
ABSTRACT
Introduction
Artificial intelligence (AI) encompasses a wide range of algorithms with risks when used to support decisions about diagnosis or treatment, so professional and regulatory bodies are recommending how they should be managed.
Areas covered
AI systems may qualify as standalone medical device software (MDSW) or be embedded within a medical device. Within the European Union (EU) AI software must undergo a conformity assessment procedure to be approved as a medical device. The draft EU Regulation on AI proposes rules that will apply across industry sectors, while for devices the Medical Device Regulation also applies. In the CORE-MD project (Coordinating Research and Evidence for Medical Devices), we have surveyed definitions and summarize initiatives made by professional consensus groups, regulators, and standardization bodies.
Expert opinion
The level of clinical evidence required should be determined according to each application and to legal and methodological factors that contribute to risk, including accountability, transparency, and interpretability. EU guidance for MDSW based on international recommendations does not yet describe the clinical evidence needed for medical AI software. Regulators, notified bodies, manufacturers, clinicians and patients would all benefit from common standards for the clinical evaluation of high-risk AI applications and transparency of their evidence and performance.
1. Introduction
Artificial intelligence (AI) is an oxymoron. In their seminal proposal, John McCarthy and his colleagues suggested that a machine could be made to simulate ‘every aspect of learning or any other feature of intelligence’ [Citation1], but such ‘general’ AI remains an elusive goal that for most medical applications may always be inappropriate. Even in non-clinical contexts the name is misleading: computers process binary code but they are not conscious organisms and they cannot understand its meaning [Citation2]. Nonetheless, the term artificial ‘intelligence’ is widely applied to an extensive range of software algorithms with varying functions, sophistication and transparency, including natural language processing and machine learning (ML).
Multiple administrative, scientific, diagnostic and therapeutic applications in healthcare already depend on some variant of AI to perform a myriad of tasks. In essence, most can be considered advanced statistical methods for interpreting large datasets or performing complex analyses [Citation3]. It seems unscientific to lump such a wide array of techniques together under a single umbrella term ‘AI’ and to try to find a precise definition for AI that covers all the possibilities. From clinical and regulatory perspectives, AI should not be the objective but rather always a means to an end. Irrespective of whether an analytical algorithm (AI software) constitutes a medical device in its own right or is an integral part of a medical device, it should be evaluated for regulatory approval according to the ultimate purpose claimed by its manufacturer. The level of clinical evidence and the criteria by which it will be evaluated will be determined by the clinical risks inherent in the AI application if it reaches an erroneous conclusion for the individual patient or directs an inappropriate action within the context of its use.
Despite the lack of precision and frequent nonspecificity of use of the term AI, many task forces or working groups have defined it and developed classifications as a prelude to formulating standards or regulatory guidance for clinical evaluation. An alternative framework for stratifying medical device software (MDSW) according to the risks associated with its use was recommended by the International Medical Device Regulators Forum (IMDRF) [Citation4] and adopted with some modifications by the EU [Citation5]. Its approach can guide proportionate assessment of AI medical devices in the European Union (EU), determined by existing legislation but taking more account of expert scientific guidance. To contribute to the development of appropriate methodologies for clinical evaluation, we have reviewed and here summarize:
Regulatory definitions and terms relating to AI
Consensus recommendations from professional and academic experts
Standards relevant to medical AI proposed by international standards organizations
Projects by regulatory bodies to develop requirements for clinical evaluation of AI
European laws with relevance to clinical uses of AI, and
Transparency and interpretability of AI and ML methodologies.
This is a narrative rather than a systematic review, which summarizes the results of literature searches made using combinations of terms related to artificial intelligence, machine learning, standards, and regulations. We also searched the websites of international regulators and standards organizations, and followed up sources mentioned in reference lists and official publications. defines some of the common terms that are used in this paper [Citation6–11].
Table 1. Definitions.
Table 2. Definitions of artificial intelligence in reports and regulatory documents with relevance for medical devices.
2. Background and the CORE-MD project
In the European Union (EU), medical devices are governed by Regulation (EU) 2017/745 on Medical Devices (MDR) which has been applicable since May 2021. Article 2(1) defines a medical device as ‘Any instrument, apparatus, appliance, software, implant, reagent, material or other article, intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes […] diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease [or] investigation, replacement or modification of the anatomy or of a physiological or pathological process or state’ [Citation12] (our emphases).
The term ‘software’ was added to the description in 2007 under the previous EU medical device directives [Citation13], while prediction and prognosis were first included among the purposes of a medical device by the MDR. The Medical Device Coordination Group (MDCG) of the European Commission published guidance in 2019 about what should qualify as MDSW and how it should be classified [Citation5], and in 2020 it added recommendations for clinical evaluation (of software in general) [Citation14].
The explosion of interest in harnessing the power of AI to accelerate medical research and exploit its capacity when addressing specific clinical challenges, has produced many applications already approved by regulators without their scientific basis being transparent. In the USA in 2017–2018, for example, the Food and Drug Administration (FDA) approved 11 out of 14 AI software products through their 510(k) pathway, which means on the basis of equivalence to existing devices and without new clinical evidence [Citation15]. Concerning 222 AI medical devices approved between 2015 and 2020, 92% had been assessed through the 510(k) pathway [Citation16]. In another study, only 9 out of 64 AI medical devices had undergone de novo or premarket approval [Citation17].
In Europe, peer-reviewed literature was available for only 36 of 100 AI medical device software products that were CE-marked for use in diagnostic radiology, and the published evidence addressed diagnostic accuracy rather than clinical efficacy or impact [Citation18]. Out of 240 devices approved under the previous EU medical device directives, only 1% were considered high-risk (class III) [Citation16]. At that time, most medical device software was considered Class I and self-certified without external review, whereas under the MDR the majority will likely fall under Class IIa or higher. Application of the MDR will hopefully lead to an increase in clinical data supporting the safety and performance of these devices.
There is skepticism about some claims for medical applications of AI that are insufficiently supported by evidence, when systematic reviews have demonstrated that they can currently replicate the results of experts rather than exceed their performance. For example, external validation of diagnostic imaging tools resulted in a pooled sensitivity of 87%, compared with 92.5% for evaluation by experts [Citation19]. In 2018 a Cochrane Collaboration review of 42 studies that used ML to diagnose skin cancer in adults found high sensitivity but low and variable specificity; the evidence base was considered too poor to understand any impact of the results on clinical decision-making, particularly in unselected subjects with a low prevalence of disease [Citation20]. Applying AI clinical prediction models in 71 studies was equivalent to established statistical methods using logistic regression [Citation21].
There are few prospective randomized trials of ML and the risk of bias is high [Citation22–24]. Most reports do not include an analysis of performance errors [Citation25]. A majority of studies have involved only a few centers, and when external validation has been undertaken it has shown a significant drop in the accuracy of the software [Citation23,Citation24]. To date, there is sparse evidence that the use of ML-based clinical decision support systems [Citation26] is associated with improved clinician diagnostic performance [Citation27]. The use of AI for predictive modeling is increasing but often insufficient details are published to support their external validation [Citation28].
Similar questions could be raised about the reliability of clinical measurements and diagnoses made by humans. Their decisions may be based on incomplete, inaccurate or biased information [Citation29], or taken when affected by fatigue or other human factors. For particular indications, AI medical devices may overcome some of these limitations and provide support for making diagnostic decisions. The critical question is whether the combination of human and machine (or more specifically, an AI algorithm) performs better and more efficiently than a human alone, for an intended clinical purpose.
There is widespread recognition that standards are needed to guide what evidence from clinical evaluation is needed for submissions for regulatory approval of AI applications in healthcare. A proposal from the EU Horizon 2020 program that was published in 2019 stated that new medical technologies such as AI ‘pose additional challenges and opportunities for developers to generate high-quality clinical evidence’ [Citation30]. It called for research to ‘identify gaps to be filled’ and to ‘derive recommendations for the choice of clinical investigation methodology to obtain sufficient evidence’ [Citation30]. An overview of the concerted action now addressing that call has been published (CORE-MD, Coordinating Research and Evidence for Medical Devices) [Citation31]. This review of regulatory initiatives has been conducted as part of the CORE-MD task on AI.
3. Definitions of artificial intelligence
In 2013 the IMDRF defined Software as a Medical Device (SaMD) in very general terms as any software designed for a medical purpose [Citation32] (). Software utilizing AI is covered by the definition but only if it is not an essential component of a hardware device. In all its reports, the IMDRF Working Group on SaMD has stated that medical software does not meet its definition of SaMD if its intended medical purpose is to drive a hardware medical device – for software that is embedded in a medical device, other provisions apply. The IMDRF also does not mention the functions of prediction and prognosis in its definition of a medical device, although it states that SaMD may be an aid to prediction or prognosis (paragraph 5.2.3) [Citation32].
Some regulatory agencies use the term SaMD which may be designated as ‘stand-alone’ although strictly speaking that is a misnomer since the software is always connected to or integrated within a digital device, computer or IT platform. Instead of SaMD, EU guidance uses the term MDSW [Citation5] for software applications including those that incorporate AI or ML algorithms. The FDA differentiates between SaMD and SiMD (Software in a Medical Device) [Citation49] ().
Figure 1. Comparison of regulatory definitions of medical device software and their scope.
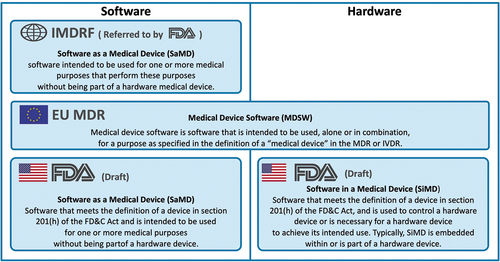
Early definitions of AI in Europe were developed for general rather than for healthcare applications. The European AI strategy refers to ‘systems that display intelligent behaviour by analysing their environment and taking action – with some degree of autonomy – to achieve specific goals’ [Citation50]. Emphasis on autonomy is probably not relevant for most clinical AI applications since human agency and oversight are considered paramount [Citation47] (although for some medical AI systems they may not be feasible or wanted).
For the objective of providing guidance to manufacturers and regulators on the clinical evaluation of AI in medical devices, we think that it is preferable not to restrict definitions according to the physical context in which particular software operates. In our opinion, it is more inclusive and useful for most purposes to refer to ‘AI systems,’ which is the term that was recommended by the EU High-Level Expert Group on AI (HLEG) in 2018 in its first report to the European Commission; an AI system was defined as any AI-based component, whether software and/or hardware, usually embedded as a component of a larger system [Citation35].
In 2018 the European Commission established a knowledge service called AI Watch, managed by the EU Joint Research Centre from its site in Seville, which has responsibility for monitoring the development, uptake and impact of AI. One of its first reports referred to the absence of any standard definition, so AI Watch adopted the definition from HLEG (see ) as the starting point for proposing a detailed taxonomy [Citation6]. The document has been updated and now includes a comprehensive and highly informative summary of 69 definitions of AI that have been applied in various countries or proposed by academic authors [Citation51].
The draft EU Regulation on Artificial Intelligence (AI Act) that was proposed in 2021 is now being reviewed by the European Parliament and Council [Citation37]. The AI Act will apply across all sectors, but AI that qualifies as a medical device will also need to satisfy the requirements of the MDR. Neither the AI Act nor the MDR refers specifically to medical AI systems. The introduction to the AI Act states that uniformity and ‘a single future-proof definition of AI’ are needed. It is unclear to us if that will be achieved by this legislative definition since it specifies particular methodologies (in an annex) and those will evolve, but amendments have been proposed that if accepted would bring the EU AI Act definition in line with that proposed by OECD.
The most recent contribution to this debate within the EU has come from the scientific advisory service to the European Parliament (Science and Technical Options Assessment, STOA), which reverted to a historical definition [Citation38] (see ). On a wider stage, the Organisation for Economic Co-operation and Development (OECD) has an AI Policy Observatory that has published several reports, which give overlapping definitions. OECD is a voluntary collaboration of 38 member countries with no legal authority, yet its papers are influential [Citation52] – the OECD definition has been cited by the World Health Organization (WHO) [Citation47]. Note that the definition does not mention ‘intelligence’ (see : OECD Council on AI).
A consensus may develop but current definitions tend to be overelaborate and they have not yet converged into a version accepted by regulators worldwide. One of the most straightforward definitions, given by the AI pioneer Marvin Minsky in 1968 as ‘the science of making machines do things that would require intelligence if done by men’ [Citation6], is appealing but begs the question of how to define intelligence. Features of human cognitive ability that are considered to correlate with intelligence include reasoning, memory, and processing speed [Citation53,Citation54]. Many recent advances in AI have been in the sub-discipline of ML, which involves building systems that can learn to solve problems without being explicitly programmed to do so. Another short and useful definition that would apply particularly to ML, was included in the executive summary of WHO guidance on the ethics and governance of AI: ‘the ability of algorithms to learn from data so that they can perform automated tasks’ (see for the full quotation) [Citation47].
3.1. Definitions of risks of AI medical devices
Suppose it is agreed that specifying each individual purpose of an AI system is much more important than having a unifying definition for AI MDSW. In that case, it is essential to establish criteria that should be used to identify which AI systems need more in-depth clinical investigation and most evidence of benefit before they are certified by a notified body and placed on the EU market. In 2014 the IMDRF guidance on SaMD identified two main dimensions of risk, which can be simplified as the function of the software and the severity of the disease [Citation4]. SaMD should be designated in the highest-risk groups (called categories III and IV in that document) if it provides information used to diagnose or treat a disease, and if the healthcare situation is serious or critical [Citation4]. The full list of factors identified in the IMDRF report as influencing risk, is given in . EU guidance from the MDCG gave illustrative examples for classification [Citation5].
Table 3. Factors influencing the risks of software as a medical device (SaMD).
These documents have concerned MDSW in general, while recent guidance from the Chinese regulatory agency referred to AI-based MDSW specifically. It states that an AI system with ‘low maturity in medical applications,’ meaning that its safety and effectiveness have not been fully established, should be managed as a Class III medical device [defined as a high-risk medical device implanted into the human body, and used to support or sustain life] if it is going to be used ‘to assist decision-making, such as providing lesion feature identification, lesion nature determination, medication guidance, and treatment plan formulation’ [Citation55]. If it is not used to aid decision-making, but rather for data processing or measurement that provides clinical reference information, then it will be managed as a Class II medical device. The UK regulatory agency envisages using a similar mechanism, called the ‘airlock classification rule,’ to assign class III to SaMD with a poorly understood risk profile [Citation56]. The challenge for UK and Chinese policymakers is to translate ‘sufficiently understood’ or ‘fully established safety’ into a predictable and objective legal instrument. Equivalent clarity from all IMDRF regulators would be helpful.
In the EU, Rule 11 in paragraph 6.3 of Annex VIII of the MDR, concerning classification, states that ‘Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause: death or an irreversible deterioration of a person’s state of health, in which case it is in class III; or a serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as class IIb. Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb. All other software is classified as class I’ [Citation12].
4. Expert consensus recommendations
Ideally, regulatory and scientific requirements for evaluation should align. When appraising new technology, however, the focus of regulators and healthcare professionals is not always the same. Several academic initiatives have emerged to fill a perceived gap in guidance for evaluating AI robustly from a clinical perspective. They can be divided into three broad groups: methodological recommendations, tools to assess the risk of bias and/or quality, and reporting guidelines. The most authoritative guidelines so far were developed using Delphi processes and have focused on reporting, whereas methodological guidance has come from smaller expert groups or single research laboratories. Overall, the trend is toward a staged approach to evaluating AI through several preclinical and clinical assessment steps.
The first consensus statements on how to report AI studies came from the CONSORT-AI and SPIRIT-AI working groups in 2020. Their checklists focus respectively on randomized controlled trials and their protocol [Citation57,Citation58]. Then DECIDE-AI was published in May 2022, with an emphasis on the early-stage evaluation of AI decision-support systems when used in clinical settings where their recommendations have an impact on the patient’s care [Citation59]. These three checklists were developed by multi-stakeholder groups that included regulators in their feedback rounds and consensus meetings. They incorporate items that are needed for regulatory reviews, such as a clear statement of the intended use, reporting of safety issues, and a description of the data-management process.
Two more cross-specialty AI reporting guidelines are being developed: TRIPOD-AI focusing on the development and validation of predictive models using AI [Citation60], and STARD-AI concerning the diagnostic accuracy of AI-based diagnostic tools [Citation61]. The TRIPOD-AI authors are also preparing the PROBAST-AI extension, which will be a ‘risk of bias’ assessment tool adapted specifically to AI studies [Citation60].
gives an overview of these current and forthcoming cross-specialty AI guidelines based on large consensus processes. Other initiatives based on systematic consensus methodology have published specialty-specific guidance [Citation62–64] (). Reporting guidelines that are based on expert opinion, and additional recommendations for quality control of AI clinical tools, are also included in [Citation62–78], but the list is not exhaustive. For up-to-date information on existing and planned reporting guidelines, the EQUATOR network remains the source of choice [Citation79].
Table 4. Cross-specialty recommendations on medical AI, developed using a systematic expert consensus methodology.
Table 5. Cross-specialty or specialty-specific recommendations on medical AI, published by medical associations or groups of investigators.
Methodological development pathways have been proposed [Citation63,Citation65–70] but there is not yet a broad consensus on the most appropriate options. Overall, the suggested pathways often include an initial phase of in silico internal validation, followed by an external in silico validation, and then a phase of off-line or ‘shadow mode’ fine-tuning to the context in which the AI system will be implemented. Thereafter, the performance of the AI should be assessed in the deployment settings without impacting patient care (i.e. in parallel but independently from standard clinical decision-making), before a phase of early live clinical evaluation (meaning in a situation where the recommendations of the AI systems have an actual impact on patient care). The recommended later stages would then be a large and possibly multicentric comparative evaluation, and finally, a phase of scaling up with adequate long-term monitoring, also coined by some as ‘algorithmovigilance.’ summarizes the key components present in these consensus-based guidelines, which were also reviewed by Shelmerdine et al. [Citation80].
Table 6. Key components of consensus-based guidelines.
Even if not aimed explicitly at medical AI, some other recommendations are worth noting. The framework from the Medical Research Council in the UK for developing and evaluating complex interventions [Citation81], as well as the IDEAL framework for surgical innovation [Citation82,Citation83] and especially its extension to medical devices [Citation84], offer transferable guidance for the evaluation of clinical AI. A German medical device consultancy with expertise in medical software has developed guidance with colleagues from a German notified body; their detailed suggestions overlap with those of the professional consensus statements [Citation85]. In the biological and life sciences, Heil et al. proposed reproducibility standards for ML [Citation86] and Luo et al. published guidelines for developing and reporting ML predictive models [Citation87]. A shared registry has been established, called AIMe, for investigators to provide transparent details of their AI biomedical applications [Citation88].
A recent systematic review of 503 clinical diagnostic studies that had employed deep learning for medical imaging, found considerable heterogeneity between studies and extensive variation in methodology, terminology, and outcome measures [Citation89]. The authors concluded that ‘there is an immediate need for the development of AI-specific EQUATOR guidelines.’ This need has now been answered, at least partially, since the professional recommendations listed in can be consulted and applied to inform best practice in clinical AI. Their shared features (see ) could be considered by medical device regulators for incorporation into guidance or common specifications.
5. Guidance, standards, and common specifications for AI medical devices
Medical device legislation provides high-level requirements. The MDR contains ‘General safety and performance requirements’ but it would be impractical to cover the wide variety of possible technologies and purposes individually. Instead, legislators expect that guidance, standards, or technical specifications will provide detailed advice for implementation.
For particular types of devices the EU relies on recommendations prepared by the global standards organizations – especially the ISO (International Organization for Standardization) and the IEC (International Electrotechnical Commission) – that were originally established to meet the needs of industry. Selected standards are harmonized by their equivalent European standards organizations CEN and CENELEC (Comité Européen de Normalization; and Comité Européen de Normalization Electrotechnique), which verify that they are concordant with any relevant EU laws. Their titles are then published in the Official Journal of the EU (OJEU); the first list of harmonized standards relating to the MDR appeared in July 2021 [Citation90] and there have been two updates. So far, all the harmonized medical device standards are general; none relates specifically to AI.
Compliance with ISO standards and harmonized CEN and CENELEC standards is voluntary, although a legal case in 2016 established that harmonized standards that have been cited in the OJEU are a measure of EU law [Citation91]. A manufacturer receives a ‘presumption of conformity’ with EU legal requirements for its device if it meets the specifications of the relevant harmonized standard. If international standards are not available, the European standards organizations may develop European-specific standards. In the absence of standards suitable for harmonization, the European Commission can issue common specifications. For medical devices, this implies that the MDCG has the authority to write common specifications with input from expert panels and expert laboratories. The inter-relationships between standards organizations are illustrated in , which also lists relevant initiatives engaged with the interoperability of information technology.
Figure 2. A simplified landscape of the inter-relationships of European and global bodies engaged in the development of standards for AI medical devices.
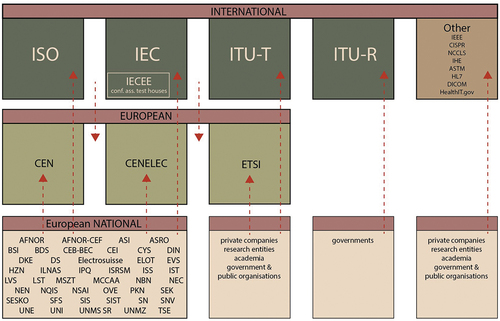
5.1. International and European standardization bodies
The ISO and the IEC established a joint project on AI in 2018; formally, it is conducted by Sub-Committee 42 (on AI) of Joint Technical Committee 1 (on information technology), or ISO/IEC JTC 1/SC 42, with the secretariat being provided by the American National Standards Institute (ANSI). Working Groups (WG) of this committee have published 8 standards (), and at least 25 more are at different stages of preparation (). These foundational standards deal with general issues such as definitions (WG 1). None is focused on medical applications of AI but some may have relevance for medical devices, such as the first report from WG3 in 2020 on trustworthiness. In future, WG3 will work on methods for verifying the performance of neural networks [Citation92].
Table 7. Information technology standards relating to general aspects of AI, that have already been published.
Table 8. General standards under joint development by the International Standardization Organization (ISO) and the International Electrotechnical Commission (IEC), related to artificial intelligence.
For general guidance relating to medical devices, the EU regulatory system applies ISO 13485 about quality management systems [Citation93], ISO standard 14,155 which describes the clinical investigation of medical devices [Citation94] (or more correctly their equivalents EN ISO 13485:2016, and EN ISO 14155:2020, but note that only the first of these has been harmonized to the MDR), and ISO 14971 on risk management [Citation95]. ISO 14155 recommends principles for clinical evaluation and mentions types of studies (in Annex I) but it does not prescribe particular methodologies or clinical trials whether for AI systems or any other type of medical device. A new and more specific ISO standard concerning clinical evaluation for medical devices has been proposed, to recommend methodologies for collecting and appraising clinical data across the full lifecycle of a device. If formally adopted, then responsibility for developing and producing the standard within 3 years will be passed to the same working group that prepared ISO 14155 (namely ISO/TC 194 WG4).
The European bodies CEN and CENELEC also have a joint technical committee on AI (Joint CEN-CENELEC Technical Committee 21 or CEN-CLC/JTC 21) that was created in 2021. A third EU standardization organization, called the European Telecommunications Standards Institute (ETSI), is the European equivalent of the Telecommunications sector of the International Technology Union (ITU-T). In addition to telecommunications and broadcasting, ETSI is concerned with standards for other electronic communication networks and services, and it has an Industry Specification Group on ‘Securing Artificial Intelligence’ and an Operational Coordination Group on AI. ETSI is recognized by the EU as the preferred partner for ICT standards supporting EU strategic objectives relating to health.
While the legislative process on the AI Act is continuing, the European Commission is seeking to speed up the development of harmonized standards and have them available by the time the regulation is applicable, which could be in late 2024 or early 2025. To this aim, on 20 May 2022 the Commission issued a draft standardization request to CEN, CENELEC, and ETSI in support of safe and trustworthy AI [Citation96]. CEN and CENELEC have responded by setting up a Standardization Request Ad Hoc Group (SRAHG) on AI.
A comprehensive overview and comparison of the contents and recommendations of international standards relevant to AI was prepared for AI Watch in the EU [Citation97].
5.2. The Institute of Electrical and Electronics Engineers (IEEE)
The division of IEEE that is most relevant for medicine is the IEEE Engineering in Medicine and Biology Society (EMB). It has a Standards Committee (EMB-SC) which recommends engineering practices to be followed by electrical and electronic manufacturers and health care providers. IEEE prefers to use the term autonomous and intelligent systems (A/IS), rather than AI which it considers too vague [Citation10]. There is a standard that specifies taxonomy and definitions for human augmentation, which refers to technologies including many implants and wearables that enhance human capability. IEEE also has a global initiative on the ethics of A/IS, whether they are physical robots or software such as medical diagnostic systems [Citation10]. lists some IEEE projects that are developing guidance on medical AI.
Table 9. Selected regulatory initiatives and proposals under development.
6. Initiatives of medical device regulators and intergovernmental organizations
The promotion of AI has become a political priority, with countries competing to become global leaders [Citation98,Citation99]. The trend has been accompanied by a growing recognition that AI needs regulation, so now international and national regulatory bodies, academic institutions, and other organizations are developing standards and guidance. A voluntary registry maintained by IEEE, called OCEANIS (Open Community for Ethics in Autonomous and Intelligent Systems) currently lists 77 documents [Citation100]. Many initiatives are not explicitly related to medical devices and those that mainly provide general recommendations overlap significantly. We understand that practical elements to guide the manufacturer, procurer, and user of medical AI systems are most clear in IMDRF, FDA, and NMPA (National Medical Products Administration of China) documents.
The following paragraphs and provide an overview of some current initiatives, with a focus on global projects and those of particular relevance to the European Union.
6.1. International Medical Device Regulators Forum
The IMDRF Working Group on SaMD published its first report in 2013, on definitions, and then a framework for regulatory evaluation, in 2014 [Citation4]. It states that the transparency of inputs used and the technological characteristics ‘do not influence the determination of the category of SaMD’ [Citation4]. Its recommendation that regulators should consider SaMD according to the context in which it is used and the risks of its use has become widely accepted. The two criteria that should be applied were summarized in their 2017 recommendations on clinical evaluation, as the ‘intended medical purpose’ (whether to treat or diagnose/drive clinical management/or inform clinical management) and the ‘targeted healthcare situation’ (whether critical/serious/or non-serious) [Citation33]. For the verification of product performance, the IMDRF specifies under analytical and technical validation ‘accuracy, reliability, (and) precision’; and for clinical validation, it refers to ‘sensitivity, specificity, (and) odds ratio.’
The SaMD Working Group completed its tasks but has been reinstated so that it can revise the IMDRF guidance. Another IMDRF working group, on Artificial Intelligence-based Medical Devices, has considered key terms and definitions for ML-enabled medical devices. The EU participates through delegates from the Danish, German, French and Portuguese national regulatory agencies, with a policy officer from the European Commission.
6.2. World Health Organization
The WHO secretariat and members have agreed on six principles that should be followed to ensure that AI works for the public interest in all countries, limiting risks and maximizing the opportunities intrinsic to its use for health. For AI regulation and governance, these include protecting human autonomy and ensuring transparency, explainability and intelligibility [Citation47].
There is a joint initiative between WHO and ITU, called the ITU-WHO Focus Group on Artificial Intelligence for Health (FG-AI4H). The topics on its agenda include regulatory best practices and specification of requirements. The group ‘has tasked itself’ to work toward creating evaluation standards [Citation123,Citation124].
6.3. European Union
The Directorate-General for Health and Food Safety of the European Commission (DG SANTE) coordinates the implementation of the MDR. Unit D3 (Medical Devices) collaborates with national regulators through the MDCG, which has a working group on New Technologies that is composed of competent authorities (regulatory agencies) from the Member States, and which is attended by delegates from stakeholder organizations including some European medical professional associations. Its terms of reference include responsibility for advising on the regulation of MDSW [Citation125] and its work programme includes exploration of the need for guidance on MDSW that is intended to treat, and on requirements for AI devices.
The European Medicines Agency (EMA) has similar responsibilities relating to pharmaceutical products. The exploitation of AI in decision-making is one of its strategic goals, and the development of methodological guidance for AI in clinical trials is one of its priority actions (see ) [Citation126,Citation127]. EMA is a member of the International Coalition of Medicines Regulatory Authorities (ICMRA) which recommended in a report on AI in 2021 that ‘Regulatory guidelines for AI development, validation and use with medicinal products should be developed in areas such as data provenance, reliability, transparency and understandability, pharmacovigilance, and real-world monitoring of patient functioning’ [Citation128]. That agenda is remarkably similar to the tasks being set by medical device regulators. The ICMRA report even used as its example an app (rather than a drug), and it cited IMDRF documents and identified ‘a need to establish clear mechanisms for regulatory cooperation between medicines and medical device competent authorities and notified bodies to facilitate the oversight of AI-based software intended for use in conjunction with medicinal products’ [Citation128].
The role of the Scientific Foresight Unit of the European Parliament is to provide expert scientific advice to members of the European Parliament (MEPs), through their Panel for the Future of Science and Technology (STOA). At a meeting in January 2020, STOA launched its Centre for AI (C4AI) with a mandate to provide strategic advice to MEPs until the end of the current European parliamentary term in 2024 [Citation129]. Already in April 2019, STOA had proposed a governance framework for algorithmic accountability and transparency [Citation104], and during 2022 it has prepared policy options for data governance [Citation130] and a report on the applications, risks, and ethical and societal impacts of AI in healthcare [Citation38]. The C4AI publishes briefing documents, and has launched a ‘Partnership on AI’ with the OECD Global Parliamentary Network.
The EU AI Watch has been productive. Its publications include an overview of the landscape of standardization for AI in Europe [Citation97] and reports on AI applications in medicine and healthcare [Citation131] and their uptake [Citation132]. AI Watch has identified nine international standards that can support the proposed AI Act [Citation97].
The European Commission appointed 52 experts to form the High-Level Expert Group on Artificial Intelligence (HLEG). Its report on ethics made general recommendations and referred to autonomous AI for ‘deciding the best action(s)’ [Citation11]. It referred to the potential of AI to improve health and well-being, with examples (in footnotes) based on assumptions that earlier or more precise diagnosis would translate into better outcomes or more targeted treatments or more lives saved. The horizontal approach taken by the HLEG needs to be supplemented by recommendations for clinical AI tools, that should be prepared with the active involvement of medical experts and propose standards against which such assumptions could be tested. The HLEG also prepared a tool for assessing AI, with more than 100 questions on human agency and oversight, technical robustness and safety, privacy and data governance, transparency, diversity, nondiscrimination and fairness, societal and environmental well-being, and accountability [Citation133].
6.4. Organisation for Economic Co-operation and Development
The OECD AI Policy Observatory report in 2019, known as the OECD AI Principles [Citation45], was the first intergovernmental standard on AI and it provided the basis for the 2019 G20 AI Principles. They are relatively nonspecific but include transparency, explainability, and accountability. Their implementation was recently reviewed by an OECD working group on national AI policies [Citation134].
A Working Party on Artificial Intelligence Governance (WP AIGO) was created by OECD in March 2022, with members including a Legal and Policy Officer from the European Commission (Directorate-General for Communications Networks, Content and Technology, DG CNECT). The AIGO mandate until the end of 2023 includes supporting the implementation of OECD standards relating to AI, and developing tools, methods and guidance to advance the responsible stewardship of trustworthy AI [Citation135]. The tasks are not focussed on clinical applications of AI, and the working party will report to the OECD Committee on Digital Economy Policy. It will cooperate with other international organizations and initiatives with complementary activities on AI, including the Global Partnership on AI (GPAI), the Council of Europe, and the United Nations Educational, Scientific and Cultural Organization (UNESCO).
6.5. The Council of Europe
The Council of Europe is an international organization that was founded in 1949 in the wake of the second world war to uphold human rights, democracy and the rule of law. It has a broader reach than the EU and now includes 46 member states. It has a committee (Council of Ministers) that needs to be distinguished from the European Council (of ministers) of the EU.
In September 2019, the Council of Europe set up an Ad Hoc Committee on Artificial Intelligence (CAHAI), which has now been succeeded by a permanent Committee on Artificial Intelligence (CAI). In a recent commentary, one of their directors stated that ‘we clearly need regulation to leave essential decision-making to humans and not to mathematical models, whose adequacy and biases are not controlled’ [Citation136]. The strategic agenda of the Council of Europe will therefore include, by 2028, the issue of AI regulation ‘in order to find a fair balance between the benefits of technological progress and the protection of our fundamental values’ [Citation136].
Among other reports, CAHAI proposed elements for a legal framework on AI such as the right to be informed about the application of an AI system in the decision-making process; and the right to choose interaction with a human in addition to or instead of an AI system [Citation137] (see ). In 2020 the Council of Ministers of the Council of Europe adopted a recommendation that each country should develop appropriate legislative, regulatory and supervisory frameworks, and it called for effective cooperation with the academic community [Citation138]. In 2022 the Steering Committee for Human Rights in the fields of Biomedicine and Health (CDBIO) of the Council of Europe commissioned a technical report and set up a drafting group to consider AI in healthcare and in particular its impact on the doctor-patient relationship [Citation109].
6.6. National regulatory agencies
Initiatives by national regulatory agencies related to the general governance of AI systems are too numerous to mention here. They have been summarized, for example in 2018 by Access Now, which is a global nonprofit organization [Citation139], in 2019 by the OECD AI Policy Observatory which prepared ‘country dashboards’ [Citation140], and in 2022 by AI Watch which reported that 24 EU countries already have their own national policies on standards, with health a high priority [Citation141].
As an example of national approaches to AI in healthcare, UK standards for digital health technologies from the National Institute for Health and Care Excellence (NICE) have been updated [Citation142]. They are relevant for SaMD applications including AI but not for software incorporated in medical devices. The British Standards Institution (BSI) will publish guidance that it has prepared together with the Association for the Advancement of Medical Instrumentation (AAMI) in the USA, on the application of ISO standard 14,971 (risk management) to AI [Citation143]. The Medicines and Healthcare products Regulatory Agency (MHRA) with the FDA and Health Canada recently identified guiding principles for Good Machine Learning Practice for Medical Device Development [Citation144].
In the USA, the FDA has proposed a total product lifecycle (TPLC) regulatory approach to AI – and ML‒based SaMD. Its principles include initial premarket assurance of safety and effectiveness, SaMD prespecifications (SPS), an algorithm change protocol (ACP), transparency, and monitoring of real-world performance (RWP). The paper in April 2019 on a ‘Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning-Based Software as a Medical Device’ [Citation48] has been influential. After public consultation, the FDA published its response in 2021 with an action plan that includes a tailored regulatory framework for AI–ML‒based SaMD and the design of regulatory science methods ‘to eliminate algorithm bias and increase robustness’ [Citation145]. A detailed review of US legislation relevant to AI medical devices was published by the National Academy of Medicine [Citation146].
The Therapeutic Goods Administration (TGA) in Australia revised its approach, in 2021. The new guidance does not specify applications using AI or ML separately from other software as a medical device [Citation147]. It reinforced essential principles for the clinical evaluation of high-risk devices and added special criteria such as transparency and version tracking. Any SaMD applications that are autonomous or which do not allow interpretation or oversight by a clinician of diagnoses or therapeutic recommendations are designated as high-risk [Citation148].
The Chinese government recognizes the development of AI as an essential component of its national strategy [Citation149] and it is developing a national AI standardization subcommittee (SAC/TC28/SC42) led by the China Electronic Standardization Institute (CESI) under the Ministry of Industry and Information Technology (MIIT). This subcommittee participates in ISO/IEC JTC1 SC42 activities on behalf of China [Citation150]. There are three approaches to the general governance of AI: the Cyberspace Administration of China is responsible for rules for online algorithms, the China Academy of Information and Communications Technology for tools for testing and certification of ‘trustworthy AI’ systems, and the Ministry of Science and Technology for establishing AI ethics principles and review boards. Regulatory documents and standards have been published for AI medical devices by the NMPA, but they are not provided in English translations. The implementation of software risk management activities shall be based on the intended use of software (target disease, clinical use, degree of importance, and degree of urgency), usage scenarios (target population, target users, place of use, clinical procedures), and core functions (objects of processing, data compatibility, type of function) and shall be performed throughout the life cycle of the software.
7. European laws with relevance to AI medical devices
Several EU laws may impact how medical AI systems can be used or how much information can or must be disclosed. Their provisions may have to be considered when developing EU guidance on reporting clinical evidence for AI as a medical device.
7.1. Freedom of access to information
The principle that information about clinical evidence for medical devices must be publicly accessible [Citation151] was mentioned as a key element of the reform of the EU regulations. Recital 43 of the MDR states that ‘transparency and adequate access to information […] are essential in the public interest, to protect public health, to empower patients and healthcare professionals and to enable them to make informed decisions’ [Citation12]. Since EU notified bodies are independent companies, however, they are excluded from the provisions of the EU legislation on freedom of access to documents [Citation152].
The opinions of the Expert Panels which provide independent scientific review of the Clinical Evaluation Assessment Reports prepared by notified bodies, are now being published by the European Commission with the name of the individual medical device and parts of the content redacted – despite the fact that the EMA, which is managing the panels, is an agency of the European Commission and therefore subject to the law on freedom of access. The specific name of a device is published later, but only if it has been granted a certificate of conformity. Further legal opinion and clarification of the law may be needed, or this interpretation may need to be contested.
7.2. The AI act proposal
The EU AI Act proposes to regulate high-risk AI systems that could have an adverse impact on the safety or fundamental rights of people [Citation153]. Unless its text is amended, providers of high-risk AI systems (most likely manufacturers of AI medical devices [Citation154]) will have to register them in the EU database before placing them on the market. As part of their conformity assessment for the MDR, AI medical devices will have to comply with requirements dealing with risk management and quality criteria concerning the training, validation, and testing of data sets. Manufacturers will have to provide technical documents describing compliance with applicable rules and keep records that ensure an appropriate level of traceability of the system’s functioning including accuracy, robustness, and cybersecurity throughout its lifecycle. This is intended to enable users to interpret the output and have human oversight of an AI system. An AI medical device will fall within the Act’s scope if a notified body is involved in its conformity assessment under sectorial legislation.
7.3. Data laws
The EU General Data Protection Regulation (GDPR) [Citation155] sets horizontal rules, which apply to medical devices that process personal data. Guidance from the EU Data Protection Board has clarified how the GDPR relates to the analysis of clinical data for research [Citation156]. The rules of the GDPR that are most relevant to the processing of personal data by AI systems in healthcare include Article 9 concerning the processing of special categories of personal data (which include health data and data concerning health) and Articles 13–15 which establish rights to ‘meaningful information about the logic involved’ in automated decisions made by AI.
The European Commission has recently initiated reforms concerning the regulation of data, including the Data Governance Act (DGA) [Citation157] and proposals for a European Health Data Space (EHDS) [Citation158] and a Data Act [Citation159]. The DGA was approved in 2022 and will become applicable from September 2023; its objective is to foster the availability of data for use in the economy and society, including in healthcare settings. The EHDS proposal was published in May 2022; its objectives include regulating the sharing of electronic health data. It sets additional personal rights and mechanisms designed to complement those already provided under the GDPR for electronic health data. The Data Act proposal is aimed to apply horizontally also to medical and health devices (Recital 14) [Citation160]; the proposal includes harmonized rules on fair access to and use of (health) data for the so-called relationships of ‘business to consumer,’ ‘business to business,’ and ‘business to government.’ Transposed to the medical device environment, it implies that new rules could govern access to data and its sharing between medical device manufacturers and hospitals or patients, among manufacturers themselves, or from manufacturers to public health bodies.
7.4. Intellectual property rights
There is a European Parliament resolution of 20 October 2020 on intellectual property rights for AI technologies [Citation161]. The European Patent Organization (EPO) manages a long- and well-established system in Europe that has a category known as computer-implemented inventions (CII). Mathematical models and algorithms are not patentable under the European Patent Convention, but technical aspects of AI inventions are generally patentable as a subgroup of CII [Citation162]. Source code could be copyrighted. Trade secrets may provide the broadest scope of IP protection. There may be tensions between some of these legal provisions and the need for transparency about the function and outcomes of AI applications used in clinical practice.
7.5. Liability laws
Liability laws protect individuals from defects in manufactured products. In the EU, this has been regulated since 1985 through Directive 85/374/EEC on liability for defective products [Citation163]. EU Directives as legal instruments require implementation by the Member States, so liability schemes usually depend on national laws and their doctrinal and judicial orientations. The Directive sets general rules of strict liability that are relevant to medical devices, and it has served as an interpretative basis for claims by patients [Citation164,Citation165]. It is under review after the European Commission set up an expert group to advise on liability law and challenges relating to the complexity, openness, autonomy, predictability, data-drivenness or vulnerability of new technologies [Citation166].
With regard to the risks created by emerging digital technologies, the EU Expert Group noted in its 2019 report that the existing rules on liability offer solutions which do ‘not always seem appropriate’ [Citation167]. Later, the European Commission issued a ‘Report on the safety and liability implications of artificial intelligence, the internet of things and robotics’ which highlighted legal fragmentation across Europe concerning liability paradigms as applied to AI, the complexity of digital ecosystems, and the plurality of actors involved [Citation168]. It stated that this situation ‘could make it difficult to assess where a potential damage originates and which person is liable for it’ [Citation168]. Further studies have commented on possible shortcomings of AI liability and on the European Commission’s reports and initiatives [Citation166,Citation169]. Following a public consultation, the Commission has proposed a new AI Liability Directive to harmonize national liability rules [Citation170].
7.6. Cybersecurity of AI medical devices
Ensuring the cybersecurity of AI medical devices and the healthcare systems of which they are a part is crucial to maintaining patients’ health and safety [Citation171]. In 2019 the MDCG issued the first EU guidance on medical device cybersecurity [Citation172], but the first EU-wide general cybersecurity legislation was the Network and Information Security Directive (NIS Directive) of 2016 [Citation173]. Its aim is to enhance the security of entities providing digital or essential services, which includes healthcare service providers that Member States have recognized as Operators of Essential Services (OES). The EU has proposed to recast the NIS Directive to expand the scope of its application to include medical devices, in which case security measures and risk management obligations would apply to medical device manufacturers. In addition, in 2019 the Cybersecurity Act (CSA) strengthened the role and tasks of the EU Agency for Cybersecurity (known as ENISA) and established a new cybersecurity certification framework for products and services [Citation174]. The Radio Equipment Directive also applies in certain circumstances to medical devices [Citation171,Citation175] and Article 32 of the GDPR is relevant to the security of data processing [Citation171].
8. Transparency and interpretability of AI medical device software
The transparency and interpretability of AI systems are key determinants of risk that should be factored into regulatory processes and decisions, but neither has a universally accepted definition, and the terms are often used interchangeably in the literature. Transparency was one of the seven key requirements for trustworthy AI identified by the EU HLEG [Citation11].
8.1. Transparency
Transparency may be defined as the availability of information about each component of an AI system, namely the training data, the model structure, the algorithms used for training, deployment, pre- and post-processing of data, and the objective function of the AI system. Transparency has been proposed as a fundamental principle for the ethical use of AI [Citation176] since it allows the performance of an AI system to be characterized across a range of conditions and it enables both regulatory audits and the identification of vulnerabilities and biases [Citation104].
A system might be made fully transparent by openly publishing all relevant details including the data and programme code, but sometimes that may be unnecessary and/or unfeasible. The privacy of patients’ clinical data must be protected, for example by ensuring secure encryption before data are transmitted or by using a distributed or federated analysis when data are combined for reporting the performance of an AI device. A compromise may need to be struck for commercial applications to protect intellectual property. Transparency is important and can be necessary for patients, so it would be useful to have consensus on when they should be informed that AI is being used [Citation177].
Regulators who assess the clinical evidence presented for an AI system should focus firstly on the transparency of the data and methods, which is vital for determining the strength of the research methodology and the results presented, and secondly on how thoroughly it has been evaluated. It may not be essential to understand every aspect of the detailed methods for specific measurement tasks, as long as the training data are characterized for example with details of inclusion and exclusion criteria, demographic and relevant physiological characteristics, and as long as the model’s performance is fully reported. A review of open-access datasets used to develop AI tools for diagnosing eye diseases showed poor availability of sufficient metadata [Citation178].
8.2. Interpretability
Full transparency of methods is a precondition for interpretability [Citation179]. We consider an interpretable system to be one that a human with relevant knowledge and experience can understand. The degree of interpretability ranges from a global understanding of the inner workings of a system, to the ability to understand each individual prediction made by the AI system. A more interpretable model poses less risk as it could allow human oversight and easier identification of errors. Consider, for example, an AI system for measuring the volume of tumors on a computed tomography (CT) scan. An end-to-end solution that presents only a numeric value lacks interpretability, but if intermediate results are presented such as an illustration of the AI delineation (segmentation) of the tumor, then it is more easily interpretable by a clinician. This approach also allows the assignment of accountability to the interpreting clinician.
Many factors influence the overall interpretability of an AI system, but the complexity of the underlying model is a key determinant. Some AI algorithms, such as decision trees, are easy to understand and have predictable deterministic performance, while artificial neural networks can have up to 175 billion parameters [Citation180] which obfuscates their interpretation. Even simple decision trees lack interpretability when they are used as part of ensemble learning, where many decision trees are combined to form stochastic algorithms such as random forests or gradient-boosted trees [Citation181]. In such cases additional aids, post-hoc analysis, or surrogate models can sometimes explain the AI system’s results [Citation9]. Feature importance scoring, using methods such as SHAPely additive explanation [Citation182], is another approach that may increase system interpretability.
A model that reports the uncertainty associated with each prediction aids the usability of an algorithm, without necessarily improving interpretability and transparency. If a set of possible outcomes and their associated probabilities are presented, rather than a single dichotomous classification, then clinicians can integrate this information into their clinical decision-making [Citation183].
8.3. Explainability
Many authors state that transparency for a medical AI system that uses a neural network (deep learning) needs to include the ability of the user to understand the structure of the network and the features that it has identified; interpretability then refers to the ability to understand the inner mechanics of the model. For example, if ‘the associations between the individual features and the outcome remain hidden, the predictions are difficult to scrutinize and to question […] underlining the importance of explicability, also to avoid incorrectly inferring that the strongest associations between features and outcome indicate causal relations’ [Citation184].
There is a contrary view that the benefits of understanding the detailed internal operation of a neural network are overrated [Citation185]. Even if a ‘black box’ is impenetrable, the performance of an algorithm can always be understood and interpreted at a higher level; ‘rather than seeking explanations for each behaviour of a computer system, policies should formalize and make known the assumptions, choices, and adequacy determinations associated with a system’ [Citation186]. Markus and colleagues provide a useful summary of the components of explainability and interpretability [Citation187]. These authors suggest that for clinical applications, criteria such as the quality of the input data, the robustness or reproducibility of the algorithm, and the results of external validation, are more important. Explainability would then be defined as the ability to explain the behavior of an ML model in human terms.
8.4. Implications for regulatory guidance
There is considerable heterogeneity between AI models even within the same class of algorithm, and providing a taxonomy of AI methods linked to their regulatory implications would be challenging. Nonetheless within each category of ML algorithms, it may be helpful to understand which methods are more or less transparent since those criteria will impact on how the evidence is assessed. Many gradings of software are qualitative and subjective, but a pragmatic approach could provide a summary of simple criteria of AI systems that notified bodies and regulators can assess.
9. Conclusions
Medical devices incorporating AI are being deployed rapidly and are already widely used. On 5 October 2022 the FDA listed 521 AI-enabled medical devices that it has approved since 1997 [Citation188]. Regulatory guidance needs to keep pace with this fast progress and with technological developments. It would make sense for regulatory efforts to be concentrated on gaps in advice [Citation36] or on challenges that are unique to AI devices, such as how to assure that software is applied only in the circumstances for which and for individuals in whom its use has been validated, how to cope with the need for approval of iterative changes in software that may be self-learning, and how to conduct appropriate post-market surveillance. Special provisions for AI-related aspects of each class of medical devices would best be an integral part of their overall evaluation requirements, ideally described in harmonized standards or, when lacking, common specifications. The primary focus should always be on the purpose of the AI rather than the technology.
10. Expert opinion
We have identified considerable concordance between recommendations about AI, and a remarkable plethora of stakeholders involved in developing guidance and regulations (). Their varying constitutions, authority, responsibilities and acronyms (such as FG-AI4H, C4AI, WP AIGO, GPAI, CAI, and SRAHG; all explained in this review) have become confusing and their collective but apparently uncoordinated engagement is probably counterproductive. There are likely to be other and more recent initiatives of which we are unaware. Unless they are all coordinated, there is a real risk of over-regulation.
Figure 3. Overview of European and global institutions and organizations engaged in the development of regulations for medical AI systems.

Principle 2.5 from the OECD, on international cooperation for trustworthy AI, states that ‘Governments should promote the development of multi-stakeholder, consensus-driven global technical standards for interoperable and trustworthy AI.’ A report from the Council of Europe says that ‘robust testing and validation standards should be an essential pre-deployment requirement for AI systems in clinical care contexts’ [Citation109]. Many other organizations reached similar conclusions. We would add that for medical AI systems as for all high-risk medical devices, standards should be based on scientific evidence and proportionate to the clinical risks. Within the EU there should be collaboration between medical device regulators and EMA, and at the international level there should be collaboration between IMDRF and ICMRA. A concrete and practical initiative for global regulatory convergence would be sensible.
10.1. Concordance needed for scope of definitions
Precise regulatory definitions of what qualifies as an AI system that will need approval as a medical device vary between jurisdictions. In the USA, the FDA does not consider software that has the purpose (only) to ‘inform’ to be a medical device, if the software is used by a healthcare professional who is expected to be able to review the evidence underlying the information, independently [Citation189]. Similarly, Australian regulations exclude clinical decision support software with limited functions that does not replace the judgment of a healthcare professional [Citation190]. The scope of FDA regulations on SaMD and ML has been further narrowed by excluding software that does not receive, analyze or otherwise process a medical image or signal from a medical device or an in vitro diagnostic medical device or from any other signal acquisition system, and by specifying that a healthcare professional must be able to understand the basis of its recommendations (but not necessarily the exact processes) [Citation191].
All low-risk software that used to be class I was deregulated under the US 21st Century Cures Act of 2016 [Citation192]. In the EU, software with a search function does not qualify [Citation5] but AI algorithms that calculate a prognostic score are designated as medical devices. Logistic regression software for routine checkups by healthcare professionals does not qualify as SaMD in the USA, while it can be considered as MDSW in the EU. Concordance of scope and regulatory requirements would be preferable.
10.2. Rationale for standards
The personal risks arising from autonomous systems that employ AI for non-medical applications appear greater than occur currently with the vast majority of AI applications in healthcare, and thus they indeed merit public safeguards as envisaged by the EU AI Act. Specific standards for AI medical devices, however, should be determined by their particular clinical risks. That has been the focus of the IMDRF documents, and more recently of the ICMRA report since it concurred that ‘regulators may need to apply a risk-based approach to assessing and regulating AI’ [Citation128]. Standards for assessing AI as a medical device should consider both the components of the system and the context in which it is deployed [Citation193]. The need for transparency as the foundation for evidence-based clinical practice extends to the standards applied for AI devices. European harmonized standards undergo public consultation, but it is unsatisfactory that ISO and IEC standards are developed without open public consultation and available only for purchase.
Concepts have become clouded by the inappropriate use of anthropomorphic language to describe functions of AI algorithms that their developers and users have determined, and by the tendency to refer to ‘AI’ as a single technology. AI tools do not replicate the thought processes employed by clinicians in practice [Citation194] so it might be wiser to refer to human intelligence augmented by computer algorithmic tools. Current vocabulary leads to the idea that AI somehow is different – but the basic principles for its regulation in healthcare are not. All high-risk medical devices should be evaluated to establish evidence of clinical benefit and minimal risk before they are approved. It is noteworthy that the fields that are common to all the professional recommendations (see : data acquisition, preprocessing, model, study population, performance, benchmarking, and data availability) are relevant not only for studies of AI medical devices but also for all clinical research studies and statistical analyses.
10.3. Determinants of risk for classifying software
Expert review by scientists and clinicians would clarify which AI systems should be regarded as high-risk (class IIb or class III) medical devices, and which methodologies would then be indicated for clinical evaluation to demonstrate their scientific validity. The CORE-MD project will consider those questions and prepare recommendations separately, taking note of relevant questions that have been suggested or are being prepared by other authors [Citation36,Citation76,Citation195–197]. Although it has not been a focus of this review, similar considerations would apply to the designation of in vitro diagnostic medical devices (IVDs) that use AI, either as Class C or Class D [Citation198].
Declarations of interest
AGF is Chairman of the Regulatory Affairs Committee of the Biomedical Alliance in Europe, and Scientific Coordinator of the CORE-MD consortium; he attends the Clinical Investigation and Evaluation Working Group of MDCG. He has no financial conflicts of interest. EC has received honoraria as consultant or as speaker from Medtronic, Daiki-Sankyo, and Novartis. KC is an employee of Koninklijke Philips N.V.; he is Chair of the Software Focus Group at COCIR and DITTA, and delegate to the IMDRF working groups on Software as a Medical Device and AI-based Medical Devices. He is a Belgian expert member contributing to international standards committees at IEC/TC62 Electrical equipment in medical practice, ISO/TC215 Health Informatics, ISO/TC210 Quality management, ISO/IEC JTC 1/SC 42 Artificial Intelligence, and CEN/CENELEC JTC21 Artificial Intelligence. RHD has share options in Mycardium.AI. SHG declares no non-financial interests; he is an advisor/consultant for Ada Health GmbH and holds share options; and he has consulted for Una Health GmbH, Lindus Health Ltd. and Thymia Ltd. LH is an employee of Elekta; he is chair of the clinical committees of COCIR and DITTA; industry stakeholder at IMDRF working groups on Clinical Investigation, Evaluation, PMCF and IVD Performance Evaluation; industry stakeholder at ISPOR (WHO) Health Technology Assessment; and industry stakeholder at the MDCG Working Groups on Clinical Investigation & Evaluation, and Borderline and Classification (Software and Risk Classification). GMcG is a member of the Medical Device Coordination Group (MDCG) working group on Clinical Investigation and Evaluation (CIE), and of the International Standardization Organisation (ISO) technical committee for implants for surgery (ISO/TC 150). GO’C is a member of the Clinical Investigation and Evaluation subgroup of MDCG. ZK is an employee of Dedalus HealthCare GmbH and represented industry in European Task Force on Clinical Evaluation of MDSW (MDCG2020-1). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewers disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
Acknowledgments
We thank Paul Piscoi and Nada Alkhayat, Unit D3, DG SANTE, European Commission for their advice, Piotr Szymanski for his suggestions, and Yijun Ren for help in accessing the documents of the NMPA.
Additional information
Funding
References
- McCarthy J, Minsky M, Rochester N, et al. A proposal for the dartmouth summer research project on artificial intelligence. August 1955. Available from: file:///C:/Users/wcgagf/Downloads/McCarthy,%20Minsky,%20Rochester,%20Shannon%20(1955)%20-%20A%20Proposal%20for%20the%20Dartmouth%20Summer%20Research%20Project%20on%20Artificial%20Intelligence.pdf (Accessed 2022 May 19.
- Bishop JM. Artificial intelligence is stupid and causal reasoning will not fix it. Front Psychol. 2021; 11: 2603. www.frontiersin.org/article/10.3389/fpsyg.2020.513474.
- Faes L, Sim DA, van Smeden M, et al. Artificial intelligence and statistics: just the old wine in new wineskins? Front Digit Health. 2022;4:833912.
- International Medical Device Regulators Forum. Software as a Medical Device (SaMD) Working Group. “Software as a Medical Device”: possible framework for risk categorization and corresponding considerations. IMDRF/SaMD WG/N12FINAL. 2014. Available from: https://www.imdrf.org/documents/software-medical-device-possible-framework-risk-categorization-and-corresponding-considerations.
- Medical Device Coordination Group. MDCG 2019-11: Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR. Available from: https://ec.europa.eu/health/system/files/2020-09/md_mdcg_2019_11_guidance_qualification_classification_software_en_0.pdf.
- Samoili S, López Cobo M, Gómez E, et al. AI watch. Defining artificial iIntelligence. Towards an operational definition and taxonomy of artificial intelligence. EUR 30117 EN. Luxembourg: Publications Office of the European Union; 2020.
- ISO/IEC 22989:2022 Information technology - Artificial intelligence - Artificial intelligence concepts and terminology. Available from: https://www.iso.org/standard/74296.html
- Doshi-Velez F, Kim B Towards a rigorous science of interpretable machine learning. 2017. ArXiV pre-print. Available from: https://arxiv.org/abs/1702.08608
- Rudin C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead |. Nature Mach Intell. 2019;1:206–215.
- IEEE Global Initiative on ethics of autonomous and intelligent systems. Ethically aligned design: a vision for prioritizing human well-being with autonomous and intelligent systems. First. IEEE. 2019; Available from: https://ethicsinaction.ieee.org/wp-content/uploads/ead1e.pdf
- High-Level Expert Group. European Commission, Directorate-General for Communications Networks, Content and Technology. Ethics guidelines trustworthy AI, 2019. Available from: https://data.europa.eu/doi/10.2759/346720.
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745.
- Directive 2007/47/EC of the European Parliament and of the Council of 5 September 2007 amending Council Directive 90/385/EEC on the approximation of the laws of the Member States relating to active implantable medical devices, Council Directive 93/42/EEC concerning medical devices and Directive 98/8/EC concerning the placing of biocidal products on the market. Available from: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:247:0021:0055:en:PDF
- Medical Device Coordination Group. MDCG 2020-1: guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software. March 2020. Available from: https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2020_1_guidance_clinic_eva_md_software_en_0.pdf.
- Hwang TJ, Kesselheim AS, Vokinger KN. Lifecycle regulation of artificial intelligence– and machine learning–based software devices in medicine. JAMA. 2019;322(23):2285–2286.
- Muehlematter UJ, Daniore P, Vokinger KN. Approval of artificial intelligence and machine learning-based medical devices in the USA and Europe (2015–20): a comparative analysis. Lancet Digit Health. 2021;3(3):e195–e203.
- Benjamens S, Dhunnoo P, Meskó B. The state of artificial intelligence-based FDA-approved medical devices and algorithms: an online database. Npj Digital Medicine. 2020;3(1):118.
- van Leeuwen KG, Schalekamp S, Rutten MJCM, et al. Artificial intelligence in radiology: 100 commercially available products and their scientific evidence. Eur Radiol. 2021;31:3797–3804.
- Liu X, Faes L, Kale AU, et al. A comparison of deep learning performance against health-care professionals in detecting diseases from medical imaging: a systematic review and meta-analysis. Lancet Digit Health. 2019;1:e271–e297.
- Ferrante Di Ruffano L, Takwoingi Y, Dinnes J, et al. Computer‐assisted diagnosis techniques (dermoscopy and spectroscopy‐based) for diagnosing skin cancer in adults. Cochrane Database Syst Rev. 2018;12(CD013186). DOI:10.1002/14651858.CD013186
- Christodoulou E, Ma J, Collins GS, et al. A systematic review shows no performance benefit of machine learning over logistic regression for clinical prediction models. J Clin Epidemiol. 2019;110:12–22.
- Nagendran M, Chen Y, Lovejoy CA, et al. Artificial intelligence versus clinicians: systematic review of design, reporting standards, and claims of deep learning studies. BMJ. 2020;368:m689.
- Wu E, Wu K, Daneshjou R, et al. How medical AI devices are evaluated: limitations and recommendations from an analysis of FDA approvals. Nat Med. 2021;27:582‒4.
- Kelly BS, Judge C, Bollard SM, et al. Radiology artificial intelligence: a systematic review and evaluation of methods (RAISE). Eur Radiol. 2022;32:7998‒8007.
- Plana D, Shung DL, Grimshaw AA, et al. Randomized clinical trials of machine learning interventions in health care: a systematic review. JAMA Network Open. 2022;5(9):e2233946.
- Lyell D, Coiera E, Chen J, et al. How machine learning is embedded to support clinician decision making: an analysis of FDA-approved medical devices. BMJ Health Care Inform. 2021;28:e100301.
- Vasey B, Ursprung S, Beddoe B, et al. Association of clinician diagnostic performance with machine learning-based decision support systems: a systematic review. JAMA Network Open. 2021;4:e211276.
- Yang C, Kors JA, Ioannou S, et al. Trends in the conduct and reporting of clinical prediction model development and validation: a systematic review. J Am Med Inform Assoc. 2022;29:983–989.
- Wynants L, Van Calster B, Collins GS, et al. Prediction models for diagnosis and prognosis of covid-19: systematic review and critical appraisal. BMJ. 2020;369:m1328. Update in: BMJ. 2021 Feb 3;372:n236
- European Commission. SC1-HCO-18-2020: developing methodological approaches for improved clinical investigation and evaluation of high-risk medical devices. Available from: https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-details/sc1-hco-18-2020;callCode=H2020-SC1-BHC-2018-2020;freeTextSearchKeyword=;matchWholeText=true;typeCodes=1;statusCodes=31094501,31094502,31094503;programmePeriod=null;programCcm2Id=null;programDivisionCode=null;focusAreaCode=null;destination=null;mission=null;geographicalZonesCode=null;programmeDivisionProspect=null;startDateLte=null;startDateGte=null;crossCuttingPriorityCode=null;cpvCode=null;performanceOfDelivery=null;sortQuery=sortStatus;orderBy=asc;onlyTenders=false;topicListKey=callTopicSearchTableState (Accessed 2022 Jun 16
- Fraser AG, Nelissen RGHH, Kjærsgaard-Andersen P, et al. CORE-MD Investigators. Improved clinical investigation and evaluation of high-risk medical devices: the rationale and objectives of CORE-MD (Coordinating Research and Evidence for Medical Devices). Eur Heart J Qual Care Clin Outcomes. 2022;8:249–258.
- International Medical Device Regulators Forum, SaMD Working Group. Software as a Medical Device (SaMD): Key Definitions. IMDRF/SaMD WG/N10FINAL:2013. Available from: https://www.imdrf.org/documents/software-medical-device-samd-key-definitions
- International Medical Device Regulators Forum, SaMD Working Group. Software as a medical device: clinical evaluation. IMDRF/SaMD WG/N41: 2017. Available from: https://www.imdrf.org/documents/software-medical-device-samd-clinical-evaluation.
- International Medical Device Regulators Forum, Working Group on Artificial Intelligence Medical Devices. machine learning-enabled medical devices—A subset of artificial intelligence-enabled medical devices: key terms and definitions. IMDRF/AIMD WG (PD1)/N67. Proposed document, 16 Sep 2021. Available from: https://www.imdrf.org/sites/default/files/2021-10/Machine%20Learning-enabled%20Medical%20Devices%20-%20A%20subset%20of%20Artificial%20Intelligence-enabled%20Medical%20Devices%20-%20Key%20Terms%20and%20Definitions.pdf
- High-Level Expert Group on Artificial Intelligence. A definition of AI: main capabilities and scientific disciplines. 2018. Available from: https://ec.europa.eu/futurium/en/system/files/ged/ai_hleg_definition_of_ai_18_december_1.pdf
- Larson DB, Harvey H, Rubin DL, et al. Regulatory frameworks for development and evaluation of artificial intelligence-based diagnostic imaging algorithms: summary and recommendations. J Am Coll Radiol. 2021;18:413–424.
- European Commission. Proposal for a Regulation of the European Parliament and of the Council laying down harmonised rules on artificial intelligence (Artificial Intelligence Act) And amending certain union legislative acts. Brussels. 2021;21(4):COM(2021) 206 final 2021/0106 (COD).
- Lekadir K, Quaglio G, Tselioudis Garmendia A, et al. Artificial intelligence in healthcare. Applications, risks, and ethical and societal impacts. European Parliamentary Research Service - Scientific Foresight Unit (EPRS - STOA). PE 729.512 – June 2022. Available from: https://www.europarl.europa.eu/RegData/etudes/STUD/2022/729512/EPRS_STU%282022%29729512_EN.pdf.
- International Standardisation Organisation/International Electrotechnical Commission (ISO/IEC). Joint Technical Committee 1/SC 42. Information technology — vocabulary. (2015). ISO/IEC 2382: 2015.(revision under review) Available from: https://www.iso.org/obp/ui/#iso:std:iso-iec:2382:ed-1:v1:en
- ISO/IEC JTC 1 (on information technology)/Sub-committee 42 (on AI). Information technology — artificial intelligence — artificial intelligence concepts and terminology. 2022. 2022–07 ISO/IEC 22989:2022 Available from: https://www.iso.org/obp/ui/#iso:std:iso-iec:22989:ed-1:v1:en
- ISO/IEC JTC 1 (on information technology)/Sub-committee 42 (on AI) IEEE Engineering in Medicine and Biology Society (EMBS). Artificial Intelligence Medical Devices Working Group (EMB/Stds Com/AIMDWG). Available from: https://sagroups.ieee.org/aimdwg/
- IEEE Recommended practice for the quality management of datasets for medical artificial intelligence. 2022 IEEE 2801-2022 Available from: https://standards.ieee.org/ieee/2801/7459/
- Organisation for Economic Co-operation and Development. OECD Science, Technology and Industry Scoreboard 2017: The Digital Transformation. doi: 10.1787/9789264268821-en
- OECD. Council on Artificial Intelligence. OECD, Recommendation of the Council on Artificial Intelligence, OECD/LEGAL/0449. 2019. Available from: https://legalinstruments.oecd.org/en/instruments/OECD-LEGAL-0449
- Expert OECD Group on Artificial Intelligence. Scoping the OECD AI principles: deliberations of the expert group on artificial intelligence at the OECD (AIGO). OECD digital economy Papers No. 291. OECD Publishing; Paris; 2019. doi: 10.1787/d62f618a-en
- OECD Committee on digital economy policy. OECD Framework for the classification of AI systems. (2022). OECD Digital Economy Papers, No. 323.
- World Health Organization. Ethics and governance of artificial intelligence for health. WHO guidance. Geneva (2021) https://www.who.int/publications/i/item/9789240029200.
- Food and Drug Administration, USA. Proposed regulatory framework for modifications to artificial intelligence /machine learning (AI/ML)-based software as a medical device (SaMD). 2019. Available from: https://www.fda.gov/files/medical%20devices/published/US-FDA-Artificial-Intelligence-and-Machine-Learning-Discussion-Paper.pdf.
- U.S. Food & Drug Administration. Content of premarket submissions for device software functions - draft guidance for industry and food and drug administration staff November 4, 2021. Available from: https://www.fda.gov/media/153781/download
- European Commission. Communication from the Commission to the European Parliament, the European Council, the Council, the European Economic and Social Committee and the Committee of the Regions. Artificial Intelligence for Europe. COM/2018/237 final. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=COM%3A2018%3A237%3AFIN
- Samoili S, López Cobo M, Delipetrev B, et al., AI Watch. Defining Artificial Intelligence 2.0. Towards an operational definition and taxonomy for the AI landscape, EUR 30873 EN, Publications Office of the European Union, Luxembourg, 2021.
- Schmitt L. Mapping global AI governance: a nascent regime in a fragmented landscape. AI and Ethics. 2022;2:303–314.
- Salthouse TA. Localizing age-related individual differences in a hierarchical structure. Intelligence. 2004;32:10.1016.
- Deary IJ, Penke L, Johnson W. The neuroscience of human intelligence differences. Nat Rev Neurosci. 2010;11:201–211.
- National Medicine Products Administration, People’s Republic of China. Technical guideline on AI-aided software. 1 July 2021 [in Mandarin] https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20210708111147171.html (accessed 2022 Aug 4
- Medicines and Healthcare products Regulatory Agency. Government response to consultation on the future regulation of medical devices in the United Kingdom, 26 June 2022. https://www.gov.uk/government/consultations/consultation-on-the-future-regulation-of-medical-devices-in-the-united-kingdom
- Liu X, Rivera SC, Moher D. et al; SPIRIT-AI and CONSORT-AI Working Group. Reporting guidelines for clinical trial reports for interventions involving artificial intelligence: the CONSORT-AI Extension. BMJ. 2020;Vol. 370:m3164.
- Rivera SC, Liu X, Chan AW, et al. SPIRIT-AI and CONSORT-AI Working Group. Guidelines for clinical trial protocols for interventions involving artificial intelligence: the SPIRIT-AI Extension. BMJ. 2020;370:m3210.
- Vasey B, Nagendran M, Campbell B, et al. DECIDE-AI expert group. Reporting guideline for the early stage clinical evaluation of decision support systems driven by artificial intelligence: DECIDE-AI. BMJ. 2022;377:e070904.
- Collins GS, Dhiman P, Andaur Navarro CL, et al. Protocol for development of a reporting guideline (TRIPOD-AI) and risk of bias tool (PROBAST-AI) for diagnostic and prognostic prediction model studies based on artificial intelligence. BMJ Open. 2021;11(7):e048008.
- Sounderajah V, Ashrafian H, Golub RM, et al. STARD-AI steering committee. Developing a reporting guideline for artificial intelligence-centred diagnostic test accuracy studies: the STARD-AI protocol. BMJ Open. 2021;11(6):e047709.
- Kitaguchi D, Watanabe Y, Madani A, et al. Computer Vision in Computer Vision in Surgery International Collaborative. Artificial intelligence for computer vision in surgery: a call for developing reporting guidelines. Ann Surg. 2022;275:e609–e611.
- Schwendicke F, Singh T, Lee JH, et al. IADR E-oral health network and the ITU WHO focus group AI for Health. Artificial intelligence in dental research: checklist for authors, reviewers, readers. J Dent. 2021;107:103610.
- Daneshjou R, Barata C, Betz-Stablein B, et al. Checklist for evaluation of image-based artificial intelligence reports in dermatology: CLEAR derm consensus guidelines from the international skin imaging collaboration artificial intelligence working group. JAMA Dermatol. 2022;158:90–96.
- Park Y, Jackson GP, Foreman MA, et al. Evaluating artificial intelligence in medicine: phases of clinical research. JAMIA Open. 2020;3:326–331.
- Sendak MP, D’Arcy J, Kashyap S, et al. A path for translation of machine learning products into healthcare delivery. EMJ Innov. 2020. DOI:10.33590/emjinnov/19-00172.
- McCradden MD, Stephenson EA, Anderson JA. Clinical research underlies ethical integration of healthcare artificial intelligence. Nat Med. 2020;26:1325–1326.
- Higgins D, Madai VI. From bit to bedside: a practical framework for artificial intelligence product development in healthcare. Adv Intell Sys. 2020;2(10):2000052.
- Sengupta PP, Shrestha S, Berthon B, et al. Proposed Requirements for Cardiovascular Imaging-Related Machine Learning Evaluation (PRIME): a checklist: reviewed by the American College of Cardiology Healthcare Innovation Council. JACC Cardiovasc Imaging. 2020;13:2017–2035.
- Lekadir K, Osuala R, Gallin C, et al. FUTURE-AI: guiding principles and consensus recommendations for trustworthy artificial intelligence in medical imaging. https://arxiv.org/pdf/2109.09658.pdf (accessed 2022 Aug 18).
- Norgeot B, Quer G, Beaulieu-Jones BK, et al. Minimum information about clinical artificial intelligence modeling: the MI-CLAIM checklist. Nat Med. 2020;26:1320–1324.
- Hernandez-Boussard T, Bozkurt S, Ioannidis JPA, et al. MINIMAR (MINimum Information for Medical AI Reporting): developing reporting standards for artificial intelligence in health care. J Am Med Inform Assoc. 2020;27:2011–2015.
- Sendak MP, Gao M, Brajer N, et al. Presenting machine learning model information to clinical end users with model facts labels. NPJ Digit Med. 2020;3:41.
- Olczak J, Pavlopoulos J, Prijs J, et al. Presenting artificial intelligence, deep learning, and machine learning studies to clinicians and healthcare stakeholders: an introductory reference with a guideline and a Clinical AI Research (CAIR) checklist proposal. Acta Orthop. 2021;92:513–525.
- Mongan J, Moy L, Kahn CE Jr. Checklist for Artificial Intelligence in Medical Imaging (CLAIM): a Guide for Authors and Reviewers. Radiol Artif Intell. 2020;2:e200029.
- Vollmer S, Mateen BA, Bohner G, et al. Machine learning and artificial intelligence research for patient benefit: 20 critical questions on transparency, replicability, ethics, and effectiveness. BMJ. 2020;368(l):6927.
- Scott I, Carter S, Coiera E. Clinician checklist for assessing suitability of machine learning applications in healthcare. BMJ Health & Care Informatics. 2021;28:e100251.
- Petzold A, Albrecht P, Balcer L, et al. IMSVISUAL, ERN-EYE Consortium. Artificial intelligence extension of the OSCAR-IB criteria. Ann Clin Transl Neurol. 2021;8:1528–1542.
- EQUATOR Network. Available from: https://www.ndorms.ox.ac.uk/research/research-groups/equator-network
- Shelmerdine SC, Arthurs OJ, Denniston A, et al. Review of study reporting guidelines for clinical studies using artificial intelligence in healthcare. BMJ Health Care Inform. 2021;28:e100385.
- Skivington K, Matthews L, Simpson SA, et al. A new framework for developing and evaluating complex interventions: update of medical research council guidance. BMJ. 2021;374:n2061.
- McCulloch P, Altman DG, Campbell WB, et al. No surgical innovation without evaluation: the IDEAL recommendations. Lancet. 2009;374:1105–1112.
- Hirst A, Philippou Y, Blazeby J, et al. No surgical innovation without evaluation: evolution and further development of the IDEAL framework and recommendations. Ann Surg. 2019;269:211–220.
- Sedrakyan A, Campbell B, Merino JG, et al. IDEAL-D: a rational framework for evaluating and regulating the use of medical devices. BMJ. 2016;353:i2372.
- Johner C, Molnar C, Purde A, et al. Guideline for AI for medical products. Available from: https://github.com/johner-institut/ai-guideline/blob/master/Guideline-AI-Medical-Devices_EN.md
- Heil BJ, Hoffman MM, Markowetz F, et al. Reproducibility standards for machine learning in the life sciences. Nat Methods. 2021;18:1132–1135.
- Luo W, Phung D, Tran T, et al. Guidelines for developing and reporting machine learning predictive models in biomedical research: a Multidisciplinary View. J Med Internet Res. 2016;18(12):e323.
- Matschinske J, Alcaraz N, Benis A, et al. The AIMe registry for artificial intelligence in biomedical research. Nat Methods. 2021;18:1128–1131.
- Aggarwal R, Sounderajah V, Martin G, et al. Diagnostic accuracy of deep learning in medical imaging: a systematic review and meta-analysis. NPJ Digit Med. 2021;4:65.
- Commission Implementing Decision (EU) 2021/1182 of 16 July 2021 on the harmonised standards for medical devices drafted in support of Regulation (EU) 2017/745 of the European Parliament and of the Council. C/2021/5210. OJ L 256: 100–102. Available from: http://data.europa.eu/eli/dec_impl/2021/1182/oj
- European Court of Justice. Judgment of the Court (Third Chamber) of 27 October 2016. James Elliott construction limited v Irish asphalt limited (Case C613/14). ECLI:EU:C:2016:821. https://curia.europa.eu/juris/document/document.jsf;jsessionid=D54C832F0D6E8B4AAAC47ECD004A5733?text=&docid=184891&pageIndex=0&doclang=EN&mode=lst&dir=&occ=first&part=1&cid=1528615
- Zielke T. Is artificial intelligence ready for standardization? In: Yilmaz M, Niemann J, Clarke P, et al., editors. Systems, software and services process improvement. EuroSPI 2020. Communications in computer and information science. 2020;1251:259–274. Springer: Cham.
- ISO 13485: 2016. Medical devices — quality management systems — requirements for regulatory purposes. Available from: https://www.iso.org/standard/59752.html
- ISO 14155:2020. Clinical investigation of medical devices for human subjects — good clinical practice. Available from: https://www.iso.org/standard/71690.html
- ISO 14971:2019. Medical devices — application of risk management to medical devices. Available from: https://www.iso.org/standard/72704.html
- ETSI. Standardization request in support of safe & trustworthy artificial intelligence. Available from: https://www.etsi.org/newsroom/blogs/entry/standardization-request-in-support-of-safe-trustworthy-artificial-intelligence
- Nativi S, De Nigris S. AI standardisation landscape: state of play and link to the EC proposal for an AI regulatory framework, EUR 30772 EN. Luxembourg: Publications Office of the European Union; 2021.
- Joint Research Centre, European Commission. Science for policy brief: how can Europe become a global leader in AI in health? 26 January 2021. https://ai-watch.ec.europa.eu/publications/science-policy-brief-how-can-europe-become-global-leader-ai-health_en
- Righi R, Lopez Cobo M, Samoili S, et al. Science for policy brief. EU in the global artificial intelligence landscape. Eur Commission, 2021, JRC125613.
- OCEANIS. Global AI standards repository. Available from: https://ethicsstandards.org/repository/#:~:text=The%20Global%20AI%20Standards%20Repository,compilation%20of%20user%20submitted%20entries
- AI Watch. Joint Research Centre, European Commission, Seville. Available from: https://ai-watch.ec.europa.eu/publications_en
- European Medicines Agency (EMA). Regulatory science, research needs (2021) Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf
- EMA with HMA (Heads of Medicines Agencies). Accelerating clinical trials in the EU (ACT EU) (2022). Available from: https://www.ema.europa.eu/en/news/accelerating-clinical-trials-eu-act-eu-better-clinical-trials-address-patients-needs
- Koene A, Clifton C, Hatada Y, et al. A governance framework for algorithmic accountability and transparency. European Parliament Directorate General for Parliamentary Research Services, Scientific Foresight Unit (STOA). PE 624.262 April 2019. https://www.europarl.europa.eu/RegData/etudes/STUD/2019/624262/EPRS_STU(2019)624262_EN.pdf.
- OECD.AI Policy Observatory. Available from: https://oecd.ai/en/
- Council of Europe Ad Hoc Committee on AI (CAHAI). Available from: https://www.coe.int/en/web/artificial-intelligence/cahai
- Committee on Artificial Intelligence (CAI). Available from: https://www.coe.int/en/web/artificial-intelligence/cai
- Council of Europe. Steering Committee for Human Rights in the fields of Biomedicine and Health (CDBIO). Available from: https://www.coe.int/en/web/artificial-intelligence
- Mittelstadt B. The impact of artificial intelligence on the doctor-patient relationship. Report commissioned by the steering committee for human rights in the fields of biomedicine and health. Council of Europe, 2021. Available from: https://www.coe.int/en/web/bioethics/report-impact-of-ai-on-the-doctor-patient-relationship
- CEN-CENELEC Joint Technical Committee 21 ‘Artificial intelligence.’ Available from: https://www.cencenelec.eu/areas-of-work/cen-cenelec-topics/artificial-intelligence/
- CEN-CENELEC Work Programme 2022. Available from: https://www.cencenelec.eu/media/CEN-CENELEC/News/Publications/2022/cen-cenelec_work_programme2022.pdf
- WHO/ITU-T Focus Group on Artificial Intelligence for Health (FG-AI4H). Available from: https://www.itu.int/en/ITU-T/focusgroups/ai4h/Documents/FG-AI4H-ToR.pdf
- Working Groups include Regulatory considerations on AI for health, and Clinical evaluation of AI for health. Available from: https://www.itu.int/en/ITU-T/focusgroups/ai4h/Pages/default.aspx
- International Standardization Organisation ISO. ISO/IEC JTC 1/SC 42 Artificial Intelligence. Available from: https://www.iso.org/committee/6794475.html
- International Electrotechnical Commission (IEC). 62/411/NP: testing of Artificial Intelligence/Machine Learning-enabled Medical Devices. Available from: https://www.iec.ch/dyn/www/f?p=103:23:612016022541470::::FSP_ORG_ID,FSP_LANG_ID:1245,25
- International Electrotechnical Commission (IEC). Artificial intelligence/machine learning-enabled medical device – performance evaluation process. Available from: https://www.iec.ch/ords/f?p=103:38:7211217005198::::FSP_ORG_ID,FSP_APEX_PAGE,FSP_PROJECT_ID:1245,23,107066
- IEEE P2673 Intelligence Augmentation for Medical Imaging Working Group. Available from: https://sagroups.ieee.org/2673/
- EMBS Artificial Intelligence Medical Devices Working Group. Available from: https://sagroups.ieee.org/aimdwg/
- IEEE 2801-2022. Recommended practice for the quality management of datasets for medical artificial intelligence. 2022 Available from: https://standards.ieee.org/ieee/2801/7459/
- IEEE 2801-2022. Recommended practice for the quality management of datasets for medical artificial intelligence. 2022. Available from: https://sagroups.ieee.org/aimdwg/wp-content/uploads/sites/23/2018/12/P2801-PAR.pdf
- Artificial Intelligence Medical Devices Working Group. Standard for the performance and safety evaluation of artificial intelligence based medical device: terminology. Available from: https://sagroups.ieee.org/aimdwg/wp-content/uploads/sites/23/2018/12/P2802-PAR.pdf
- New IEEE standard (IEEE P2802) Available from: https://development.standards.ieee.org/myproject-web/public/view.html#pardetail/6755
- Wiegand T, Lee N, Pujari S, et al. Whitepaper for the ITU/WHO Focus Group on Artificial Intelligence for Health. Available from: https://www.itu.int/en/ITU-T/focusgroups/ai4h/Documents/FG-AI4H_Whitepaper.pdf
- Wenzel MA, Wiegand T Towards international standards for the evaluation of artificial intelligence for health. 2019 ITU Kaleidoscope: ICT for Health: networks, Standards and Innovation (ITU K), 2019: 1–10. doi: 10.23919/ITUK48006.2019.8996131.
- European Commission. Terms of reference of the MDCG Working Group on New Technologies. Available from: https://health.ec.europa.eu/system/files/2020-09/md_tor_wg7_nt_en_0.pdf
- European Medicines Agency. EMA Regulatory science to 2025. Strategic reflection. 2020. Available from: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf
- European Medicines Agency. Accelerating clinical trials in the EU (ACT EU) - Delivering an EU clinical trials transformation initiative. 2022. https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/accelerating-clinical-trials-eu-act-eu-delivering-eu-clinical-trials-transformation-initiative_en.pdf
- ICMRA Informal Innovation Network. Horizon Scanning Assessment Report – artificial Intelligence. 6 August 2021. Available from: https://www.icmra.info/drupal/sites/default/files/2021-08/horizon_scanning_report_artificial_intelligence.pdf
- European Parliament, Panel for the Future of Science and Technology (STOA). Centre for Artificial Intelligence (C4AI). Available from: https://www.europarl.europa.eu/stoa/en/centre-for-AI
- Lopez Solano J, Martin A, de Souza S, et al. Governing data and artificial intelligence for all. Models for sustainable and just data governance. European Parliamentary Research Service - Scientific Foresight Unit (EPRS - STOA). PE 729.533. July 2022. Available from: https://www.europarl.europa.eu/RegData/etudes/STUD/2022/729533/EPRS_STU(2022)729533_EN.pdf.
- Gómez-González E, Artificial GE. Intelligence in medicine and healthcare: applications, availability and societal impact, EUR 30197 EN. AI Watch. Luxembourg: Publications Office of the European Union; 2020. Available from: https://idus.us.es/bitstream/handle/11441/105744/jrc120214_ai_in_medicine_and_healthcare_report-aiwatch_v50.pdf?sequence=1&isAllowed=y.
- De Nigris S, Craglia M, Nepelski D, et al. AI Watch: AI uptake in health and healthcare 2020, EUR 30478 EN. Luxembourg: Publications Office of the European Union; 2020. doi: 10.2760/948860
- High-Level Expert Group on Artificial Intelligence. The assessment list for trustworthy artificial intelligence (ALTAI) for self assessment. 17 July 2020. https://ec.europa.eu/newsroom/dae/document.cfm?doc_id=68342.
- OECD. State of implementation of the OECD AI principles. Insights from national AI policies. OECD Digital Economy Papers, June 2021: No. 311. Available from: https://www.oecd-ilibrary.org/docserver/1cd40c44-en.pdf?expires=1660206152&id=id&accname=guest&checksum=D3BBC087730E06114B8883D76B8B965F
- OECD. Mandate of the Working Party on Artificial Intelligence Governance. Available from: https://oecdgroups.oecd.org/Bodies/AdvancedSearch.aspx?Title=artificial+intelligence&Match=false
- Council of Europe. How to regulate the development of artificial intelligence? Kleijssen J. Available from: https://www.coe.int/en/web/artificial-intelligence/director-jan-kleijssen
- Council of Europe. Ad Hoc Committee on Artificial Intelligence (CAHAI). Possible elements of a legal framework on artificial intelligence, based on the Council of Europe’s standards on human rights, democracy and the rule of law. Strasbourg, 3 December 2021. CAHAI(2021)09rev. Available from: https://rm.coe.int/cahai-2021-09rev-elements/1680a6d90d
- Council of Europe. Recommendation CM/Rec(2020)1 of the Committee of Ministers to member States on the human rights impacts of algorithmic systems (Adopted by the Committee of Ministers on 8 April 2020). Available from: https://search.coe.int/cm/pages/result_details.aspx?objectid=09000016809e1154
- Access Now. Mapping regulatory proposals for artificial intelligence in Europe. (2018) Available from: https://www.accessnow.org/cms/assets/uploads/2018/11/mapping_regulatory_proposals_for_AI_in_EU.pdf (accessed 2022 Jul 31
- OECD. National AI policies & strategies. Available from: https://oecd.ai/en/dashboards/overview
- Jorge Ricart R, Van Roy V, Rossetti F, et al. AI Watch - National strategies on artificial intelligence: a European perspective, 2022. Vol. EUR 31083 EN. Luxembourg: Publications Office of the European Union; 2022
- National Institute for Health and Care Excellence (NICE). Evidence standards framework for digital health technologies. 09 August 2022. https://www.nice.org.uk/corporate/ecd7
- British Standards Institution (BSI). BS AAMI 34971 Guidance on the Application of ISO 14971 to artificial intelligence and machine learning. Available from: https://standardsdevelopment.bsigroup.com/projects/2020-02770#/section
- US Food & Drug Administration, Health Canada, Medicines and Healthcare Products Regulatory Agency. good machine learning practice for medical device development: guidingPrinciples. October 2021. Available from: https://www.fda.gov/media/153486/download.
- U.S. Food & Drug Administration. Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) action plan. January 2021. Available from: https://www.fda.gov/media/145022/download
- McNair D, Price WN. In: Health care AI: law, regulation, and policy. In: artificial intelligence in health care: the hope, the hype, the promise, the peril (Chapter 7). MS M, ST I, Ahmed M, et al.,editors.Washington DC:National Academy of Medicine; 2019.
- Australian Government, Department of Health. Therapeutic Goods Administration. Regulatory changes for software based medical devices. Version 1.2, August 2021. https://www.tga.gov.au/sites/default/files/regulatory-changes-software-based-medical-devices.pdf
- Australian Government, Department of Health. Therapeutic Goods Administration. Clinical decision support software. Scope and examples Version 1.1, October 2021. https://www.tga.gov.au/sites/default/files/clinical-decision-support-software.pdf
- Wu F, Lu C, Zhu M, et al. Towards a new generation of artificial intelligence in China. Nat Mach Intell. 2020;2(6):312–316.
- Ning SX, Wu H AI, machine learning and big data laws and regulations 2022, China. GLI Global Legal Insights. Available from: https://www.globallegalinsights.com/practice-areas/ai-machine-learning-and-big-data-laws-and-regulations/china
- Fraser AG, Butchart EG, Szymański P, et al., The need for transparency of clinical evidence for medical devices in Europe. Lancet. 2018;392(10146): 521–530.
- Regulation (EC) No 1049/2001 of the European Parliament and of the Council of 30 May 2001 regarding public access to European Parliament, Council and Commission documents. Available from: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex:32001R1049
- Madiega T Artificial intelligence act. European Parliamentary Research Service briefing, PE 698.792 – January 2022. Available from: https://www.europarl.europa.eu/RegData/etudes/BRIE/2021/698792/EPRS_BRI(2021)698792_EN.pdf
- Biasin E, Kamenjasevic E. Cybersecurity of medical devices: new challenges arising from the AI Act and NIS 2 Directive proposals. International Cybersecurity Law Review. 2022;3(1):163–180.
- Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016. on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC (General Data Protection Regulation). Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32016R0679
- European Data Protection Board. EDPB Document on response to the request from the European Commission for clarifications on the consistent application of the GDPR, focusing on health research. 2 February 2021. Available from: https://edpb.europa.eu/sites/default/files/files/file1/edpb_replyec_questionnaireresearch_final.pdf
- Regulation (EU) 2022/868 of the European Parliament and of the Council of 30 May 2022 on European data governance and amending Regulation (EU) 2018/1724 (Data Governance Act). Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32022R0868
- European Commission. Proposal for a regulation of the European Parliament and of the Council on the European Health data space. COM(2022) 197 final. 2022. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A52022PC0197
- European Commission. Proposal for a Regulation of the European Parliament and of the Council on harmonised rules on fair access to and use of data (Data Act). COM(2022) 68 final. 2020 Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=COM%3A2022%3A68%3AFIN
- Biasin E The data act will concern ehealth apps and medical devices. KU Leuven CiTiP Blog. 2022. Available from: https://www.law.kuleuven.be/citip/blog/the-data-act-will-concern-ehealth-apps-and-medical-devices/
- European Parliament resolution of 20 October 2020 on intellectual property rights for the development of artificial intelligence technologies (2020/2015(INI)). TA-9-2020-0277_EN. Available from: https://www.europarl.europa.eu/doceo/document/TA-9-2020-0277_EN.html
- Zaichuk M. IP Protection of Artificial Intelligence in Europe: tailor-made solutions required. April 2020. Available from: https://www.jonesday.com/en/insights/2020/04/ip-protection-of-artificial-intelligence-in-europe
- Directive 85/374/EEC of 25 July 1985 on the approximation of the laws, regulations and administrative provisions of the Member States concerning liability for defective products. OJ L210: 29-33. Available from: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A31985L0374
- European Court of Justice. Judgement of the Court (First Chamber) of 16 February 2017. Elisabeth Schmitt v TÜV Rheinland LGA Products GmbH. Case C‑219/15. ECLI:EU:C:2017:1z
- European Court of Justice. Judgment of the Court (Fourth Chamber) of. 5 March 2015. In: Boston Scientific Medizintechnik GmbH v AOK Sachsen-Anhalt – die Gesundheitskasse and Betriebskrankenkasse RWE. Joined Cases C-503/13 and C-504/13. ECLI:EU:C:2015. Vol. 148.
- European Parliament. Artificial intelligence and civil liability. Study requested by the JURI committee 2020. Available from: https://www.europarl.europa.eu/RegData/etudes/STUD/2020/621926/IPOL_STU(2020)621926_EN.pdf
- European Parliament. Liability for artificial intelligence and other emerging digital technologies. Report from the expert group on liability and new technologies (2019). Available from: https://www.europarl.europa.eu/meetdocs/2014_2019/plmrep/COMMITTEES/JURI/DV/2020/01-09/AI-report_EN.pdf
- Report from the Commission to the European Parliament, the Council and the European Economic and Social Committee. Report on the safety and liability implications of Artificial Intelligence, the Internet of Things and Robotics. Brussels, 19.2.2020. COM/2020/64 final. Available from: https://eur-lex.europa.eu/legal-content/en/TXT/?qid=1593079180383&uri=CELEX%3A52020DC0064
- Twigg-Flesner C. Guiding principles for updating the product liability directive for the digital age. ELI (European Law Institute) Innovation Paper Series, 2021. Available from: https://europeanlawinstitute.eu/fileadmin/user_upload/p_eli/Publications/ELI_Guiding_Principles_for_Updating_the_PLD_for_the_Digital_Age.pdf
- European Commission. Proposal for a Directive of the European Parliament and of the Council on adapting non-contractual civil liability rules to artificial intelligence (AI Liability Directive). Brussels. 2022;28(9):COM(2022) 496 final 2022/0303 (COD). Available from: https://ec.europa.eu/info/files/proposal-directive-adapting-non-contractual-civil-liability-rules-artificial-intelligence_en
- Biasin E, Kamenjasevic E. Cybersecurity of medical devices: regulatory challenges in the European Union. In: Cohen IG, Minssen T, Price WN, et al, editors. The future of medical device regulation: innovation and protection. Chapter 4, pp 51-62. Cambridge, Cambridge University Press; 2020.
- Medical Device Coordination Group. MDCG 2019-16: Guidance on cybersecurity for medical devices. December 2019. Available from: https://ec.europa.eu/docsroom/documents/41863
- Directive (EU) 2016/1148 of the European Parliament and of the Council of 6 July 2016 concerning measures for a high common level of security of network and information systems across the Union. OJ L 194. 2016;19(7):1–30.
- Regulation (EU) 2019/881 of the European Parliament and of the Council of 17 April 2019 on ENISA (the European Union Agency for Cybersecurity) and on information and communications technology cybersecurity certification and repealing Regulation (EU) No 526/2013 (Cybersecurity Act). OJ L 151. 2019;7(6):15–69. http://data.europa.eu/eli/reg/2019/881/oj
- In: Directive 2014/53/EU of the European Parliament and of the Council of 16 April 2014 on the harmonisation of the laws of the Member States relating to the making available on the market of radio equipment and repealing Directive 1999/5/EC. Official Journal of the European Union, L 153, 22 May 2014; 57:62-104. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=OJ:L:2014:153:TOC
- Jobin A, Ienca M, Vayena E. The global landscape of AI ethics guidelines. Nat Mach Intell. 2019;1:389–399.
- Cohen G. Informed consent and medical artificial intelligence: what to tell the patient? Georgetown Law J. 2020;108:1425–1469.
- Khan SM, Liu X, Nath S, et al. A global review of publicly available datasets for ophthalmological imaging: barriers to access, usability, and generalisability. Lancet Digit Health. 2021;3:e51–e66.
- Lipton ZC. The mythos of model interpretability: in machine learning, the concept of interpretability is both important and slippery. Queue. 2018;16:31–57.
- Brown TB, Mann B, Ryder N, et al. Language models are few-shot learners. in advances in neural information processing systems. Curran Associates Inc. 2020;33:1877–1901.
- Breiman L. Random forests. Mach Learn. 2001;45:5–32.
- Lundberg SM, Lee S-I. A unified approach to interpreting model predictions. in advances in neural information processing systems. Curran Associates Inc. 2017;30:4768–4777.
- Gill CJ, Sabin L, Schmid CH. Why clinicians are natural bayesians. BMJ. 2005;330:1080–1083.
- Wilkinson J, Arnold KF, Murray EJ, et al. Time to reality check the promises of machine learning-powered precision medicine. Lancet Digit Health. 2020;2:e677–e680.
- Babic B, Gerke S, Evgeniou T, et al. Beware explanations from AI in health care. Science. 2021;373:284–286.
- Kroll JA. The fallacy of inscrutability. Phil.Trans.R.Soc. 2018; A376: 20180084.
- Markus AF, Kors JA, Rijnbeek PR. The role of explainability in creating trustworthy artificial intelligence for health care: a comprehensive survey of the terminology, design choices, and evaluation strategies. J Biomed Inform. 2021;113:103655.
- U.S. Food & Drug Administration. Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices. Available from: https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-aiml-enabled-medical-devices
- U.S. Food & Drug Administration. Clinical decision support software guidance for industry and Food and Drug Administration Staff. Document issued on September 28, 2022. Available from: https://www.fda.gov/media/109618/download
- Australian Government, Therapeutic Goods Administration. Exemption for certain clinical decision support software. Guidance on the exemption criteria. Version 1.0, August 2022. Available from: https://www.tga.gov.au/sites/default/files/2022-08/exemption-for-certain-clinical-decision-support-software.pdf
- Wiens J, Saria S, Sendak M, et al. Do no harm: a roadmap for responsible machine learning for health care. Nat Med. 2019;25:1337–1340.
- U.S. 21st century cures act. Public Law 114–255—DEC. 13, 2016. Available from: https://www.congress.gov/114/plaws/publ255/PLAW-114publ255.pdf
- Gerke S, Babic B, Evgeniou T, et al. The need for a system view to regulate artificial intelligence/machine learning-based software as medical device. NPJ Digit Med. 2020;3:53.
- Harada T, Shimizu T, Kaji Y, et al. A perspective from a case conference on comparing the diagnostic process: human diagnostic thinking vs. artificial intelligence (AI) decision support tools. Int J Environ Res Public Health. 2020; 17: 6110.
- Challen R, Denny J, Pitt M, et al. Artificial intelligence, bias and clinical safety. BMJ Qual Saf. 2019;28:231–237.
- Unsworth H, Wolfram V, Dillon B, et al. NICE Artificial intelligence evidence standards working group. Building an evidence standards framework for artificial intelligence-enabled digital health technologies. Lancet Digit Health. 2022;4:e216–e217.
- van Smeden M, Heinze G, Van Calster B, et al. Critical appraisal of artificial intelligence-based prediction models for cardiovascular disease. Eur Heart J. 2022;43:2921–2930.
- Dombrink I, Lubbers BR, Simulescu L, et al. Critical implications of IVDR for innovation in diagnostics: input from the BioMed Alliance diagnostics task force. Hemasphere. 2022;6:e724.