ethnic variability in aspirin metabolism being reported [Citation80,Citation89–91]. Salicylic acid clearance was determined to be 41% higher in females taking oral contraceptives compared to a control female group [Citation90]. The observed variation can be influenced by the availability of the co-factors (ATP, CoA, and glycine), genetic variation in ACSM2B and GLYAT, expression level of ACSM2B and GLYAT, as well as diseases [Citation92–97].
2.4.1. Availability of the co-factors – glycine, CoA and ATP – can limit glycine conjugation
The availability of glycine may be a limiting factor in glycine conjugation of benzoate [Citation94,Citation98,Citation99]. It can influence the rate of detoxification as was demonstrated by the dose dependent increase in benzoylglycine (hippurate) formation after administration of glycine [Citation94]. Benzoylglycine is formed from benzoate and is the preferred substrate for detoxification by the glycine conjugation pathway. However, even though the formation of salicylate has been found to be capacity-limited at therapeutic doses [Citation69], oral administration of glycine does not increase the excretion of salicyluric acid. This indicates that glycine is not the limiting co-factor. However, the available amount of ATP for the reaction can be reduced by salicylate itself as it uncouples oxidation and phosphorylation [Citation65]. This then limits the rate of the reaction.
In a study investigating the effect of orally administered glycine on salicylate metabolism during aspirin overdose, it was found that the plasma glycine was lower in the overdose patients suggesting depletion of available glycine. Orally administered glycine increased the plasma glycine concentration while the fraction of total salicylate recovered as salicyluric acid remained the same. This supported previous studies, however, orally administered glycine did shorten the time it took to reach the maximum rate of salicyluric acid excretion [Citation100].
2.4.2. Genetic variation in ACSM2 and GLYAT can affect glycine conjugation
A study on twins suggested that there is a genetic component to the variation seen in the glycine conjugation rate with identical twins having a more similar conjugation rate [Citation97]. Both ACSM2B [Citation101] and GLYAT [Citation102] are highly conserved, but previous studies on GLYAT enzyme kinetics have shown that rare variants can have a deleterious effect on enzyme function. A correlation exists between haplotype frequency and relative enzyme activity with GLYAT haplotypes that have a high haplotype frequency also exhibiting a higher enzyme activity. A single nucleotide polymorphism (SNP) can reduce the enzyme activity of GLYAT to 12.3% of that of the wildtype enzyme [Citation102–104]. It is, therefore, important to investigate the effect of genetic variants (specifically haplotypes) on aspirin pharmacokinetics. At present, studies on these genes with relation to aspirin detoxification are very limited, and no variants in either of these genes have been associated with the pharmacokinetics of aspirin.
2.4.3. Age affects the expression level of GLYAT
Newborns have a very limited glycine conjugation capacity [Citation79,Citation105,Citation106], probably due to the low level of expression of GLYAT below 18 months of age [Citation107]. However, the level of expression of ACSM2 in children is not known which might contribute to the limited glycine conjugation ability.
The rate of glycine conjugation is also reduced in the elderly [Citation108,Citation109]. It has not been confirmed whether this might be due to a general decline in the detoxification ability of the liver [Citation110] as the expression level of ACSM2B and GLYAT have not been determined in older individuals.
Therefore, the level of expression of the genes in the glycine conjugation pathway can also affect the excretion rate of salicyluric acid.
2.4.4. The effect of diseases on the rate of glycine conjugation
The expression of ACSM2 significantly declines compared to normal conditions in kidney injury models such as acute kidney injury, partial unilateral ureteral obstruction, and chronic kidney diseases [Citation111]. This inhibition could stem from the dysregulation of mitochondrial β-oxidation by Sirtuin 5 (Sirt5), leading to reduced mitochondrial ATP production, alterations in the AMP/ATP ratio, and activation of 5’ AMP-activated protein kinase (AMPK) [Citation112]. Given ACSM2’s pivotal role in fatty acid metabolism and its association with kidney injury, changes in mitochondrial β-oxidation, under Sirt5 regulation, might modulate the availability of substrates or intermediates, thereby affecting ACSM2 function.
The rate of glycine conjugation is reduced in liver disease [Citation64,Citation113–115] obesity, Type 2 diabetes mellitus [Citation116,Citation117] and cancer [Citation118]. Downregulated expression is observed for both GLYAT [Citation118–120], and ACSM2B [Citation121,Citation122]. The low expression of GLYAT has been linked to poor prognosis and is inversely proportional to the stage and grade of cancer. Therefore, the entire pathway involving Coenzyme A activation and glycine conjugation is compromised in cancer cells. There might be a relationship between GLYAT activity, glycine availability, and cancer cell proliferation [Citation123,Citation124]. This is further elaborated on in the Expert Opinion section. No studies have been undertaken to indicate whether aspirin ingestion should be decreased in patients with these diseases.
3. Aspirin toxicity
Aspirin consumption is linked to adverse effects requiring medical care [Citation125] and deaths caused by adverse drug reactions [Citation126,Citation127]. The dose of aspirin ingested determines the level of toxicity with mild toxic effects associated with doses <125 mg/kg, moderate toxicity associated with doses >250 mg/kg, and severe toxicity associated with doses >500 mg/kg [Citation128].
Aspirin toxicity can occur at several points along the route of aspirin metabolism within the body i.e. i) delayed absorption [Citation129–131], ii) mechanism of action (decreased prothrombin formation, platelet adhesiveness, and platelet numbers [Citation132,Citation133]; uncoupling of oxidative phosphorylation [Citation134]), and iii) delayed/defective excretion of aspirin as its drug metabolite, salicyluric acid. This review will only focus on the latter.
In children, salicylate toxicity is sometimes caused by the accumulation of salicylate because the dose exceeds the rate at which it can be eliminated [Citation135]. An example of this was found with the chronic use of teething gel (containing the equivalent of 930 mg of aspirin) in a 17– month-old boy which caused metabolic derangement and liver impairment [Citation136]. The observed toxicity is probably due to the low expression level of GLYAT in children younger than 18 months. The half-life of salicylate elimination in children intoxicated with this drug ranges from 15 to 29 hours [Citation137].
If aspirin is given to children with viral infections, it might lead to the development of Reye’s syndrome. Individuals with Reye’s syndrome cannot metabolize fatty acids due to the depletion of free CoA and an accumulation of acyl-CoAs [Citation138–141]. It has also been shown that salicylate can inhibit the activity of β-oxidation [Citation142]. To prevent the development of Reye’s syndrome, paracetamol and ibuprofen are recommended for use in children [Citation143,Citation144]. These drugs are metabolized into inactive hydroxylated or glucuronidated products and are not detoxified by the glycine conjugation pathway [Citation144]. This might indicate that the impaired excretion of salicyluric acid by glycine conjugation is contributing to the adverse effects seen in Reye’s syndrome. This will be elaborated on in the expert opinion section.
Liver damage has been reported because of salicylate rechallenge therapy for rheumatoid arthritis, detectable with serum levels of salicylate as low as 25 mg per 100 ml [Citation145]. Moreover, the association between aspirin and hepatotoxicity has also been established in pediatric populations with the same condition of rheumatoid arthritis or even rheumatic fever [Citation146]. The mechanism of salicylate-induced liver damage involves oxidative stress, mitochondrial dysfunction, and lipid peroxidation. It commences with mitochondrial dysfunction, resulting in reduced ATP levels, ultimately culminating in significant liver cell injury via lipid peroxidation [Citation147].
4. Conclusion
The available data on the pharmacokinetics of aspirin by the major detoxification route, namely glycine conjugation, is very limited. The first enzyme involved in the pathway, namely ACSM2B, can form salicyl-CoA at a low efficiency (<1%); however, studies on bovine ACSM2B have shown an inhibitory effect of salicylate. The ACSM2 ligases and their role in xenobiotic metabolism is understudied, particularly the effect of genetic variation on enzyme activity; the identity of factors affecting ACSM ligase activity in vivo, the role of inhibitors on the conjugation ability of ACSM2B and the association between ACSM2B and detrimental health outcomes.
Glycine conjugation is saturable and can, therefore, only detoxify a limited range and concentration of substrates. The rate of glycine conjugation can be affected by the formation of the acyl-CoA (ACSM2B) and/or the conjugation of the acyl-CoA to glycine (GLYAT).
The rate of glycine conjugation can be affected by the available co-factors (ATP, CoA, and glycine); genetic variants in the ACSM2B and GLYAT genes; different expression levels of ACSM2B and GLYAT and disease states. Studies have shown that although ATP can limit glycine conjugation of salicylate, glycine concentration is not a limiting factor. Like ACSM2, GLYAT is highly conserved, and alleles predicted to have a deleterious effect on enzyme function are found at low frequencies. Genetic and functional variation in genes responsible for the detoxification of xenobiotics, such as aspirin, can have complex consequences, depending, for example, on whether enzyme structure or expression is affected. Despite the impact of genetic variation on pharmacokinetics, no studies to date have analyzed the effect of genetic variants on the enzyme activity of the enzymes involved in the glycine conjugation pathway. This lack of knowledge limits the clinical application in personalized medicine. Glycine conjugation ability is also affected by age, with both newborns and the elderly having a reduced glycine conjugation rate, while diseases such as cancer, diabetes, and liver diseases can negatively affect glycine conjugation.
Toxicity can manifest at various points along aspirin metabolism, including delayed absorption, altered mechanisms of action, and defective excretion of salicylate metabolites. Salicylate accumulation in children can lead to metabolic derangement and liver impairment, as seen in cases of chronic use of salicylate-containing products. Additionally, aspirin ingestion during viral infections in children has been linked to Reye’s syndrome, characterized by impaired fatty acid metabolism and mitochondrial dysfunction in the liver.
A 2009 paper stated: ‘Overall information indicates that pharmacogenomics knowledge in aspirin intolerance is presently in its infancy’ [Citation148]. After 15 years, not much has been added to this knowledge. We need to thoroughly understand detoxification systems to understand disease pathology and the mechanisms behind xenobiotic toxicity.
5. Expert opinion
5.1. Limitations in the pharmacokinetic studies of aspirin
5.1.1. The glycine conjugation rate is evaluated as a ’one enzyme system,’ ignoring the formation of the intermediate salicyl-CoA
In general, pharmacokinetic studies of aspirin metabolism measure the plasma concentrations of salicylate [Citation90,Citation149] and the end product of glycine conjugation [Citation64,Citation92,Citation97,Citation109,Citation150–153], namely salicyluric acid. Therefore, the pharmacokinetics of aspirin refers to the rate of the entire pathway when, in fact, salicylate first needs to be activated to a salicyl-CoA before it can be conjugated to glycine to form the glycine conjugate (). Previous studies have shown that the activation step occurs at a much slower rate than the glycine conjugation rate [Citation69,Citation74,Citation154,Citation155]. This is because salicylate is not the preferred substrate for ACSM2B [Citation70]. Even though ACSM2B plays an important role in the detoxification of salicylate no further research has been done after the preliminary substrate selectivity characterization of ACSM2B [Citation70,Citation156].
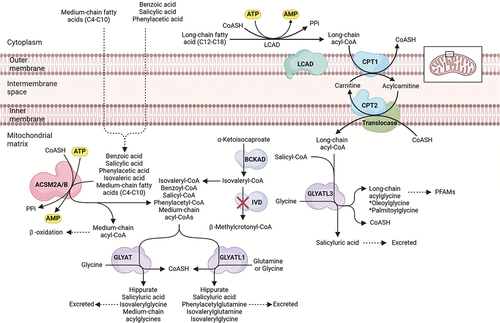
Figure 2. Paralogues of GLYAT and ACSM2B as possible detoxification enzymes of salicylic acid.
Measuring the rate of the salicyl-CoA formation, however, is challenging as this needs to be done using liver biopsies. Liver biopsies are very invasive, which prevents sampling in humans to determine the rate of acyl-CoA formation. An alternative would be to evaluate the enzyme kinetics of both ACSM2B and GLYAT using a model in which these enzymes can be expressed recombinantly. A model system will also enable the evaluation of the effect of genetic variants on the detoxification rate of glycine conjugation and further our understanding of the contribution of impaired glycine conjugation to disease pathologies. No studies have been published using an in vitro recombinantly expressed enzyme to evaluate the substrate selectivity for aspirin.
5.1.2. The interaction/competition of multiple substrates for glycine conjugation is unknown
The glycine conjugation pathway is often oversimplified, as it’s not just limited to detoxifying benzoate into hippurate. This pathway plays a crucial role in metabolizing and detoxifying various substrates, including natural compounds from food like salicylate, dietary polyphenols, and medium-chain fatty acids (MCFAs), as well as xenobiotics and metabolites from organic acidemias [Citation98,Citation101,Citation157,Citation158] (). Xenobiotics, such as salicylate and MCFAs, are first activated to acyl-CoA. Salicyl-CoA is conjugated to glycine, while MCFA acyl-CoAs are further processed by β-oxidation [Citation159]. The medium-chain acyl-CoAs are seen as glycine conjugates in urinary excretion of individuals with a Medium-chain acyl-CoA dehydrogenase deficiency (MCADD) [Citation160]. Benzoyl-CoA can also be produced by gut microorganisms from dietary polyphenols [Citation161].
The glycine conjugation pathway evolved to metabolize endogenous substrates (e.g. MCFAs), and therefore, xenobiotics such as salicylate add an additional detoxification load. It has not been determined if these multiple substrates have a deleterious effect on the rate of glycine conjugation, but it is known that the pathway can be saturated [Citation64,Citation92,Citation93,Citation97,Citation109,Citation162,Citation163]. This probably indicates that the preferred substrate will be detoxified at a rate that does not exceed a threshold concentration and that less preferred substrates will accumulate.
It is difficult comparing in vitro determined values with the in vivo determined pharmacokinetics. The greatest limitation of previous pharmacokinetic studies is that the diet (e.g. amount of salicylates ingested through plant products) of the test subjects was not strictly controlled. Furthermore, benzoate, a preservative found in many food items, can outcompete salicylate for glycine conjugation, increasing the half-life of salicylate. Microorganisms in the gut produce benzoyl-CoA from dietary polyphenols. This contributes a very large load that needs to be metabolized by the glycine conjugation pathway [Citation161]. The diet of the test subjects should, therefore, be controlled in order to only determine the conjugation rate of the salicylate dose given in the experiment. Salicylate at pharmacological doses can also inhibit ACSM2B and will affect the metabolism of medium-chain fatty acids and xenobiotics [Citation73].
5.1.3. Paralogous of ACSM and GLYAT might also contribute to the detoxification of salicylate
ACSM2A is a xenobiotic/medium chain fatty acid: CoA ligase also present in the liver and has a nucleotide identity of 98.8% and an amino acid identity of 97.6% to ACSM2B [Citation164]. ACSM2A has not been characterized enzymatically, but it can be hypothesized that due to the high nucleotide and amino acid similarity of ACSM2A to ACSM2B [Citation164], these two enzymes perform the same function. Gene duplication events, as is the case for ACSM2A and ACSM2B, increase the fitness of an organism [Citation164–167]. It is possible that the duplication of ACSM2 resulted from a need to metabolize different substrates more efficiently, i.e. substrates from food (benzoate and salicylate) as well as MCFAs. It is important to characterize the substrate specificity differences between ACSM2A and ACSM2B to understand the contribution of each enzyme to the detoxification rate.
GLYATL1, GLYATL1B, GLYATL2, and GLYATL3 are paralogues of GLYAT (ensemble.org). The paralogues expressed in the liver and kidney (GLYAT, GLYATL1, and GLYATL3) [Citation168] might play a role in the detoxification of xenobiotics. There is currently no evidence that GLYATL3 plays a role in xenobiotic metabolism. Previous studies have shown that GLYAT can detoxify benzoyl-CoA, salicyl-CoA, and isovaleryl-CoA, while GLYATL1 can detoxify phenylacetyl-CoA, benzoyl-CoA, and isovaleryl-CoA () [Citation169–173]. Not all studies agree on the substrate specificity of GLYAT and GLYATL1 [Citation174–176]. This might be because partially purified enzymes from the liver were used to determine the substrate selectivity [Citation71,Citation99,Citation177]. GLYAT (296 aa) and GLYATL1 (302 aa) are very similar in size, and older studies were not able to distinguish between these enzymes. Although a recombinantly expressed purified GLYAT enzyme was characterized recently using the preferred substrates benzoyl-CoA and glycine [Citation103,Citation104,Citation118,Citation178–180], nothing is currently known regarding the substrate selectivity of GLYAT, GLYATL1 and GLYATL3 for salicyl-CoA.
5.1.4. The effect of genetic variants on the conjugation rate has not been studied
Both the ACSM2A and ACSM2B genes are highly conserved, and the majority of the haplotypes are rare [Citation101]. It is not known whether these rare haplotypes negatively affect the enzyme activity as well as what the effect of the haplotype variants is on aspirin metabolism. Previous pharmacokinetic studies did not determine the variants in the ACSM2B and GLYAT genes, and therefore, no correlation can be made between the conjugation rate and genetic variants. This is puzzling as several studies investigating the effect of variants on CYP450 detoxification have been published, e.g [Citation181,Citation182]. It is unclear why this avenue of research is not being pursued for the glycine conjugation pathway.
Previous enzyme characterization studies did not determine the sequence of the enzymes that were characterized. It has been shown in vitro that variants within GLYAT can affect enzyme activity [Citation103,Citation104,Citation179] and the level of cooperative binding of substrates [Citation104]. In vivo, it was shown that the level of excretion of p-aminobenzoic acid varied significantly (16–56%, hippurate ratio) even in individuals who had the same GLYAT variant [Citation183]. Future studies should use the reference haplotype, namely S156 GLYAT [Citation102–104,Citation178], to compare the effect of variants on enzyme activity.
Individuals with variants that have a deleterious effect on the glycine conjugation ability can experience adverse health effects if a diet high in salicylates, benzoates and MCFAs are consumed.
Therefore, to more thoroughly understand the pharmacokinetics of aspirin several gaps in the current research still needs to be addressed namely: i) What is the role of the competition of various substrates on the detoxification rate of aspirin; ii) What role do the paralogous of ACSM2 and GLYAT play in detoxification; and iii) What is the effect of genetic variants on the detoxification ability of individuals? These are all basic research questions that can easily be answered using available research methodologies. Advances in this research could significantly impact real-world outcomes, such as improving diagnosis and treatment guidelines for aspirin toxicity. Understanding the contributions of the individual enzymes involved in the glycine conjugation pathway could lead to personalized treatment strategies, reducing toxicity risks in susceptible individuals. However, implementation in clinical practice is currently hindered by the lack of comprehensive models and the need for further research to validate findings.
5.2. Impaired glycine conjugation might contribute to aspirin toxicity
Previous studies have shown that impaired detoxification systems can contribute to negative reactions to pharmaceutical drugs as well as the symptoms seen in multifactorial diseases [Citation184,Citation185]. Aspirin toxicity is still observed in many patients, but the mechanism related to the toxicity is unknown. There are a few possibilities whereby an impaired glycine conjugation system can contribute to aspirin toxicity.
5.2.1. Toxicity due to an inability to activate salicylate
The first step in the glycine conjugation pathway is the activation of salicylate to a salicyl-CoA by ACSM2B. If this step can’t occur or activation of salicylate is delayed it can lead to toxicity. This is illustrated by the example of aspirin toxicity in felids. Aspirin is used in cats for acute pain or more chronically as an anti-thrombotic [Citation186]. Cats injected with 44 mg/kg sodium salicylate exhibited a plasma half-life of 37.6 hours [Citation187] and dosages comparable to that used for humans are lethal in cats [Citation186]. Felids cannot activate salicylate to salicyl-CoA as they do not have an orthologue for ACSM2B. Cats do have an orthologue for human ACSM1 that has a very low substrate selectivity for salicylate (<0.004%) [Citation70]. Therefore, felids can form salicyluric acid but at a very low rate, resulting in the accumulation of salicylate [Citation187]. This might explain the observed toxicity of salicylate in cats although further research is needed. The same might be true for humans as it has not been shown that ACSM2 can effectively activate salicylate to salicyl-CoA [Citation70], possibly leading to toxicity.
In Reye’s syndrome patients, virus infections can downregulate the expression of CYP450 enzymes [Citation188] by affecting Kupffer cells and increasing proinflammatory cytokine release [Citation189,Citation190]. It is not known if the enzymes of glycine conjugation (ACSM2B and GLYAT) are also downregulated in these patients. This will explain the toxicity of salicylate as it cannot be detoxified during a virus infection.
In hepatocellular carcinoma the glycine conjugation pathway and its paralogues are downregulated i.e. GLYAT, GLYATL1, ACSM2A and ACSM2B [Citation118–120,Citation191–194]. This means that in these cells, salicylates cannot be detoxified leading to possible toxicity.
5.2.2. Substrate competition can lead to toxicity
Various substrates and co-factors are involved in the glycine conjugation pathway (). Substrate competition and a deficiency of the co-factors can lead to toxicity. This is confirmed by the observation that the glycine conjugation pathway can be saturated, and as a result, the half-life of salicylate elimination is increased with increasing dose [Citation109,Citation195]. Furthermore, the formation of salicyluric acid reaches a maximum rate at higher doses of aspirin (1 g or more) [Citation153].
Humans are consuming and increasing concentration of substrates that need to be detoxified by the glycine conjugation pathway. These include natural sources (e.g. salicylate found in berries [Citation158]), polyphenols [Citation196], and xenobiotics [Citation197,Citation198]. Therefore, the ability of an individual to detoxify salicylate will also depend on the type of diet they follow, as a diet high in preservatives (benzoate) will reduce the amount of salicyluric acid that is formed. Salicyluric acid formation can be inhibited for long periods if even small amounts of benzoate are repeatedly ingested [Citation199]. This can cause toxicity and liver damage due to the accumulation of salicylate, specifically in young children [Citation146,Citation200].
5.2.3. Sequestration of CoA and inhibition of ACSM2B
The salicyl-CoA intermediate can lead to toxicity if it is not effectively conjugated to glycine by GLYAT. This is because the salicyl-CoA sequesters CoA [Citation201], which means that it is no longer available for other cellular processes. Salicyl-CoA also inhibits ACSM2B, which means that ACSM2B can no longer activate MCFAs for use during β-oxidation.
It seems that the conjugation of salicyl-CoA to glycine does not function effectively in Reye’s patients. This is evident from the fact that paracetamol (acetaminophen) and ibuprofen can be detoxified by forming glucuronidated products [Citation143,Citation144], while salicylate (detoxified by glycine conjugation) seems to have a toxic effect. Monitoring the concentration of salicyluric acid in these patients is crucial as reduced levels of salicyluric acid may indicate impaired detoxification of salicylate, leading to the accumulation of toxic intermediates like salicyl-CoA. It was further found that in some Reye’s patients, a metabolic disturbance is exacerbated by a virus infection because these patients have an unidentified inborn error in metabolism [Citation202]. This can cause an increase in toxic metabolites (additional acyl-CoA’s) that need to be detoxified by glycine conjugation, resulting in an overload of substrates for the detoxification pathway. An example is isovaleric acidemia where the toxic metabolite isovaleryl-CoA is also detoxified by GLYAT [Citation157].
Further research holds great potential, with the goal of elucidating the complete pharmacokinetic profile of aspirin and its detoxification pathways. This knowledge could lead to safer aspirin formulations and improved therapeutic protocols. In 5 to 10 years, it is anticipated that standard procedures for evaluating aspirin pharmacokinetics will evolve to include more sophisticated models and comprehensive genetic screenings. This evolution will likely result in more accurate predictions of individual responses to aspirin, enhancing patient safety and treatment efficacy.
5.3. Hypothesis: mechanism for the anticancer activity of aspirin
The metabolism of salicylic acid in cancer patients are severely limited [Citation153] and it has been shown that the GLYAT gene is transcriptionally downregulated in hepatocellular carcinoma [Citation118,Citation203]. Cancer cells have a high demand for glycine and serine for growth [Citation204]. The inhibition of glycine uptake or biosynthesis can impair cancer cell growth [Citation123]. Therefore, the GLYAT gene is downregulated in cancer cells to prevent the enzyme from depleting hepatic glycine, which would inhibit the rapid proliferation of cancer cells [Citation118,Citation123,Citation124,Citation205,Citation206]. It is possible that aspirin may exert its anticarcinogenic properties via the induction of expression of GLYAT which then leads to the depletion of hepatic glycine and inhibition of cancer cell growth. It has been shown that the long-term use of salicylate may increase the production of salyciluric acid by induction [Citation97,Citation207,Citation208].
Article highlights
Available aspirin pharmacokinetics studies quantified a 2-step enzyme reaction (i.e. activation by the ligase and conjugation by the transferase) effectively as one enzyme reaction.
Therefore, the contribution of each of the enzymes to the kinetics is unknown.
Furthermore, paralogues of these enzymes might also contribute to detoxification of salicylate.
Glycine conjugation, essential in aspirin and drug metabolism, can be influenced by substrate competition and co-factor deficiencies, potentially leading to toxicity.
The adverse effects seen in Reye’s syndrome, associated with aspirin use in children with viral infections, might result from impaired salicyluric acid excretion via glycine conjugation.
Limited data on aspirin pharmacokinetics through glycine conjugation underscores the need for further research on the roles of ACSM2B and GLYAT enzymes in xenobiotic metabolism and disease pathology.
Abbreviation
| 15 R-HETE | = | 15(R)-hydroxyeicosatetraenoic acid |
| ACSM2A | = | Xenobiotic/medium chain fatty acid: CoA ligase |
| ACSM2B | = | Xenobiotic/medium chain fatty acid: CoA ligase |
| ADP | = | Adenosine diphosphate |
| AMP | = | Adenosine monophosphate |
| AMPK | = | 5’ AMP-activated protein kinase |
| ATP | = | Adenosine triphosphate |
| BCKAD | = | Branched-chain α-ketoacid dehydrogenase |
| cGMP | = | Cyclic guanosine monophosphate |
| CoASH or CoA | = | Coenzyme A |
| COX-1 | = | Cyclooxygenase-1 |
| COX-2 | = | Cyclooxygenase-2 |
| CPT1 | = | Carnitine O-palmitoyltransferase 1 |
| CPT2 | = | Carnitine O-palmitoyltransferase 2 |
| eNOS | = | Endothelial nitric oxide synthase |
| GLYAT | = | Glycine N-acyltransferase |
| GLYATL1 | = | Glutamine N-acyltransferase-like 1 |
| GLYATL3 | = | Glycine N-acyltransferase-like 3 |
| LCAD | = | Long-chain acyl-CoA dehydrogenase |
| IVA | = | Isovaleric academia |
| IVD | = | Isovaleryl-CoA dehydrogenase |
| MCADD | = | Medium-chain acyl-CoA dehydrogenase deficiency |
| MCFAs | = | Medium-chain fatty acids |
| NSAID(s) | = | Non-steroidal anti-inflammatory drug(s) |
| PFAMs | = | Primary fatty acid amides |
| PGG2 | = | Prostaglandin G2 |
| PGs | = | Prostaglandins |
| PGI2 | = | Prostacyclin |
| PPi | = | Pyrophosphate |
| Sirt5 | = | Sirtuin 5 |
| SNP | = | Single nucleotide polymorphism |
| T2D | = | Type 2 diabetes |
| Translocase | = | Carnitine/acylcarnitine translocase |
| TxA2 | = | Thromboxane A2 |
| UGTs | = | Uridine 5’-diphosphoglucuronosyltransferases |
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or material discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or mending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Wells J. Poppy juice and willow bark: advances in their use for the 21st century. The pain web for health professionals [WWW document]. 2003 [cited 2004 Aug]. Available from: http://wwwthepainwebcom/doclib/topics/000009htm
- Nunn JF. Ancient Egyptian medicine. Norman, OK: Norman University of Oklahoma Press; 1996.
- Murder JM, Campbell WC. Magic, and medicine. J Hist Med Allied Sci. 1994;49(1):147–149. doi: 10.1093/jhmas/49.1.147
- Jack DB. One hundred years of aspirin. The Lancet. 1997;350(9075):437–439. doi: 10.1016/S0140-6736(97)07087-6
- Gross M, Greenberg LA, Haggard HW. The Salicylates, a critical bibliographic review. Anesth & Analg. 1948;27(6):137–138.
- Riddle JM. Historical data as an aid in pharmaceutical prospecting and drug safety determination. J Alternative Complementary Med. 1998;5(2):195–201. doi: 10.1089/acm.1999.5.195
- Leroux H. Discovery of salicine. J Chim Med. 1830;6:340–432.
- Rainsford KD. Aspirin and the Salicylates. Amsterdam: Elsevier; 2013.
- Sneader W. The discovery of aspirin. Pharm J. 1997;259(6964):614–617.
- Mahdi J, Mahdi A, Mahdi A, et al. The historical analysis of aspirin discovery, its relation to the willow tree and antiproliferative and anticancer potential. Cell Prolif. 2006;39(2):147–155. doi: 10.1111/j.1365-2184.2006.00377.x
- Childs PE. The centenary of aspirin: wonder drug of the twentieth century. Chem Action. 1999;59:43–45.
- GarcíaRodríguez LA, Huerta-Alvarez C. Reduced incidence of colorectal adenoma among long-term users of nonsteroidal antiinflammatory drugs: a pooled analysis of published studies and a new population-based study. Epidemiology. 2000;11(4):376–381. doi: 10.1097/00001648-200007000-00003
- Rao CV, Reddy BS. NSAIDs and chemoprevention. Curr Cancer Drug Targets. 2004;4(1):29–42. doi: 10.2174/1568009043481632
- Zha S, Yegnasubramanian V, Nelson WG, et al. Cyclooxygenases in cancer: progress and perspective. Cancer Lett. 2004;215(1):1–20. doi: 10.1016/j.canlet.2004.06.014
- Voelker R. USPSTF: low-dose aspirin may help reduce risk of preeclampsia. JAMA. 2014;311(20):2055–2055. doi: 10.1001/jama.2014.5390
- Smith BK, Ford RJ, Desjardins EM, et al. Salsalate (salicylate) uncouples mitochondria, improves glucose homeostasis, and reduces liver lipids independent of AMPK-β1. Diabetes. 2016;65(11):3352–3361. doi: 10.2337/db16-0564
- Bellosillo B, Piqué M, Barragán M, et al. Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells. Blood, The J Am Soc Hematol. 1998;92(4):1406–1414. doi: 10.1182/blood.V92.4.1406.416k17_1406_1414
- Din F, Dunlop M, Stark L. Evidence for colorectal cancer cell specificity of aspirin effects on NFκB signalling and apoptosis. Br J Cancer. 2004;91(2):381–388. doi: 10.1038/sj.bjc.6601913
- Wong B, Zhu G, Lam S. Aspirin induced apoptosis in gastric cancer cells. Biomed Pharmacother. 1999;53(7):315–318. doi: 10.1016/S0753-3322(00)88503-0
- Klampfer L, Cammenga J, Wisniewski H-G, et al. Sodium salicylate activates caspases and induces apoptosis of myeloid leukemia cell lines. Blood, J Am Soc Hematol. 1999;93(7):2386–2394. doi: 10.1182/blood.V93.7.2386
- Marra DE, Liao JK. Salicylates and vascular smooth muscle cell proliferation: molecular mechanisms for cell cycle arrest. Trends Cardiovasc Med. 2001;11(8):339–344. doi: 10.1016/S1050-1738(01)00133-5
- Brooks G, Yu X-M, Wang Y, et al. Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit vascular smooth muscle cell proliferation via differential effects on the cell cycle. J Pharm Pharmacol. 2003;55(4):519–526. doi: 10.1211/002235702775
- Shiff SJ, Koutsos MI, Qiao L, et al. Nonsteroidal antiinflammatory drugs inhibit the proliferation of colon adenocarcinoma cells: effects on cell cycle and apoptosis. Exp Cell Res. 1996;222(1):179–188. doi: 10.1006/excr.1996.0023
- von Weiss JF, Lever WF. Percutaneous salicylic acid intoxication in psoriasis. Arch Dermatol. 1964;90(6):614–619. doi: 10.1001/archderm.1964.01600060080013
- Myers EN, Bernstein JM. Salicylate ototoxicity: a clinical and experimental study. Archiv Otolaryngology - Head And Neck Surg. 1965;82(5):483–493. doi: 10.1001/archotol.1965.00760010485006
- Myers EN, Bernstein JM, Fostiropolous G. Salicylate ototoxicity: a clinical study. N Engl J Med. 1965;273(11):587–590. doi: 10.1056/NEJM196509092731104
- Boettcher FA, Salvi RJ. Salicylate ototoxicity: review and synthesis. Am J Otolaryngol. 1991;12(1):33–47. doi: 10.1016/0196-0709(91)90071-M
- Pearlman BL, Gambhir R. Salicylate intoxication: a clinical review. Postgrad Med. 2009 Jul 01;121(4):162–168. doi: 10.3810/pgm.2009.07.2041
- Kowalski ML, Asero R, Bavbek S, et al. Classification and practical approach to the diagnosis and management of hypersensitivity to nonsteroidal anti‐inflammatory drugs. Allergy. 2013;68(10):1219–1232. doi: 10.1111/all.12260
- Raithel M, Baenkler H, Naegel A, et al. Significance of salicylate intolerance in diseases of the lower gastrointestinal tract. J Physiol Pharmacol. 2005;56 Suppl 5:89–102.
- Levine MS, Verstandig A, Laufer I. Serpiginous gastric erosions caused by aspirin and other nonsteroidal antiinflammatory drugs. Am J Roentgenol. 1986;146(1):31–34. doi: 10.2214/ajr.146.1.31
- Trost LC, Lemasters JJ. The mitochondrial permeability transition: a new pathophysiological mechanism for Reye’s syndrome and toxic liver injury. J Pharmacol Exp Ther. 1996;278(3):1000–1005.
- Lewis HD Jr, Davis JW, Archibald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in men with unstable angina: results of a veterans administration cooperative study. N Engl J Med. 1983;309(7):396–403. doi: 10.1056/NEJM198308183090703
- Levine M, Wong D, Brown DF, et al. Chest pain and arthritis. J Emerg Med. 2005;29(1):91–95. doi: 10.1016/j.jemermed.2005.04.001
- Malakar S, Gibson PR, Barrett JS, et al. Naturally occurring dietary salicylates: a closer look at common Australian foods. J Food Composition Anal. 2017;57:31–39. doi: 10.1016/j.jfca.2016.12.008
- Kęszycka PK, Szkop M, Gajewska D. Overall content of salicylic acid and salicylates in food available on the European market. J Agric Food Chem. 2017;65(50):11085–11091. doi: 10.1021/acs.jafc.7b04313
- Lawrence J, Peter R, Baxter G, et al. Urinary excretion of salicyluric and salicylic acids by non-vegetarians, vegetarians, and patients taking low dose aspirin. J Clin Pathol. 2003;56(9):651. doi: 10.1136/jcp.56.9.651
- Cashman J. Routes of administration. Clin Pain Manag Second Ed: Acute Pain. 2008;2:201–216.
- Feldman M, Cryer B. Aspirin absorption rates and platelet inhibition times with 325-mg buffered aspirin tablets (chewed or swallowed intact) and with buffered aspirin solution. Am J Cardiol. 1999;84(4):404–409. doi: 10.1016/S0002-9149(99)00324-0
- Needs CJ, Brooks PM. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet. 1985;10(2):164–177. doi: 10.2165/00003088-198510020-00004
- Hogben CAM, Schanker LS, Tocco DJ, et al. Absorption of drugs from the stomach. II. The human. J Pharmacol Exp Ther. 1957;120(4):540–545.
- Schanker LS, Shore PA, Brodie BB, et al. Absorption of drugs from the stomach I. The Rat J Pharmacol Exp Ther. 1957;120(4):528–539.
- Schanker LS, Tocco DJ, Brodie BB, et al. Absorption of drugs from the rat small intestine. J Pharmacol Exp Ther. 1958;123(1):81–88.
- Hollander D, Dadufalza VD, Fairchild PA. Intestinal absorption of aspirin: influence of pH, taurocholate, ascorbate, and ethanol. J Lab And Clin Med. 1981;98(4):591–598.
- Meshkinpour H, Hollander D, Harmon D. Colonic absorption of acetylsalicylic acid in the rat. Gener Pharmacol. 1984;15(1):55–58. doi: 10.1016/0306-3623(84)90081-8
- Lecomte M, Laneuville O, Ji C, et al. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J Biol Chem. 1994;269(18):13207–13215. doi: 10.1016/S0021-9258(17)36820-5
- Roth GJ, Stanford N, Majerus PW. Acetylation of prostaglandin synthase by aspirin. Proc Natl Acad Sci USA. 1975;72(8):3073–3076. doi: 10.1073/pnas.72.8.3073
- Vane J, Flower R, Salmon J. Inhibitors of arachidonic acid metabolism, with especial reference to the aspirin-like drugs. Prostaglandins Relat Lipids. 1982;2:21–45.
- Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72(8):2994–2998. doi: 10.1073/pnas.72.8.2994
- Mancini JA, O’Neill GP, Bayly C, et al. Mutation of serine‐516 in human prostaglandin G/H synthase‐2 to methionine or aspirin acetylation of this residue stimulates 15‐R‐HETE synthesis. FEBS Lett. 1994;342(1):33–37. doi: 10.1016/0014-5793(94)80579-2
- Zidar N, Odar K, Glavac D, et al. Cyclooxygenase in normal human tissues–is COX‐1 really a constitutive isoform, and COX‐2 an inducible isoform? J Cell Mol Med. 2009;13(9b):3753–3763. doi: 10.1111/j.1582-4934.2008.00430.x
- Patrono C, Ciabattoni G, Patrignani P, et al. Clinical pharmacology of platelet cyclooxygenase inhibition. Circulation. 1985;72(6):1177–1184. doi: 10.1161/01.CIR.72.6.1177
- Burch JW, Stanford N, Majerus PW. Inhibition of platelet prostaglandin synthetase by oral aspirin. J Clin Invest. 1978;61(2):314–319. doi: 10.1172/JCI108941
- Jaffe EA, Weksler BB. Recovery of endothelial cell prostacyclin production after inhibition by low doses of aspirin. J Clin Invest. 1979;63(3):532–535. doi: 10.1172/JCI109332
- Rinder CS, Student LA, Bonan JL, et al. Aspirin does not inhibit adenosine diphosphate-induced platelet alpha-granule release. 1993.
- Xu X-M, Sansores-Garcia L, Chen X-M, et al. Suppression of inducible cyclooxygenase 2 gene transcription by aspirin and sodium salicylate. Proc Natl Acad Sci USA. 1999;96(9):5292–5297. doi: 10.1073/pnas.96.9.5292
- Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA. 1995;92(21):9475–9479. doi: 10.1073/pnas.92.21.9475
- Paul-Clark MJ, Van Cao T, Moradi-Bidhendi N, et al. 15-epi-lipoxin A4–mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J Exp Med. 2004;200(1):69–78. doi: 10.1084/jem.20040566
- Chiang N, Bermudez EA, Ridker PM, et al. Aspirin triggers antiinflammatory 15-epi-lipoxin A4 and inhibits thromboxane in a randomized human trial. Proc Natl Acad Sci USA. 2004;101(42):15178–15183. doi: 10.1073/pnas.0405445101
- Taubert D, Berkels R, Grosser N, et al. Aspirin induces nitric oxide release from vascular endothelium: a novel mechanism of action. Br J Pharmacol. 2004;143(1):159–165. doi: 10.1038/sj.bjp.0705907
- Francis SH, Busch JL, Corbin JD, et al. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62(3):525–563. doi: 10.1124/pr.110.002907
- De Caterina R, Libby P, Peng H-B, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96(1):60–68. doi: 10.1172/JCI118074
- Bardal SK, Waechter JE, Martin DS. Chapter 2 - Pharmacokinetics. Elsevier Health Sci. 2011.
- Dorne JLCM, Walton K, Renwick AG. Human variability for metabolic pathways with limited data (CYP2A6, CYP2C9, CYP2E1, ADH, esterases, glycine and sulphate conjugation). Food Chem Toxicol. 2004;42(3):397–421. doi: 10.1016/j.fct.2003.10.003
- Bedford C, Cummings AJ, Martin BK. A kinetic study of the elimination of the salicylate in MAN. Br J Pharmacol Chemother. 1965 Apr;24(2):418–431. doi: 10.1111/j.1476-5381.1965.tb01729.x
- Heavner JE. Pharmacology of analgesics. Anesthesia and analgesia in laboratory animals. San Diego, CA: Elsevier; 1997. p. 43–56.
- Alabi QK, Adeyemi WJ. Vernonia amygdalina (Del) as an antioxidant, aspirin toxicity, and oxidative stress. Toxicology: Elsevier; 2021. p. 491–504.
- Gibson T, Zaphiropoulos G, Grove J, et al. Kinetics of salicylate metabolism. Br J Clin Pharmacol. 1975;2(3):233–238. doi: 10.1111/j.1365-2125.1975.tb01581.x
- Levy G. Pharmacokinetics of salicylate in man. Drug Metab Rev. 1979;9(1):3–19. doi: 10.3109/03602537909046431
- Vessey DA, Kelley M, Warren RS. Characterization of the CoA ligases of human liver mitochondria catalyzing the activation of short- and medium-chain fatty acids and xenobiotic carboxylic acids. Biochim et Biophys Acta (BBA) - Gener Subj. [1999 Aug 5];1428(2–3):455–462. doi: 10.1016/S0304-4165(99)00088-4
- Kelley M, Vessey D. Characterization of the acyl-CoA: amino acid N-acyltransferases from primate liver mitochondria. J Biochem Toxicol. 1994;9(3):153–158. doi: 10.1002/jbt.2570090307
- Killenberg PG, Davidson ED, Webster LT Jr. Evidence for a medium-chain fatty acid: coenzyme a ligase (adenosine monophosphate) that activates salicylate. Mol Pharmacol. 1971 May;7(3):260–268.
- Vessey DA, Hu J, Kelley M. Interaction of salicylate and ibuprofen with the carboxylic acid: CoA ligases from bovine liver mitochondria. J Biochem Toxicol. 1996;11(2):73–78. doi: 10.1002/(SICI)1522-7146(1996)11:2<73:AID-JBT4>3.0.CO;2-R
- Vessey DA, Hu J. Isolation from bovine liver mitochondria and characterization of three distinct carboxylic acid: CoA ligases with activity toward xenobiotics. J Biochem Toxicol. 1995;10(6):329–337. doi: 10.1002/jbt.2570100608
- Kasuya F, Igarashi K, Fukui M, et al. Purification and characterization of a medium chain acyl-coenzyme a synthetase. Drug metabolism and disposition. Drug Metab Dispos. 1996;24(8):879–883.
- van der Sluis R, Knights KM. Enzymology of mitochondrial amino acid conjugation reactions. 2023.
- Kasuya F, Hiasa M, Kawai Y, et al. Inhibitory effect of quinolone antimicrobial and nonsteroidal anti-inflammatory drugs on a medium chain acyl-CoA synthetase. Biochem Pharmacol. 2001;62(3):363–367. doi: 10.1016/S0006-2952(01)00667-0
- Amsel LP, Levy G. Drug biotransformation interactions in man II: a pharmacokinetic study of the simultaneous conjugation of benzoic and salicylic acids with glycine. J Pharm Sci. 1969;58(3):321–326. doi: 10.1002/jps.2600580307
- Pacifici G, Mogavero S, Giulian L, et al. Conjugation of benzoic acid with glycine in the human fetal and adult liver and kidney. Dev Pharmacol Ther. 1991;17(1–2):52–62. doi: 10.1159/000457499
- Lares-Asseff I, Juarez-Olguin H, Flores-Perez J, et al. Pharmacokinetics and metabolic rates of acetyl salicylic acid and its metabolites in an Otomi ethnic group of Mexico. Biol Pharm Bull. 2004 May;27(5):706–709. doi: 10.1248/bpb.27.706
- Hutt AJ, Caldwell J, Smith RL. The metabolism of aspirin in man: a population study. Xenobiotica. 1986 Mar;16(3):239–249. doi: 10.3109/00498258609043527
- Bigler J, Whitton J, Lampe JW, et al. CYP2C9 and UGT1A6 genotypes modulate the protective effect of aspirin on colon adenoma risk. Cancer Res. [2001 May 1];61(9):3566–3569.
- MacDonald CJ, Ciolino HP, Yeh GC. The drug salicylamide is an antagonist of the aryl hydrocarbon receptor that inhibits signal transduction induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Cancer Res. [2004 Jan 1];64(1):429–434. doi: 10.1158/0008-5472.CAN-03-0974
- Patel DK, Notarianni LJ, Bennett PN. Comparative metabolism of high doses of aspirin in man and rat. Xenobiotica. 1990 Aug;20(8):847–854. doi: 10.3109/00498259009046898
- Caldwell J, O’Gorman J, Smith R. Inter‐individual differences in the glycine conjugation of salicylic acid [proceedings]. Br J Clin Pharmacol. 1980;9(1):114P–114P. doi: 10.1111/j.1365-2125.1980.tb04816.x
- Dorne J, Walton K, Renwick A. Human variability for metabolic pathways with limited data (CYP2A6, CYP2C9, CYP2E1, ADH, esterases, glycine and sulphate conjugation). Food Chem Toxicol. 2004;42(3):397–421. doi: 10.1016/j.fct.2003.10.003
- Menguy R, Desbaillets L, Masters YF, et al. Evidence for a sex-linked difference in aspirin metabolism. Nature. [1972 Sep 8];239(5367):102–103. doi: 10.1038/239102a0
- Emudianughe T, Oduleye S, Ebadan J, et al. Sex differences in salicylic acid metabolism in Nigerian subjects. Xenobiotica. 1986;16(2):177–179. doi: 10.3109/00498258609043520
- Kalow W. Interethnic variation of drug metabolism. Trends in pharmacological sciences. Trends Pharmacol Sci. 1991 Mar;12(3):102–107. doi: 10.1016/0165-6147(91)90516-U
- Miners JO, Grgurinovich N, Whitehead AG, et al. Influence of gender and oral contraceptive steroids on the metabolism of salicylic acid and acetylsalicylic acid. Br J Clin Pharmacol. 1986 Aug;22(2):135–142. doi: 10.1111/j.1365-2125.1986.tb05240.x
- Navarro SL, Saracino MR, Makar KW, et al. Determinants of aspirin metabolism in healthy men and women: effects of dietary inducers of udp-glucuronosyltransferases. Lifestyle Genom. 2011;4(2):110–118. doi: 10.1159/000327782
- Knights KM, Sykes MJ, Miners JO. Amino acid conjugation: contribution to the metabolism and toxicity of xenobiotic carboxylic acids. Expert Opin Drug Metab Toxicol. 2007;3(2):159–168. doi: 10.1517/17425255.3.2.159
- Knights KM, Miners JO. Amino acid conjugation: a novel route of xenobiotic carboxylic acid metabolism in man. In: Lyubimov A, editor. Encyclopedia of Drug metabolism and interactions). 1st ed. Hoboken, NJ: John Wiley & Sons, Inc.; 2012.
- Gregus Z, Fekete T, Varga F, et al. Dependence of glycine conjugation on availability of glycine: role of the glycine cleavage system. Xenobiotica; the fate of foreign compounds in biological systems. Xenobiotica. 1993;23(2):141–153. doi: 10.3109/00498259309059370
- Gregus Z, Fekete T, Varga F, et al. Effect of valproic acid on glycine conjugation of benzoic acid. J Pharmacol Exp Ther. 1993;267(3):1068–1075.
- Krieger I, Tanaka K. Therapeutic effects of glycine in Isovaleric Acidemia. Pediat Res. 1976;10(1):25–29. doi: 10.1203/00006450-197601000-00005
- Furst DE, Gupta N, Paulus HE. Salicylate metabolism in twins. Evidence suggesting a generic influence and induction of salicylurate formation. J Clin Invest. 1977;60(1):32–42. doi: 10.1172/JCI108766
- Tanaka K, Isselbacher KJ. The isolation and identification of N-isovalerylglycine from urine of patients with isovaleric acidemia. JBiolChem. 1967;242(12):2966–2972. doi: 10.1016/S0021-9258(18)99599-2
- van der Westhuizen FH, Pretorius PJ, Erasmus E. The utilization of alanine, glutamic acid, and serine as amino acid substrates for glycine N-acyltransferase. J Biochem Mol Toxicol. 2000;14(2):102–109. doi: 10.1002/(SICI)1099-0461(2000)14:2<102:AID-JBT6>3.3.CO;2-8
- Patel D, Ogunbona A, Notarianni L, et al. Depletion of plasma glycine and effect of glycine by mouth on salicylate metabolism during aspirin overdose. Hum Exp Toxicol. 1990;9(6):389–395. doi: 10.1177/096032719000900606
- van der Sluis R. Analyses of the genetic diversity and protein expression variation of the acyl: CoA medium-chain ligases. Mol Genet Genomics. 2018 Oct;293(5):1279–1292. doi: 10.1007/s00438-018-1460-3
- van der Sluis R, Badenhorst CP, Erasmus E, et al. Conservation of the coding regions of the glycine N-acyltransferase gene further suggests that glycine conjugation is an essential detoxification pathway. Gene. [2015 Oct 15];571(1):126–134.
- van der Sluis R, Badenhorst CP, van der Westhuizen FH, et al. Characterisation of the influence of genetic variations on the enzyme activity of a recombinant human glycine N-acyltransferase. Gene. [2013 Feb 25];515(2):447–453.
- Rohwer JM, Schutte C, van der Sluis R. Functional characterisation of three glycine N-Acyltransferase variants and the effect on glycine conjugation to benzoyl–CoA. Int J Mol Sci. [2021 Mar 18];22(6):3129. doi: 10.3390/ijms22063129
- Vest MF, Salzberg R. Conjugation reactions in the newborn infant: the metabolism of para-aminobenzoic acid. Arch Dis Child. 1965;40(209):97. doi: 10.1136/adc.40.209.97
- Irjala K. Synthesis of p-aminohippuric, hippuric, and salicyluric acids in experimental animals and man. Ann Academiae Scientiarum Fennicae Ser A5, Medica. 1972;154:1–40.
- Mawal Y, Paradis K, Qureshi IA. Developmental profile of mitochondrial glycine N-acyltransferase in human liver. JPediatr. 1997;130(6):1003–1007. doi: 10.1016/S0022-3476(97)70293-2
- Stern K, Hinds EG, Askonas BA. AGEING and DETOXICATION: studies in hippuric acid synthesis during psychoses of the involutional and Old age group. AJP. 1945;102(3):325–329. doi: 10.1176/ajp.102.3.325
- Temellini A, Mogavero S, Giulianotti PC, et al. Conjugation of benzoic acid with glycine in human liver and kidney: a study on the interindividual variability. Xenobiotica. 1993;23(12):1427–1433. doi: 10.3109/00498259309059451
- Gieseler RK, Schreiter T, Canbay A. The aging human liver: the weal and woe of evolutionary legacy. Z für Gastroenterologie. 2023;61(1):83–94. doi: 10.1055/a-1955-5297
- Watanabe H, Paxton RL, Tolerico MR, et al. Expression of Acsm2, a kidney-specific gene, parallels the function and maturation of proximal tubular cells. Am J Physiol-Renal Physiol. 2020;319(4):F603–F611. doi: 10.1152/ajprenal.00348.2020
- Goetzman ES, Bharathi SS, Zhang Y, et al. Impaired mitochondrial medium-chain fatty acid oxidation drives periportal macrovesicular steatosis in sirtuin-5 knockout mice. Sci Rep. 2020;10(1):18367. doi: 10.1038/s41598-020-75615-3
- Duffy LF, Kerzner B, Seeff L, et al. Preliminary assessment of glycine conjugation of para-aminobenzoic acid as a quantitative test of liver function. ClinBiochem. 1995;28(5):527–530. doi: 10.1016/0009-9120(95)00036-9
- Furuya KN, Durie PR, Roberts EA, et al. Glycine conjugation of para-aminobenzoic acid (PABA): a quantitative test of liver function. Clin Biochem. 1995;28(5):531–540. doi: 10.1016/0009-9120(95)00040-G
- Lebel S, Nakamachi Y, Hemming A, et al. Glycine conjugation of para-aminobenzoic acid (PABA): a pilot study of a novel prognostic test in acute liver failure in children. J Pediatr Gastroenterol Nutr. 2003;36(1):62–71. doi: 10.1002/j.1536-4801.2003.tb07959.x
- Okekunle AP, Li Y, Liu L, et al. Abnormal circulating amino acid profiles in multiple metabolic disorders. Diabetes Res Clin Pract. 2017 Oct;132:45–58. doi: 10.1016/j.diabres.2017.07.023
- Alves A, Bassot A, Bulteau A-L, et al. Glycine metabolism and its alterations in obesity and metabolic diseases. Nutrients. 2019;11(6):1356. doi: 10.3390/nu11061356
- Matsuo M, Terai K, Kameda N, et al. Designation of enzyme activity of glycine-N-acyltransferase family genes and depression of glycine-N-acyltransferase in human hepatocellular carcinoma. BiochemBiophysrescommun. 2012;420(4):901–906. doi: 10.1016/j.bbrc.2012.03.099
- Nwosu ZC, Megger DA, Hammad S, et al. Identification of the consistently altered metabolic targets in human hepatocellular carcinoma. Cellular and molecular gastroenterology and hepatology. Cell Mol Gastroenterol Hepatol. 2017;4(2):303–323. e1. doi: 10.1016/j.jcmgh.2017.05.004
- Wu J-M, Skill NJ, Maluccio MA. Evidence of aberrant lipid metabolism in hepatitis C and hepatocellular carcinoma. HPB. 2010;12(9):625–636. doi: 10.1111/j.1477-2574.2010.00207.x
- Xing X, Hu E, Ouyang J, et al. Integrated omics landscape of hepatocellular carcinoma suggests proteomic subtypes for precision therapy. Cell Rep Med. 2023;4(12):101315. doi: 10.1016/j.xcrm.2023.101315
- Zhang H-Y, Zhu J-J, Liu Z-M, et al. A prognostic four-gene signature and a therapeutic strategy for hepatocellular carcinoma: construction and analysis of a circRNA-mediated competing endogenous RNA network. Hepatobiliary & Pancreat Dis Int. 2023. doi: 10.1016/j.hbpd.2023.06.009
- Jain M, Nilsson R, Sharma S, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336(6084):1040–1044. doi: 10.1126/science.1218595
- Renwick SB, Snell K, Baumann U. The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy. Structure. 1998;6(9):1105–1116. doi: 10.1016/S0969-2126(98)00112-9
- Bloom BS. Direct medical costs of disease and gastrointestinal side effects during treatment for arthritis. Am J Med. [1988 Feb 22];84(2A):20–24. doi: 10.1016/0002-9343(88)90250-1
- Pirmohamed M, James S, Meakin S, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ (Clin Res Ed). [2004 Jul 3];329(7456):15–19. doi: 10.1136/bmj.329.7456.15
- Gummin DD, Mowry JB, Spyker DA, et al. 2016 annual report of the American Association of Poison Control Centers’ national poison data system (NPDS): 34th annual report. Clin Toxicol. 2017;55(10):1072–1254. doi: 10.1080/15563650.2017.1388087
- Proudfoot AT. Toxicity of salicylates. Am J Med. 1983;75(5):99–103. doi: 10.1016/0002-9343(83)90239-5
- Kaufman FL, Dubansky AS. Darvon poisoning with delayed salicylism: a case report. Pediatrics. 1972;49(4):610–611. doi: 10.1542/peds.49.4.610
- Wortzman DJ, Grunfeld A. Delayed absorption following enteric-coated aspirin overdose. Ann Emerg Med. 1987;16(4):434–436. doi: 10.1016/S0196-0644(87)80366-9
- Gonzalez-Conejero R, Rivera J, Corral J, et al. Biological assessment of aspirin efficacy on healthy individuals: heterogeneous response or aspirin failure? Stroke. 2005;36(2):276–280. doi: 10.1161/01.STR.0000151362.65339.f9
- Scharf RE, editor. Drugs that affect platelet function. Seminars in thrombosis and hemostasis. New York, NY: Thieme Medical Publishers; 2012.
- Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov. 2003;2(1):15–28. doi: 10.1038/nrd985
- Ehrlich GE. Therapeutic applications of nsaids: subpopulations and new formulations. Arthritis and Rhematology. 1992;36(5):727–728.
- Tschetter pn. salicylamide toxicity and salicylism-Reply. Arch Pediatr Adolesc Med. 1964;108(1):109–110. doi: 10.1001/archpedi.1964.02090010111018
- Nguyen T, Cranswick N, Rosenbaum J, et al. Chronic use of teething gel causing salicylate toxicity. J Paediatr Child Health. 2018;54(5):576–578. doi: 10.1111/jpc.13861
- Done AK. Salicylate intoxication: significance of measurements of salicylate in blood in cases of acute ingestion. Pediatrics. 1960;26(5):800–807. doi: 10.1542/peds.26.5.800
- Hall S, Plaster P, Glasgow J, et al. Preadmission antipyretics in Reye’s syndrome. Arch Dis Child. 1988;63(7):857. doi: 10.1136/adc.63.7.857
- McGovern M, Glasgow J, Stewart M. Reye’s syndrome and aspirin: lest we forget. BMJ (Clin Res Ed). 2001;322(7302):1591–1592.
- Thabet F, Durand P, Chevret L, et al. Syndrome de Reye sévère : à propos de 14 cas pris en charge dans une unité de réanimation pédiatrique pendant 11 ans. Archives de Pédiatrie. 2002;9(6):581–586. doi: 10.1016/S0929-693X(01)00924-1
- Glasgow JF. Reye’s syndrome: the case for a causal link with aspirin. Drug Saf. 2006;29(12):1111–1121. doi: 10.2165/00002018-200629120-00003
- Glasgow JF, Middleton B, Moore R, et al. The mechanism of inhibition of β-oxidation by aspirin metabolites in skin fibroblasts from Reye’s syndrome patients and controls. Biochim Et Biophys Acta (BBA)-Mol Basis Disease. 1999;1454(1):115–125. doi: 10.1016/S0925-4439(99)00025-3
- Prescott LF. Paracetamol: past, present, and future. Am J Ther. 2000;7(2):143–148. doi: 10.1097/00045391-200007020-00011
- Olive G. Analgesic/Antipyretic treatment: ibuprofen or acetaminophen? An update. Therapie. 2006;61(2):151–160. doi: 10.2515/therapie:2006034
- O’Gorman T, Koff RS. Salicylate hepatitis. Gastroenterology. 1977;72(4):726–728. doi: 10.1016/S0016-5085(77)80162-5
- Levy G, Yaffe SJ. Clinical implications of salicylate-induced liver damage. Arch Pediatr Adolesc Med. 1975 Dec;129(12):1385–1386. doi: 10.1001/archpedi.1975.02120490003001
- Doi H, Horie T. Salicylic acid-induced hepatotoxicity triggered by oxidative stress. Chem Biol Interact. 2010;183(3):363–368. doi: 10.1016/j.cbi.2009.11.024
- Agundez JA, Martinez C, Perez-Sala D, et al. Pharmacogenomics in aspirin intolerance. Curr Drug Metab. 2009 Nov;10(9):998–1008. doi: 10.2174/138920009790711814
- Ho PC, Triggs EJ, Bourne DW, et al. The effects of age and sex on the disposition of acetylsalicylic acid and its metabolites. Br J Clin Pharmacol. 1985 May;19(5):675–684. doi: 10.1111/j.1365-2125.1985.tb02695.x
- Campbell L, Wilson HK, Samuel AM, et al. Interactions of m-xylene and aspirin metabolism in man. Br J Ind Med. 1988 Feb;45(2):127–132. doi: 10.1136/oem.45.2.127
- Sakuma T. Alteration of urinary carnitine profile induced by benzoate administration. Arch Dis Child. 1991;66(7):873–875. doi: 10.1136/adc.66.7.873
- Vest MF, Salzberg R. Conjugation reactions in the newborn infant: the metabolism of para-aminobenzoic acid arch dis childh. Arch Dis Child. 1965;40(209):97–105. doi: 10.1136/adc.40.209.97
- Levy G. Pharmacokinetics of salicylate elimination in man. J Pharm Sci. 1965;54(7):959–967. doi: 10.1002/jps.2600540703
- Forman WB, Davidson ED, Webster LT. Enzymatic conversion of salicylate to salicylurate. Mol Pharmacol. 1971;7(3):247–259.
- Huckle K, Tait G, Millburn P. Species variations in the renal and hepatic conjugation of 3-phenoxybenzoic acid with glycine. Xenobiotica. 1981;11(9):635–644. doi: 10.3109/00498258109045875
- Vessey DA, Lau E, Kelley M, et al. Isolation, sequencing, and expression of a cDNA for the HXM-A form of xenobiotic/medium-chain fatty acid: CoA ligase from human liver mitochondria. J Biochem Mol Toxicol. 2003;17(1):1–6. doi: 10.1002/jbt.10056
- Bartlett K, Gompertz D. The specificity of glycine-N-acylase and acylglycine excretion in the organicacidaemias. BiochemMed. 1974;10(1):15–23. doi: 10.1016/0006-2944(74)90004-0
- Del Olmo A, Calzada J, Nunez M. Benzoic acid and its derivatives as naturally occurring compounds in foods and as additives: uses, exposure, and controversy. Crit Rev Food Sci Nutr. [2017 Sep 22];57(14):3084–3103. doi: 10.1080/10408398.2015.1087964
- Lemarie F, Beauchamp E, Legrand P, et al. Revisiting the metabolism and physiological functions of caprylic acid (C8: 0) with special focus on ghrelin octanoylation. Biochimie. 2016 Jan;120:40–48. doi: 10.1016/j.biochi.2015.08.002
- Kølvraa S, Gregersen N, Christensen E, et al. In vitro fibroblast studies in a patient with C6-C10-dicarboxylic aciduria: evidence for a defect in general acyl-CoA dehydrogenase. Clinica (Rome) Acta. 1982;126(1):53–67. doi: 10.1016/0009-8981(82)90361-8
- Rechner AR, Kuhnle G, Bremner P, et al. The metabolic fate of dietary polyphenols in humans. Free Radical Biol Med. [2002 Jul 15];33(2):220–235. doi: 10.1016/S0891-5849(02)00877-8
- Glasgow JFT, Middleton B. Reye syndrome—insights on causation and prognosis. Arch Dis Child. 2001;85(5):351–353. doi: 10.1136/adc.85.5.351
- Williams HRT, Cox IJ, Dg W, et al. Differences in gut microbial metabolism are responsible for reduced hippurate synthesis in Crohn's disease. BMC Gastroenterol. 2010;10(1):108. doi: 10.1186/1471-230X-10-108
- Watkins PA, Maiguel D, Jia Z, et al. Evidence for 26 distinct acyl-coenzyme a synthetase genes in the human genomes. J Lipid Res. 2007;48(12):2736–2750. doi: 10.1194/jlr.M700378-JLR200
- Martin J, Han C, Gordon LA, et al. The sequence and analysis of duplication-rich human chromosome 16. Nature. [2004 Dec 23];432(7020):988–994. doi: 10.1038/nature03187
- Loftus BJ, Kim UJ, Sneddon VP, et al. Genome duplications and other features in 12 mb of DNA sequence from human chromosome 16p and 16q. Genomics. [1999 Sep 15];60(3):295–308. doi: 10.1006/geno.1999.5927
- Qian W, Zhang J. Genomic evidence for adaptation by gene duplication. Genome Res. 2014 Aug;24(8):1356–1362. doi: 10.1101/gr.172098.114
- Fagerberg L, Hallström BM, Oksvold P, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014 Feb;13(2):397–406. doi: 10.1074/mcp.M113.035600
- Kelley M, Vessey DA. Interaction of 2,4-dichlorophenoxyacetate (2,4-D) and 2,4,5-trichlorophenoxyacetate (2,4,5-T) with the acyl-CoA: amino acid N-acyltransferase enzymes of bovine liver mitochondria. BiochemPharmacol. 1986;35(2):289–295. doi: 10.1016/0006-2952(86)90528-9
- Kelley M, Vessey D. The effects of ions on the conjugation of xenobiotics by the aralkyl-CoA and arylacetyl-CoA N-acyltransferases from bovine liver mitochondria. J Biochem Toxicol. 1990;5(2):125–135. doi: 10.1002/jbt.2570050208
- Nandi DL, Lucas SV, Webster LT Jr. Benzoyl-coenzyme A: glycine N-acyltransferase and phenylacetyl-coenzyme A: glycine N-acyltransferase from bovine liver mitochondria. Purif Charact J Biol Chem. 1979;254(15):7230–7237. doi: 10.1016/S0021-9258(18)50309-4
- Webster LT, Siddiqui UA, Lucas SV, et al. Identification of separate acyl- CoA: glycine and acyl-CoA: L-glutamine N-acyltransferase activities in mitochondrial fractions from liver of rhesus monkey and man. J Biol Chem. 1976;251(11):3352–3358. doi: 10.1016/S0021-9258(17)33444-0
- Kühn S, Williams ME, Dercksen M, et al. The glycine N-acyltransferases, GLYAT and GLYATL1, contribute to the detoxification of isovaleryl-CoA - an in-silico and in vitro validation. Comput Struct Biotechnol J. 2023;21:1236–1248. doi: 10.1016/j.csbj.2023.01.041
- Moldave K, Meister A. Synthesis of phenylacetylglutamine by human tissue. J Biol Chem. 1957 Nov;229(1):463–476. doi: 10.1016/S0021-9258(18)70632-7
- Schachter D, Taggart JV. Glycine N-acylase: purification and properties. J Biol Chem. 1954;208(1):263–275. doi: 10.1016/S0021-9258(18)65643-1
- Moldave K, Meister A. Enzymatic synthesis of phenylacetyl-l-glutamine (human liver, kidney). In: Colowick SP, Kaplan NO, editors. Methods in Enzymology. Vol. 17. New York, NY: Elsevier; 1970. p. 946–950.
- Mawal YR, Qureshi IA. Purification to homogeneity of mitochondrial acyl coa: glycine n-acyltransferase from human liver. Biochem Biophys Res Commun. 1994;205(2):1373–1379. doi: 10.1006/bbrc.1994.2817
- van der Sluis R, Ungerer V, Nortje C, et al. New insights into the catalytic mechanism of human glycine N-acyltransferase. J Biochem Mol Tox. 2017 Nov;31(11). doi: 10.1002/jbt.21963
- Schulke D, Sass JO. Frequent sequence variants of human glycine N-acyltransferase (GLYAT) and inborn errors of metabolism. Biochimie. [2021 Feb 7];183:30–34. doi: 10.1016/j.biochi.2021.02.002
- Dempsey DR, Bond JD, Carpenter A-M, et al. Expression, purification, and characterization of mouse glycine N-acyltransferase in Escherichia coli. Protein Expr Purif. 2014;97:23–28. doi: 10.1016/j.pep.2014.02.007
- Bains RK. African variation at cytochrome P450 genes: evolutionary aspects and the implications for the treatment of infectious diseases. Evol Med Public Health. 2013;2013(1):118–134. doi: 10.1093/emph/eot010
- Carrascal-Laso L, Isidoro-García M, Ramos-Gallego I, et al. Review: influence of the CYP450 Genetic Variation on the treatment of psychotic disorders. J Clin Med. 2021;10(18):4275. doi: 10.3390/jcm10184275
- Nortje C, van der Sluis R, van Dijk AA, et al. The Use of p-Aminobenzoic Acid as a Probe Substance for the Targeted Profiling of Glycine Conjugation. J Biochem Mol Toxicol. 2016 Mar;30(3):136–147. doi: 10.1002/jbt.21772
- Nebert D, McKinnon R, Puga A. Human drug-metabolizing enzyme polymorphisms: effects on risk of toxicity and cancer DNA and cell biol. DNA Cell Biol. 1996;15(4):273–280. doi: 10.1089/dna.1996.15.273
- Wallig MA. Glucuronidation and susceptibility to chemical carcinogenesis. Toxicological Sci. 2004;78(1):1–2. doi: 10.1093/toxsci/kfh068
- Court MH. Feline drug metabolism and disposition: pharmacokinetic evidence for species differences and molecular mechanisms. The Vet clinics of North Am Small Anim Pract. 2013 Sep;43(5):1039–1054. doi: 10.1016/j.cvsm.2013.05.002
- Davis LE, Westfall BA. Species differences in biotransformation and excretion of salicylate. Am J Vet Res. 1972 Jun;33(6):1253–1262.
- Prandota J. Important role of prodromal viral infections responsible for inhibition of xenobiotic metabolizing enzymes in the pathomechanism of idiopathic Reye’s syndrome, Stevens-Johnson syndrome, autoimmune hepatitis, and hepatotoxicity of the therapeutic doses of acetaminophen used in genetically predisposed persons. Am J Ther. 2002;9(2):149–156. doi: 10.1097/00045391-200203000-00009
- Larrick J, Kunkel S. Is Reye’s syndrome caused by augmented release of tumour necrosis factor? The Lancet. 1986;328(8499):132–133. doi: 10.1016/S0140-6736(86)91947-1
- Glasgow J, Middleton B. Reye syndrome—insights on causation and prognosis. Archives of disease in childhood. Arch Dis Child. 2001;85(5):351–353. doi: 10.1136/adc.85.5.351
- Muñoz JP, Calaf GM. Downregulation of Glycine N-Acyltransferase in Kidney Renal Clear Cell Carcinoma: A Bioinformatic-Based Screening. Diagnostics. 2023;13(23):3505. doi: 10.3390/diagnostics13233505
- Shi S, Ke J, Xu Q, et al. Integrated network analysis to identify the key genes, transcription factors, and microRNAs involved in hepatocellular carcinoma. Neoplasma. 2018;65(1):66–74. doi: 10.4149/neo_2018_161215N642
- Ma X, Zhou L, Zheng S. Transcriptome analysis revealed key prognostic genes and microRNAs in hepatocellular carcinoma. PeerJ. 2020;8:e8930. doi: 10.7717/peerj.8930
- Ru B, Hu J, Zhang N, et al. A novel metabolism-related gene signature in patients with hepatocellular carcinoma. PeerJ. 2023;11:e16335. doi: 10.7717/peerj.16335
- Knights KM, Sykes MJ, Miners JO. Amino acid conjugation: contribution to the metabolism and toxicity of xenobiotic carboxylic acids. Expert Opin on Drug Metab Toxicol. 2007;3(2):159–168. doi: 10.1517/17425255.3.2.159
- Beyoglu D, Idle JR. The glycine deportation system and its pharmacological consequences. Pharmacol Ther. 2012 Aug;135(2):151–167. doi: 10.1016/j.pharmthera.2012.05.003
- Leth T, Christensen T, Larsen IK. Estimated intake of benzoic and sorbic acids in Denmark. Food additives & contaminants Part A. Chem, Anal, Control, Exposure Risk Assess. 2010 Jun;27(6):783–792. doi: 10.1080/19440041003598606
- Mischek D, Krapfenbauer-Cermak C. Exposure assessment of food preservatives (sulphites, benzoic and sorbic acid) in Austria. Food additives & contaminants Part A. Chem, Anal, Control, Exposure Risk Assess. 2012;29(3):371–382. doi: 10.1080/19440049.2011.643415
- Levy G, Amsel LP. Kinetics of competitive inhibition of salicylic acid conjugation with glycine in man. Biochem Pharmacol. 1966 Aug;15(8):1033–1038. doi: 10.1016/0006-2952(66)90267-X
- Zimmerman HJ. Effects of aspirin and acetaminophen on the liver. Archives of internal medicine. Archiv Intern Med. [1981 Feb 23];141(3):333–342. doi: 10.1001/archinte.141.3.333
- Badenhorst CP, van der Sluis R, Erasmus E, et al. Glycine conjugation: importance in metabolism, the role of glycine N-acyltransferase, and factors that influence interindividual variation. Expert Opin on Drug Metab Toxicol. 2013 Sep;9(9):1139–1153. doi: 10.1517/17425255.2013.796929
- Vázquez AB, Ribes Koninckx C, editors. Celiac Disease: Forty Years Later. Anales de pediatria. Barcelona, Spain: Prous Science; 2003. p. 96–102.
- Guo Y, Zhang L, Liu Y, et al. Suggestion of GLYAT gene underlying variation of bone size and body lean mass as revealed by a bivariate genome-wide association study. Hum Genet. 2013;132(2):189–199. doi: 10.1007/s00439-012-1236-5
- Amelio I, Cutruzzolá F, Antonov A, et al. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014 Apr;39(4):191–198. doi: 10.1016/j.tibs.2014.02.004
- Kastanos EK, Woldman YY, Appling DR. Role of Mitochondrial and Cytoplasmic Serine Hydroxymethyltransferase Isozymes in de NoVo Purine Synthesis in Saccharomyces cereVisiae. Biochemistry. 1997;36:14956–14964.
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9(6):447–464. doi: 10.1038/nrd3137
- Day R, Paull P, Champion G, et al. Evaluation of an enteric coated aspirin preparation. Australian and New Zealand J Med. 1976;6(1):45–50. doi: 10.1111/j.1445-5994.1976.tb03290.x
- Rumble R, Brooks P, Roberts M. Metabolism of salicylate during chronic aspirin therapy. Br J Clin Pharmacol. 1980;9(1):41–45. doi: 10.1111/j.1365-2125.1980.tb04794.x