
ABSTRACT
Introduction
The new European Union (EU) Regulations for medical devices (MDs) and health technology assessment (HTA) are welcome developments that should increase the quality of clinical evidence for MDs and reduce fragmentation in the EU market access process. To fully exploit anticipated benefits, their respective assessment processes should be closely coordinated, particularly for promising, highly innovative MDs. Accelerated approval is worth exploring for certain categories of high-risk MDs to keep the EU regulatory process competitive compared to accelerated MD approval programs elsewhere (e.g. US).
Areas Covered
Problems observed in worldwide accelerated drug and MD regulatory approval programs are reviewed, including greater uncertainty in premarket clinical evidence generation and lack of oversight for post approval evidence requirements. Implications for MD approval, HTA and coverage are explored.
Expert Opinion
Through analysis of two decades of drug and MD accelerated approval programs worldwide, recommendations for an Accelerated Access Pathway for select innovative, high-risk MDs are proposed to fit the EU context, leverage the two new regulations, increase opportunities for Expert Panels to provide timely advice regarding manufacturers’ evidence generation plans along the MD lifecycle (pre, postmarket), and safely speed patient access while promoting increased collaboration among Member States on coverage decisions.
1. Introduction
An overarching goal for medical device (MD) regulation is to provide timely access to innovative technologies that are safe and effective. Full application of the European Union Medical Devices Regulation 2017/745 (MDR) became effective in May 2021 [Citation1], and the legislative regulation of health technology assessment (HTA) in the European Union (EU) received formal approval in December 2021 (HTA Regulation 2021/2282), with full application by 2025 [Citation2]. In the EU, MDs are classified according to risk level, class I (low), class IIa (medium) and classes IIb and III (high); under the new MDR, clinical investigations are required for all implantable and class III MDs [Citation1]. The new HTA Regulation provides a framework for joint clinical assessments of health technologies, and for joint scientific consultations between manufacturers and HTA bodies, for certain high-risk medical devices (class III implantable and class IIb active devices intended to administer and/or remove a medicinal product) [Citation2]. Both of these Regulations in the EU landscape will affect approval processes for MDs as well as the quantity, type and timing of clinical evidence generation [Citation3]. They will also impact exchanges between various stakeholders, including regulatory and HTA bodies, manufacturers, and coverage decision makers [Citation4–6].
Under the new EU MDR, the approval process for MDs, including certain high-risk MDs, follows a multistep, multi-stakeholder process (), now with added ties to the HTA Regulation [Citation1,Citation2,Citation7,Citation8]. Unlike medicines, which are approved centrally through the European Medicines Agency (EMA), independent organizations known as Notified Bodies are designated by EU Member States to assess the conformity of an MD before it may be placed on the market (i.e. obtain a Conformité Européenne (CE) mark) through evaluation of the technical file, including the Clinical Evaluation Report, submitted by the manufacturer [Citation7]. The Notified Bodies in turn operate within a policy context set by the European Commission’s Directorate General for Health and Food Safety and Competent Authorities in the Member States; they must meet stringent clinical competence criteria and may now receive support in performing conformity assessments from clinical Expert Panels under the Clinical Evaluation Consultation Procedure () for the above-mentioned certain high-risk MDs, according to Article 54 [Citation1]. Expert Panels (as of March 2022 under the coordination of the EMA) evaluate the Notified Bodies’ Clinical Evaluation Assessment Reports and, if deemed appropriate, issue a scientific opinion (within 60 days), which the Notified Body is not legally obliged to accept but must justify any refusal of such advice in the final conformity assessment. In turn, according to the HTA Regulation, joint clinical assessments should be performed for MDs for which a scientific opinion has been issued within the Clinical Evaluation Consultation Procedure framework (Article 7(c)) [Citation2].
Figure 1. The current premarket development and regulatory approval pathway for high-risk* medical devices in the European Union. Reprinted (adapted) with permission from Fraser AG, Byrne RA, Kautzner J, et al. Implementing the new European Regulations on medical devices - clinical responsibilities for evidence based practice: a report from the Regulatory Affairs Committee of the European Society of Cardiology. Eur Heart J. 2020;41:2589–2596.
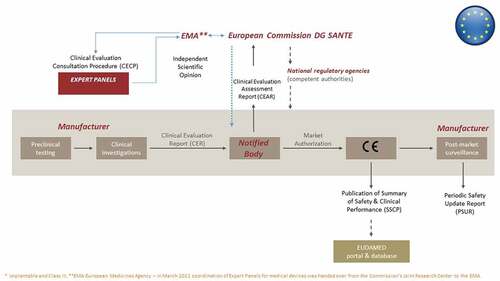
Going forward, there are two related issues for patient access to high-risk MDs. First, whereas safety and proof of efficacy for MDs are widely expected to improve [Citation3,Citation7–9], expanded requirements for clinical evidence in the MDR – and fewer certified Notified Bodies compared to before its passage – may increase the time to CE mark approval. Second, joint clinical assessments for high-risk devices under the HTA Regulation process will be centralized; however, each single member state may ask for additional data (e.g. regarding organizational and economic aspects, social implications), which may lengthen the implementation and uptake of joint clinical assessments. Since there are no guarantees that clinical evidence resulting in regulatory approval will be sufficient for HTA and coverage decisions [Citation3,Citation10,Citation11], and patient access decisions will still follow a disjointed, post CE mark, process for coverage in each Member State, this could further delay patient access in some countries. Consequently, it is critical that appropriate evidence on the clinical effectiveness of innovative high-risk (especially implantable) devices is delivered in a timely manner and that the links between regulatory approval, HTA and coverage are considered throughout the lifecycle of the MD [Citation3]. To that end, there may be classes of innovative, high-risk implantable MDs that warrant not only a prioritized premarket regulatory pathway, but also expedited HTA, pricing and reimbursement decisions in the post CE mark phase of the MD lifecycle. Such ‘deserving’ MDs should address an unmet need for a serious or life threatening condition, or offer significant improvement in safety, efficacy, clinical and cost-effectiveness relative to the current standard of care.
1.1. Objectives
Given the evolving status of accelerated approval programs around the world for medicines and MDs [Citation12–20], and in the context of the new EU MD and HTA Regulations, our objective was to examine the issues surrounding evidence generation for innovative, high-risk (particularly implantable and class III) MDs and explore the feasibility and rationale of an accelerated approval program for such MDs in the EU, with emphasis on implications for HTA and coverage. Building upon a previous initiative to provide recommendations for evidence generation throughout the lifecycle of MDs [Citation3], this project brought together an international group of experts (in health policy, health economics and management, HTA, clinical research, population health) to further examine relevant issues surrounding high-risk MD processes for CE mark authorization and beyond, and to formulate recommendations for an accelerated access pathway for MDs which links approval to postmarketing evidence requirements, HTA, and reimbursement and pricing decisions. Specifically, the following two questions were posed to the group:
What underlying principles and distinguishing features of current and previous accelerated approval programs (in Europe, the US, and elsewhere) make them fit for purpose for high-risk (especially implantable) medical devices? Are they needed, and under what circumstances?
How can the HTA community make best use of – or influence – the evidence generated though accelerated regulatory approval programs, and how and when should HTA bodies become involved? What are the implications for coverage decisions?
The paper is structured to provide, first, the underlying context of both the discussions and the recommendations, in the form of an overview of the inherent components and impact over time of accelerated approval programs, and, second, our resulting recommendations for an accelerated access pathway for high-risk MDs in the EU. Section 2 describes the structure of the discussions and salient evidence gathered to support discussions and resulting recommendations. Section 3 provides highlights from the discussions of the two research questions that led to our proposal for an Accelerated Access Pathway (3.2) and associated recommendations. The recommendations are illustrated in Section 4, followed by the conclusions (Section 5). Finally, in Section 6 – Expert Opinion – we provide our perspective on our proposed accelerated access pathway, its potential benefits and limits, and how it might change the way innovative high-risk MDs in the EU are regulated, brought to market, covered, and monitored to better serve and protect patients.
2. Background used to guide discussions and formulate recommendations
2.1. Structure of the discussions and information sharing activities
A multidisciplinary group of international experts (Appendix 1) convened for two workshops in 2021 to discuss evidence generation, approval and access pathways for high-risk implantable medical devices. To ensure all participants had equal opportunity to contribute before, during and after discussions, the Nominal Group Technique consensus development method was loosely applied [Citation21], to collect differing opinions and encourage generation of multiple ideas. This was supplemented by a modified consensus group conference method, using an informal discussion format, where each participant could equally present ideas, opinions, evidence, and ask questions, and decisions on recommendations could also include minority or alternative views [Citation21]. Workshops were carried out virtually (webinar) due to restrictions related to the COVID-19 pandemic; the encounters were recorded, and transcripts of the discussion and the comments and materials included in the online chat were shared after each workshop.
To create a common vocabulary and focus the discussions, the project leaders (RT, HB, OC) prepared briefing papers on the topics underlying the research questions, which were e-mailed prior to the workshops to all participants. The first briefing paper provided overviews of current and past global accelerated approval programs for medicines and MDs, incorporating gray literature (non-peer-reviewed reports, website information) on the various programs, and peer-reviewed literature, especially studies that defined and critically assessed the programs, measured effects or problems over time, or provided international comparisons. To support discussions, briefing papers, and drafts of the manuscript, a scoping review of the literature [Citation22] was conducted in PubMed using keywords for accelerated or expedited programs for medicines and MDs (see supplementary material for details and the list of selected publications). The purpose was to provide a check on how and whether critical issues regarding their impact brought up and discussed during the workshops have been treated in the literature.
The second briefing paper was circulated via e-mail prior to the second workshop, together with preliminary drafts of the recommendations, for comments and additions. The recommendations were debated and expanded upon during the session. Finally, a draft manuscript containing final recommendations and underlying reasoning and evidence was circulated among participants for comments and revisions, and recirculated for final approval.
2.2. Evidence from the literature informing the discussions and providing context for the recommendations
2.2.1. Accelerated approval pathways for medicines and medical devices
For medicines, priority systems have been developed over time to accelerate the process of market approval [Citation12–16,Citation20]. Quicker access to promising and innovative therapies and technologies, especially in response to an unmet need for a serious or life-threatening condition, has generally been the main impetus for developing accelerated approval pathways for medicines and other medical technologies, while the associated potential risks (physical or financial) due to uncertainty and less time to develop the necessary clinical evidence has raised serious ethical and safety issues [Citation20,Citation23]. Known collectively as accelerated or expedited approval pathways, they have taken many and changing forms over the years and are increasingly used in the United States (US), the EU, Japan and Australia [Citation15–17,Citation24–26]. A recent study reviewing new drug approvals from 2007 to 2017, found that 181 (57%) of 320 drugs approved by the US Federal Drug Administration (FDA) and 39 (15%) of 268 approved by the EMA qualified for an expedited program [Citation27].
Though specific criteria for qualifying may vary, accelerated approval programs in the US, EU and elsewhere commonly accept innovative products that address unmet medical needs, serious or life-threatening conditions, rare/orphan diseases or emergency situations, or that offer the promise of major clinical advantage and/or significant improvement in safety/efficacy, treatment where no approved alternatives exist or with significant advantage over existing alternatives, in the best interest of patients or public health [Citation12–15,Citation17,Citation19,Citation20,Citation24–26,Citation28]. Advantages for applicants include expedited development and review, flexibility regarding clinical study designs (e.g. acceptance of surrogate or intermediate endpoints, single-arm trials, approval with subsequent confirmation of results through post-marketing trials) and quality management systems (e.g. less documentation, relaxed inspection requirements), and, perhaps most importantly, enhanced interaction and early dialogue with regulatory agencies [Citation5,Citation12,Citation13,Citation24,Citation26].
Specifically for MDs, US approval processes are centralized within the FDA, and several accelerated approval programs originally developed for medicines have been adapted for MDs, such as the Breakthrough Devices Program, which replaced the Priority Review and Expedited Access Pathway programs for MDs [Citation18,Citation19,Citation29]. Accelerated (Priority, Provisional) approval is available in Australia for both medicines and MDs, while Japan, similar to US and EU programs, provides priority, conditional and orphan drug approval pathways for medicines, with an additional Sakigake (Pioneer) pathway for accelerated medicines, regenerative medical products and MDs, modeled on the US Breakthrough program and developed in 2015 for products developed in Japan and seeking first (or contemporaneous with other counties) approval in Japan [Citation24,Citation30,Citation31]. Australia’s programs for medicines are similar to US and EU programs, while the nascent MD priority program seems modeled on the US Breakthrough program; however, evidence of MDs accessing the program could not be found on website databases (https://www.tga.gov.au/ws-md-designation-notices-index) [Citation30–32]. In Europe, accelerated programs for medicines exist (e.g. Priority Medicines or PRIME, Conditional Marketing Authorization), but no specific accelerated or expedited approval processes are currently in place for MDs at the EU level [Citation12].
2.2.2. Impact of accelerated approval programs
Results and implications of accelerated approval programs have been documented in the literature, mostly for medicines but also for other medical technologies [Citation13,Citation15,Citation24,Citation33–35]. Products granted regulatory approval under accelerated programs (FDA, EMA or both) have been associated with increased decision uncertainty at filing, or lack of post-marketing confirmation of clinical outcomes, questionable quality and quantity of post-approval studies, debatable therapeutic value, or increased changes to safety labels (boxed warnings and contraindications) after approval, and in some cases voluntary manufacturer withdrawals from the approval process [Citation13,Citation27,Citation34–41].
Medicines
Although expedited approval drugs showed a greater likelihood to have high therapeutic value over standard approval drugs in a study of nearly 600 FDA and EMA approvals between 2007 and 2017, only 31% of accelerated approval drugs were rated as having high therapeutic value by independent organizations [Citation27]. A systematic review of accelerated approvals by the FDA between 2005 and 2012 additionally found that post-approval studies varied considerably in quality and quantity, with few proving post-approval confirmation of efficacy on final patient-relevant outcomes [Citation39]. While a review conducted by the FDA of its own accelerated approvals of malignant hematology and oncology drugs found that clinical benefit for most indications was indeed verified [Citation13], an independent study focusing on surrogate endpoints for 194 unique FDA cancer drug authorizations (89 accelerated) between 1992 and 2019, found no or weak association between surrogate endpoints and overall survival in 49 (23 accelerated) of 64 cases where a surrogate was used for the first time for a particular therapeutic area [Citation42].
In the EU, a report from the EMA evaluating conditional marketing authorizations (granted for one year, renewable, with associated obligations and the possibility to later convert to standard marketing authorization) between 2006 and 2016, found that 11 of 30 approvals converted to standard within four years, two were withdrawn for commercial reasons, and 17 remained conditional [Citation28]. In a study of 51 drugs approved under accelerated EMA programs, five were approved based on pivotal trials of clinical outcomes, while 90% gained approval based on surrogate endpoints; none of the surrogate endpoint approvals showed subsequent evidence of validation using patient-relevant final outcomes, such as mortality or health-related quality of life [Citation35].
Medical devices
For MDs, evidence from accelerated pathways is available only from the US. One study compared FDA priority to standard review processes for 230 (29 priority and 201 standard approval) high-risk MDs from 2006–2015 [Citation18]. The study surprisingly found higher median review times for priority review (21 vs. 14 months), probably because the novelty and complexity of devices applying for priority review led to more referrals to advisory committees, as is common in expedited review. Also, the likelihood of recall was higher for priority vs. standard review MDs, with shorter times to recall from approval for the highest risk recall class [Citation18,Citation43].
Another study evaluating eight high-risk MD approvals under the FDA’s Breakthrough MD designation from December 2016 (inception of the program) to January 2020, found shorter median (range) review times of 181.5 days (146 to 301 days) for Breakthrough MDs, compared to 260 days (180 to 429.5) for non-breakthrough MDs. However, surrogate endpoints were found to be common, and confirmatory trials with premarket cohorts were observed for half the approved products with publicly-disclosed information [Citation19].
In comparing European and US systems, pre-MDR, the EU required that clinical evidence for high-risk MDs demonstrate safety and expected performance, whereas the FDA required clinical evidence of safety and efficacy, which resulted in slower approval times in the US before accelerated MD approval pathways appeared, e.g. 510(k) for predicates, Priority Review, or Breakthrough Medical Device Designation; these programs seem to have erased a 3-year gap once thought to favor the EU over the FDA [Citation17–19].
To address criticisms regarding US expedited approvals that have emerged over time, reforms have been proposed, including: increasing transparency and consistency in selecting and reviewing surrogate endpoints; requiring more randomized controlled trials (RCTs); clearly identifying products approved through accelerated programs; enforcing requirements for confirmatory trials; limiting time periods for approval contingent on confirmatory evidence; and several reforms tied to payers (mandatory federal rebates until full approval, pricing at marginal cost to incentivize confirmatory trials, requiring outcomes-based payment contracts) [Citation44]. Some of these reforms have been proposed elsewhere for both medicines and MDs, especially with regard to surrogate endpoints [Citation45–48].
3. Expert group discussion highlights and evidence in support of the recommendations
3.1 What underlying principles and distinguishing features of current and previous accelerated approval programs (in Europe, the US, and elsewhere) make them fit for purpose for high-risk implantable medical devices? Are they needed, and under what circumstances?
In the past, the potential benefits for designing and implementing an accelerated CE marking approval process in the EU for innovative, high-risk implantable MDs before MDR were minimal, as illustrated in the literature [Citation17] and by industry consulting firms’ estimates of 12 to 16 weeks for conformity assessments from Notified Bodies for high-risk MDs [Citation49,Citation50]. However, under the more stringent clinical evidence requirements of MDR and with fewer designated Notified Bodies, timelines are likely to lengthen (some Notified Body websites now estimate at least a year [Citation51]). Under such circumstances, general agreement prevailed that an accelerated approval program, similar to those implemented over time in the US for MDs, may be warranted, as long as clear and comprehensive underlying principles and criteria are defined and met as a means to mitigate problems identified in the literature with safety and uncertainty [Citation18,Citation19,Citation27,Citation35,Citation45,Citation52–54].
Problems regarding higher uncertainty could be addressed by requiring RCTs, but carrying out RCTs in the pre-CE mark phase for MDs, even for high-risk implantable devices, may be difficult due to the sheer number of devices produced, often (up to 95% in Europe [Citation55]) by small and medium manufacturers; technical considerations unique to MDs that preclude the gathering of evidence through RCTs (e.g. short commercial lifecycles, predicates or equivalent devices, frequent modifications, inability to meet blinding requirements, challenges related to controls [Citation3,Citation11,Citation56–59]); or ethical reasons because there are no alternatives for a certain population, or preliminary clinical results promise significant improvement in outcomes or savings over current treatment [Citation23]. In this respect, the Early Feasibility Studies (EFSs) Program was introduced by the FDA in recognition of the distinctive regulatory challenges associated with early, pre-market device clinical studies, such as an iterative process in which a prototype is modified over time [Citation60]. Benefits can include faster patient access and development times, perhaps warranting a similar program in the EU, but the sustainability of associated costs for EFSs would need to be assessed [Citation60,Citation61]. Additionally, tools to decide when RCTs should be required [Citation58] and RCT methodologies such as adaptive trial design [Citation62] have been proposed for MDs.
An implication of increased decision uncertainty in pre-market space is the expectation of the need for post-market confirmatory trials once (accelerated) conditional approval has been obtained [Citation20,Citation41]. As post-market evaluations are likely to be based on a larger patient population, weak or delayed signals of harm may be detected more easily and if significant, may lead to the withdrawal of the technology [Citation58,Citation63]. In those cases where RCTs are feasible, post-market trials could address larger patient populations. In other cases, real world data and real world evidence are indicated for post-marketing surveillance, but are also of potential use in clinical study designs and adjustments in earlier phases [Citation64,Citation65]. Registry studies can be particularly important for high-risk implantable MDs, given the expected impact of ancillary technologies, surgical technique and experience development over time, as well as any design modifications or failures related to design that may develop as the device is used on a larger scale in real world settings [Citation3,Citation65,Citation66]. An important requisite is the willingness and ability to remove a product from the market should its early promise in terms of clinical efficacy and safety, not be confirmed by evidence generated during the post-market phase. However, unequivocal requirements for post-market data collection are not common [Citation20,Citation26,Citation67]. Therefore, the use of registry results in regulatory assessment may benefit from implementation guidance, e.g. on data collection and quality assurance, registry governance, and planning of benefit-risk assessments [Citation68].
3.2 How can the HTA community make best use of – or influence – the evidence generated through accelerated regulatory approval programs and how and when should HTA bodies become involved? What are the implications for coverage decisions?
There was consensus that the overall scope of an accelerated program should go beyond market approval (CE mark) to also address HTA requirements, which in turn should inform and be linked to timely local coverage or funding decisions. Based on the new MD and HTA Regulations, and mindful of the problems experienced with accelerated approval programs in the past, the discussions resulted in a proposal for an Accelerated Access Pathway for innovative high-risk MDs (), with associated recommendations outlined in Section 4. The Pathway aims to facilitate timely patient access to high-risk MDs that show innovation, promise to be potentially safe, clinically effective, and good value for money, calling for more clearly delineated roles for the European Commission, Competent Authorities, Member States, the MD Coordination Group (under MDR), the Coordination Group for HTA (under the HTA Regulation), clinical Expert Panels as well as Notified Bodies and other stakeholders (e.g. patient associations, consumer organizations, healthcare professionals, etc.), building on both Regulations’ structures, governance and interactions [Citation1,Citation2]. Such an Accelerated Access Pathway program can identify promising technologies for increased scrutiny of study design, clinical and safety data generation, as well as promoting early dialogue with clinical Expert Panels, regulators and HTA bodies [Citation5] – and with patient representatives. This streamlined process should provide the necessary clinical data for stakeholder decisions on coverage, and clinician and patient decisions regarding uptake and continued use [Citation65], addressing in part the previously fragmented, country-by-country system of entry into practice once CE marking is obtained.
Figure 2. A proposal for an Accelerated Access Pathway (AAP) in the European Union (EU) for innovative high-risk implantable medical devices (MDs).
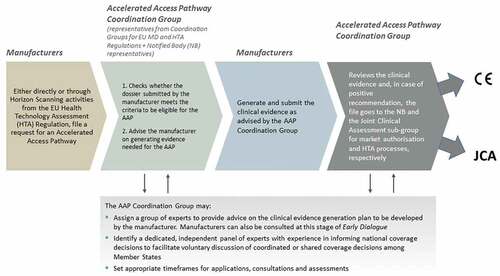
The Accelerated Access Pathway illustrates how parallel regulatory approval and HTA/coverage decision pathways could proceed simultaneously to address evidentiary needs appropriate for regulatory approval (CE Mark), as well as for HTA and coverage decisions. It builds on a key rationale for the HTA Regulation, i.e. the wasteful process of redundant and/or differing levels of HTA (in some situations) in two or more Member States at a time [Citation69]. CE Mark is often obtained relatively quickly but provides no guarantees that a MD will be reimbursed because more evidence is often required for in-country coverage decisions; the Pathway thus also seeks to mitigate delays in access for patients and higher costs to manufacturers that a decentralized process can imply.
At present, high-risk (class III implantable and relevant class IIb devices under Article 54) MDs that demonstrate sufficient innovation and promise of benefit to patients and/or the health system (or increased potential for harm) are selected under MDR to be assessed by clinical Expert Panels through activation of the Clinical Evaluation Consultation Procedure [Citation1,Citation7]. Our proposed Accelerated Access Pathway provides a mechanism for identifying potential innovative, high-risk candidates through self-selection (manufacturers apply either directly or through Horizon Scanning initiatives (HTA Regulation Article 22)), which automatically creates an Accelerated Access Pathway Coordination Group made of representatives from the MD and HTA Coordination Groups and Notified Bodies, to evaluate the application (based on defined principles and criteria) and activate a series of actions to advise the manufacturer on the appropriate clinical evidence generation plan.
The Accelerated Access Pathway Coordination Group thus provides for early – and ongoing – dialogue, pre- and post-CE mark approval, to anticipate the clinical evidence needs for regulatory approval and sufficient data for HTA to inform in-country coverage/funding decisions, anticipate any additional Member State requirements, and foster accelerated patient access. It takes advantage of provisions in both regulations earmarking innovative high-risk MDs for Expert Panel advice (MDR Article 61(2)) and manufacturer-requested joint scientific consultations (HTA Regulation Article 17) for likely joint clinical assessment candidates (HTA Regulation Article 7), and exploits the European database on MDs, EUDAMED (MDR Article 33), to automate controls on post-market evidence generation, information sharing and updates [Citation1,Citation2].
4. Recommendations for an EU innovative high-risk medical device accelerated access pathway
4.1. Recommendation 1: create a centralized Accelerated Access Pathway for innovative high-risk implantable MDs
Decide centrally within the EU about the appropriateness of an accelerated access pathway for innovative high-risk implantable MDs that covers not only market approval (CE mark), but also brings the process closer to patient access, addressing the information and evidence needs sufficient for HTA, where appropriate, and for fostering accelerated Member State decision-making processes for coverage (). This process should:
Build on the MDR and HTA Regulation roles, with support from the European Commission and cooperation among Notified Bodies and regulatory authorities (Competent Authorities) in Member States and between Member States and the European Commission for harmonized implementation.
Include a key role for the MD Coordination Group to work directly with the HTA Coordination Group and Notified Body representatives to form an Accelerated Access Pathway Coordination Group to:
Screen manufacturer applications to identify those MDs appropriate for accelerated approval, for joint clinical assessment (HTA Regulation) and accelerated patient access to innovative high-risk implantable MDs, giving priority to those technologies identified through the Horizon Scanning function of the HTA Regulation;
Expand the MDR Expert Panels to include experts identified for the Joint Scientific Consultation and Joint Clinical Assessment sub-groups from the HTA Regulation. The objective would be to provide alignment on clinical evidence requirements and advise manufacturers on generating evidence that would be sufficient for both CE mark (under MDR) and joint clinical assessments (under the HTA Regulation);
Actively consult the stakeholder network (foreseen in the HTA Regulation) for input on applications to the pathway, in particular patient associations and consumer organizations;
Be fully cognizant of the inherent differences between medicines and MDs that make the lifecycle of the product important for reasons related to mechanisms of action, setting-specific aspects, learning curve for healthcare professionals, impact on organizational efficiency, and possible modifications during and after adoption, which affect evidence generation, and generally longer timelines for follow-up.
This centralized process should thus create a system for technologies chosen for accelerated pathways that, in an early dialogue fashion, formalizes ‘early HTA’ [Citation3,Citation6,Citation70] to assess evidence needs along the access pathway that are sufficient for both regulators and HTA bodies, sharing the Clinical Evaluation Report and Summary of Safety and Clinical Performance prepared by the manufacturer, the Clinical Evaluation Assessment Report prepared by the Notified Body, and any documentation from Expert Panels through the Clinical Evaluation Consultation Procedures, where applied, from the MDR process [Citation7]. Appropriate timeframes for applications, consultations, advice, and assessments, starting from those included in the MD and HTA Regulations, should be considered and specified to avoid lengthening the process beyond the existing pathways.
Of course, Member State sovereignty mandates that final decisions on coverage remain with each country. Nevertheless, a process that brings the EU and Member States together to address in a centralized manner issues regarding clinical effectiveness and safety that affect patients equally across borders and that introduces the basis for evaluations of cost-effectiveness and organizational impact at national level as part of HTA is arguably a necessary step in the regulation and uptake of innovative technologies in Europe, and elsewhere. Market approval (CE mark) for MDs may have been faster in the EU compared to the US in the past, but patient access to the product may now actually be comparable or even slower in the EU [Citation17,Citation71], and is carried out in an uncoordinated fashion.
4.2. Recommendation 2: establish a core set of principles underlying an accelerated access pathway
Formulate and publicly share a core set of principles underlying an accelerated access pathway for innovative high-risk implantable MDs, that guarantees:
Transparency and ethics: Provide clear criteria on evidence (or evidence standards) needed to both enter the accelerated pathway and maintain approval. Publish information on how decisions are made to include an MD in the program, using the MDR database (EUDAMED), where applicable, and provide clear labeling for clinicians and patients regarding those technologies that have followed an accelerated pathway. Ensure a role for ethical committees, where appropriate.
Consistency: Guarantee consistent decisions, especially concerning withdrawing approval following negative or ambiguous confirmatory data from post-marketing studies, or the validation of surrogate endpoints.
Inclusivity: Provide appropriate opportunity for early involvement of regulatory (Competent Authorities) and HTA bodies, Expert Panels for scientific advice, patients, clinicians in discussions on the product’s development program and evidence generation, to encompass varying perspectives in an equitable manner so that decision-making can reflect the needs of society, the healthcare system and all people and entities impacted by the decision.
Impartiality: Let no influences, internal or external, exert pressure on those making decisions regarding which MDs are appropriate for an accelerated pathway for approval and/or patient access.
Commitment by beneficiaries of accelerated pathways to collect and communicate data from confirmatory trials, even in the case of negative results.
The principles underlying the accelerated access pathway and the criteria for adherence (below) to the program take inspiration from past, existing and proposed accelerated approval programs around the world for medicines and MDs [Citation12,Citation28,Citation29,Citation33,Citation72–74], early patient access or coverage with evidence development programs [Citation75–78], performance-based risk-sharing arrangements [Citation79,Citation80], and HTA [Citation81,Citation82], adapted and changed here to proactively address associated problems noted over time. Poor transparency regarding decisions on which products were deemed eligible for accelerated approval programs and inconsistencies on how such products would be followed over time was often cited during the discussions, criticisms echoed in the literature [Citation8,Citation10,Citation45,Citation83]. An ethical framework [Citation23] for all stakeholders and healthcare systems and providing opportunity for scientific advice and early dialogue, with full disclosure on labeling, have all been studied and proposed for products approved through accelerated pathways [Citation5,Citation7,Citation38,Citation83,Citation84]. Inclusivity, to consider values from all those impacted by decisions, for example, underlies an EU Erasmus+ HTA e-learning initiative with an integrative approach to training future HTA professionals to assess increasingly complex technologies [Citation85,Citation86]. Along with impartiality of experts tasked with assessing applications to the pathway (e.g. declarations of interests, confidentiality and commitment, as currently in place for MDR expert panels), these principles form a foundation for the socio-economic evaluation needed for allocation of scarce resources since accelerated pathways often involve small patient groups and high-priced technology [Citation23,Citation81]. Impartiality should not, however, mean that undue firewalls are established between different players, impeding the efficacy of early dialogue [Citation5]). Transparency, inclusivity and impartiality are also needed to avoid creating an implicit promise that agreement in early dialogue will guarantee either approval or reimbursement or even protect from market withdrawal should confirmatory evidence prove weak. In turn, manufacturers must be sufficiently attracted by the pathway – for the opportunity to better predict and prepare for HTA and coverage decision processes – but also commit to ongoing minimum sufficient evidence generation to maintain approval and coverage.
Because no newly conceived process can divorce itself from considerations regarding ethical production in an environmentally responsible manner, we also emphasize the importance of pursuing sustainable production (and disposal) for MDs and packaging, following laws and protections for worker and patient safety and to reduce contamination, pollution and energy consumption [Citation87–90].
4.3. Recommendation 3: establish clear eligibility criteria for an accelerated access pathway
According to the transparency principle mentioned above, it is important to establish criteria for eligibility for an accelerated access pathway for innovative high-risk implantable MDs based on the lifecycle of the MD. These criteria need to be debated, but could reasonably include:
The MD is intended for the prevention, diagnosis or treatment or rehabilitation of a life threatening or seriously debilitating disease or condition.
The MD addresses an unmet medical need, or a minimum yet high quality set of evidence is provided that pursuit of a standard pathway would preclude patients from access to needed treatment while guaranteeing patient safety.
There are no predicates and the device represents a novel or highly promising technology for patients in immediate need, that is, based on preliminary evidence, it offers a significant added clinical and/or non-clinical (survival benefits, significant health-related quality of life gained, reduction of morbidity, safety, care pathway efficiency, etc.) improvement for patients, captured by patient-reported or other measures, over the existing standard of care for prevention, diagnosis or treatment of a life threatening or seriously debilitating disease or condition, or a priority disease area for the healthcare system.
The technology promises significant positive impact for the individual and the healthcare system, and its sustainability and resilience and/or contributes to making care more agile and more manageable.
The MD is appropriate for iterative development, i.e. it can either be approved in stages for a restricted patient population and then extended to other patient groups over time, or conditional marketing approval can be granted based on early data and confirmed through subsequent data gathered to confirm the benefit risk balance.
Gathering of real world data (and/or additional clinical trial data, registry-based clinical trials), including patient-reported outcomes and experience data, after restricted population approval or conditional approval is a necessary condition.
Possible criteria were studied and discussed to come up with a list tailored to the specifics of high-risk implantable MDs. The innovative nature of the device must be established, clearly distinguished from predicates and in the absence of alternatives, but also with the important extension to the needs of the healthcare system to respond constantly to new and important challenges.
4.4. Recommendation 4 – set and manage expectations for evidence generation for an accelerated access pathway
Measures to mitigate uncertainty in evidence generation along the lifecycle of the high-risk implantable MDs accessing the program should be taken.
The Expert Panel(s) () should provide advice on appropriate and feasible study designs for pre-market clinical evidence generation, ideally using tools to determine whether RCTs are possible. Ongoing dialogue should address any proposed adaptations to study designs and post-market study requirements.
Where patient-relevant clinical outcome measurement is not possible, accept only validated surrogate endpoints for pre-market studies or, where unvalidated surrogates are accepted for market approval, establish clear requirements for subsequent validation and monitoring. The Accelerated Access Pathway Coordination Group (which incorporates representatives from the MD and HTA Coordination Groups and sub-groups), with Expert Panels, should develop harmonized criteria at the EU level for the acceptability and evaluation of surrogate endpoints that could be part of the open discussions with regulatory Competent Authorities for national-level adoption.
4.5. Recommendation 5 – ensure that post-market evidence generation is a necessary element for an accelerated access pathway
Tie acceptance into an accelerated access pathway to coverage with evidence development or performance-based risk-sharing agreements, so that there is a clear commitment to define, implement and regularly monitor a post-marketing evidence generation plan.
Implement and immediately employ technology using the Unique Device Identification coding and EUDAMED (from MDR) to establish mandatory post-marketing evidence generation, in the form of confirmatory trials or large observational studies or trans-national registries, and monitor its collection and findings through automated processes.
Establish controls and procedures (e.g. using EUDAMED) to easily and quickly identify a technology for which the company has failed to collect sufficient confirmatory evidence within pre-established timeframes and subsequently remove it from the market. Developing clear decision criteria for exit or removal strategies is recommended that take into consideration all stakeholders, especially physicians and patients already using the MD. Likewise, information regarding technologies that may expand indications through post-marketing evidence generation and/or real-world evidence will also be automatically updated.
5. Conclusions
In this paper, we have provided a review of issues surrounding evidence generation, approval processes, HTA and coverage decisions for high-risk MDs, and presented our perspective and recommendations on whether – and how – some technologies showing particular promise of efficacy and innovation might warrant special attention to afford more timely – i.e. accelerated – and safe patient access.
The new EU MD and HTA Regulations together are welcome developments that will hopefully increase the quality of clinical evidence for MDs and reduce fragmentation in the market access process in the EU. However, we caution that for certain types of innovative, high-risk MDs that indicate important advances in patient treatment or for healthcare systems, the new regulations may result in delays that make it unfavorable in comparison to ad-hoc accelerated approval processes in other jurisdictions (e.g. the US) and delay time to patient access. Therefore, in the interest of balancing safety and high-quality evidence on efficacy with timely market access processes, and through analysis of the lessons learned from two decades of accelerated approval programs for medicines and MDs around the world, we have developed a proposal for an Accelerated Access Pathway for innovative, high-risk MDs that can fit the EU context, leverage the two new regulations, and promote further collaboration on coverage decisions. Based on shared principles and clear eligibility criteria, the Pathway formalizes cooperation between the MD and HTA Coordination Groups from both regulations and introduces and facilitates early and ongoing expert advice on clinical evidence generation for regulatory, HTA and coverage decisions as well as for ongoing surveillance and updating indications. Thus, the role of the Accelerated Access Pathway should go beyond market approval (CE mark) to also address HTA requirements which in turn should inform and be linked to timely local coverage or funding decisions.
6. Expert opinion
Accelerated approval programs for medicines have been in place for decades in various parts of the world, a response to lengthening regulatory approval times that resulted in a marked drop in the number of new molecular entities approved, for example, by the FDA after 1990 [Citation91]. Extended in some areas to medical devices (MDs) as well, these programs have arguably resulted in making life-saving, ground-breaking medical technologies available to patient populations that might otherwise have waited considerably longer, with associated costs to patients and society. However, these programs have also been the subject of criticism for fear of reducing protections for patients or approving medical technologies based on early (often weak) evidence without subsequent proof of efficacy [Citation20,Citation38,Citation84,Citation92]. Proposing an accelerated access pathway program for MDs in the European Union (EU) at the exact moment when two new regulations have been approved, albeit not yet fully implemented, seems a timely and useful contribution to the policy debate on patient access and innovativeness in the EU.
We believe our proposal has considerable benefits for patients, manufacturers, and healthcare systems precisely because it seeks to preemptively address gaps in evidence on efficacy and patient safety observed in previous programs. It can also make the EU a more attractive market compared with other jurisdictions because it identifies where and when to start involving HTA bodies to better coordinate the evidence needs for patient access beyond CE Mark and suggests a more proactive role for the Coordination Groups for both regulations in facilitating voluntary discussion among Member States of evidence needs to support pricing and reimbursement. An important limitation in establishing an accelerated program is that there are now fewer certified Notified Bodies since the MD Regulation went into effect. However, our hope is that including Notified Body representatives and the two Coordinating Groups in one Accelerated Access Pathway Coordination Group will help to ease the burden, by supporting Notified bodies with Expert Panels to provide advice on the entire MD lifecycle and instituting early dialogue on clinical evidence that would be suitable for both CE mark and coverage decisions to serve all stakeholders.
The Accelerated Access Pathway concept for high-risk MDs in the EU recommended here will have to be further aligned with the responsibilities of the institutions implementing pricing and reimbursement regulations at the national level. Individual country sovereignty presents an obvious limitation to the strategy, but with the HTA Regulation providing for joint clinical assessments, it could be an opportune moment to provide the means for Member States to address other domains in HTA (e.g. economic, social, organizational) in a coordinated fashion to realize synergies and reduce redundancies here as well. Much as clinical guidelines have become international in nature, and CE Mark is recognized simultaneously in all EU markets, we see a natural progression (aided by improvements in data collection and analysis) toward greater pooling of analysis of these additional domains to inform coverage in several countries at once.
Another limitation might be attractiveness for manufacturers to apply for the program. Yet with less experience in clinical investigation than their medicine developer counterparts, MD manufacturers should welcome the increased opportunity for expert advice regarding pre- and post-market evidence generation needed for CE Mark, HTA and eventually coverage, in the hope of facilitating access to more than one EU market at once. The Accelerated Access Pathway could also be applied, if necessary in stages, before full implementation of both regulations (i.e. 2025) by expanding provisions in both regulations for Expert Panels with recognized expertise in evidence generation for both approval and HTA, to also include experts in pricing and reimbursement from Member States.
Thus, to make this Accelerated Access Pathway effective, it will be important to harmonize and coordinate national policies to guarantee the timely adoption of innovative, high-risk MDs that, as important advancements, merit Accelerated Access Pathway status. Our proposed Accelerated Access Pathway seeks to address inequalities in patient access and facilitate greater cooperation among EU Member States, minimize resource waste and delays, address problems observed in existing expedited regulatory approval programs, and inform MD approval, coverage and payer systems globally.
Declaration of interest
No potential conflict of interest was reported by the author(s).
Supplemental Material
Download MS Word (48.2 KB)Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/17434440.2023.2192868
Additional information
Funding
References
- European Union. Regulation (EU) 2017/745 of the European parliament and of the council of 5 April 2017 on medical devices, amending directive 2001/83/EC, regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing council directives 90/385/EEC and 93/42/EEC (Text with EEA relevance.) [Internet]. OJ L. Apr 5 2017; Available from . http://data.europa.eu/eli/reg/2017/745/oj/eng
- European Union. Regulation (EU) 2021/2282 of the European parliament and of the council of 15 December 2021 on health technology assessment and amending directive 2011/24/EU (Text with EEA relevance) [Internet]. Dec 15, 2021. Available from: http://data.europa.eu/eli/reg/2021/2282/oj/eng.
- Tarricone R, Ciani O, Torbica A, et al. Lifecycle evidence requirements for high-risk implantable medical devices: a European perspective. Expert Rev Med Devices. 2020;17:993–1006.
- Husereau D, Henshall C, Sampietro-Colom L, et al. CHANGING HEALTH TECHNOLOGY ASSESSMENT PARADIGMS? Int J Technol Assess Health Care. 2016;32:191–199.
- Blankart CR, Dams F, Penton H, et al. Regulatory and HTA early dialogues in medical devices. Health Policy. 2021;125(10):1322–1329.
- Tummers M, Kværner K, Sampietro-Colom L, et al. On the integration of early health technology assessment in the innovation process: reflections from five stakeholders. Int J Technol Assess Health Care. 2020;36:481–485.
- Fraser AG, Byrne RA, Kautzner J, et al. Implementing the new European regulations on medical devices-clinical responsibilities for evidence-based practice: a report from the Regulatory Affairs Committee of the European society of cardiology. Eur Heart J. 2020;41:2589–2596.
- Fraser AG, Nelissen RGHH, Kjærsgaard-Andersen P, et al. Improved clinical investigation and evaluation of high-risk medical devices: the rationale and objectives of CORE-MD (coordinating research and evidence for medical devices). Eur Heart J - Qual Care Clinl Outcomes. 2022;8:249–258.
- Tarricone R, Ciani O, D’Acunto S, et al. The rise of rules: will the new EU regulation of medical devices make us safer? Eur J Intern Med. 2020;80:117–120.
- Beck A, Retèl VP, Bhairosing PA, et al. Barriers and facilitators of patient access to medical devices in Europe: a systematic literature review. Health Policy. 2019;123:1185–1198.
- Tarricone R, Torbica A, Drummond M. Challenges in the assessment of medical devices: the MedtecHTA project. Health Econ. 2017;26:5–12.
- Cox EM, Edmund AV, Kratz E, et al. Regulatory Affairs 101: introduction to Expedited Regulatory Pathways. Clin Transl Sci. 2020;13:451–461.
- Beaver JA, Howie LJ, Pelosof L, et al. A 25-YEAR EXPERIENCE of US food and drug administration accelerated approval of malignant hematology and oncology drugs and biologics: a review. JAMA Oncol. 2018;4:849–856.
- Van Delm K. Accelerated access to medicinal products. Eur Pharm Law Rev. 2021;4:192–206.
- Van Norman GA. Update to drugs, devices, and the FDA: how recent legislative changes have impacted approval of new therapies. JACC Basic Transl Sci. 2020;5:831–839.
- Detela G, Lodge A. EU regulatory pathways for ATMPs: standard, accelerated and adaptive pathways to marketing authorisation. Mol Ther Methods Clin Dev. 2019;13:205–232.
- Van Norman GA. Drugs and devices: comparison of European and U.S. approval processes. JACC Basic Transl Sci. 2016;1:399–412.
- Ong C, Ly VK, Redberg RF. Comparison of priority vs standard US food and drug administration premarket approval review for high-risk medical devices. JAMA Intern Med. 2020;180:801–803.
- Johnston JL, Dhruva SS, Ross JS, et al. Early experience with the FDA’s breakthrough devices program. Nat Biotechnol. 2020;38:933–938.
- Sachs RE, Donohue JM, Dusetzina SB, et al. Taking the FDA’s concerns seriously. N Engl J Med. 2022;387:199–201.
- Murphy MK, Black NA, Lamping DL, et al. Consensus development methods, and their use in clinical guideline development [Internet]. Health Technol Assess. 1998 [cited 2021 May 20];2. Available from: https://www.journalslibrary.nihr.ac.uk/hta/hta2030.
- Munn Z, Peters MDJ, Stern C, et al. Systematic review or scoping review? Guidance for authors when choosing between a systematic or scoping review approach. BMC Med Res Methodol. 2018;18:143.
- Pace J, Ghinea N, Kerridge I, et al. An ethical framework for the creation, governance and evaluation of accelerated access programs. Health Policy. 2018;122:984–990.
- Nagai S. Flexible and expedited regulatory review processes for innovative medicines and regenerative medical products in the US, the EU, and Japan. Int J Mol Sci. 2019;20:3801.
- Bootes A, Maundu J, Golding S, et al. Fast-track pathways for drug approvals: the Australian experience so far. Aust Prescr. 2019;42:118–119.
- Jokura Y, Yano K, Yamato M. Comparison of the new Japanese legislation for expedited approval of regenerative medicine products with the existing systems in the USA and European Union. J Tissue Eng Regen Med. 2018;12:e1056–e1062.
- Hwang TJ, Ross JS, Vokinger KN, et al. Association between FDA and EMA expedited approval programs and therapeutic value of new medicines: retrospective cohort study. BMJ. 2020;371:m3434.
- European Medicines Agency. Conditional marketing authorisation. Report on ten years of experience at the European medicines agency [Internet]. 2017 [cited 2021 Oct 10]. Available from: https://www.ema.europa.eu/en/documents/report/conditional-marketing-authorisation-report-ten-years-experience-european-medicines-agency_en.pdf.
- U.S. Food and Drug Administration. Breakthrough Devices Program [Internet]. FDA. FDA; 2021 [cited 2021 May 18]. Available from: https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program.
- Therapeutic Goods Administration. Prescription medicines determination and designation notices [Internet]. Therapeutic Goods Administration (TGA). Australian Government Department of Health; [cited 2021 Nov 16]. Available from: https://www.tga.gov.au/ws-designation-notices-index.
- Therapeutic Goods Administration. Fast track approval pathways [Internet]. Therapeutic goods administration (TGA). Australian Government Department of Health; [cited 2021 Oct 19]. Available from: https://www.tga.gov.au/fast-track-approval-pathways.
- Therapeutic Goods Administration (TGA). In: Australian department of health. Priority applicant guidelines for medical devices (including IVDs) [Internet]. Australia: Therapeutic Goods Administration (TGA). Australian Government Department of Health. 2020 cited 2021 Oct 8. Available from. https://www.tga.gov.au/resource/priority-applicant-guidelines-medical-devices-including-ivds
- Tanaka M, Idei M, Sakaguchi H, et al. Achievements and challenges of the Sakigake designation system in Japan. Br J Clin Pharmacol. 2021;87:4027–4035.
- Salcher-Konrad M, Naci H, Davis C. Approval of cancer drugs with uncertain therapeutic value: a comparison of regulatory decisions in Europe and the United States. Milbank Q. 2020;98:1219–1256.
- Bruce CS, Brhlikova P, Heath J, et al. The use of validated and nonvalidated surrogate endpoints in two European medicines agency expedited approval pathways: a cross-sectional study of products authorised 2011–2018. PLoS Med. 2019;16:e1002873.
- Kim C, Prasad V. Cancer drugs approved on the basis of a surrogate end point and subsequent overall survival: an analysis of 5 years of US food and drug administration approvals. JAMA Intern Med. 2015;175:1992–1994.
- Ribeiro TB, Buss L, Wayant C, et al. Comparison of FDA accelerated vs regular pathway approvals for lung cancer treatments between 2006 and 2018. PLOS ONE. 2020;15:e0236345.
- Gyawali B, Rome BN, Kesselheim AS. Regulatory and clinical consequences of negative confirmatory trials of accelerated approval cancer drugs: retrospective observational study. BMJ. 2021;374:n1959.
- Downing NS, Shah ND, Aminawung JA, et al. Postmarket safety events among novel therapeutics approved by the US food and drug administration between 2001 and 2010. JAMA. 2017;317:1854–1863.
- Mostaghim SR, Gagne JJ, Kesselheim AS. Safety related label changes for new drugs after approval in the US through expedited regulatory pathways: retrospective cohort study. BMJ. 2017;358:j3837.
- Wallach JD, Ross JS, The NH. US Food and Drug Administration’s expedited approval programs: evidentiary standards, regulatory trade-offs, and potential improvements. Clin Trials. 2018;15:219–229.
- Chen EY, Haslam A, Prasad V. FDA acceptance of surrogate end points for cancer drug approval: 1992-2019. JAMA Intern Med. 2020;180:912–914.
- Darrow JJ, Avorn J, Kesselheim AS. FDA Regulation and. Approval of Medical Devices: 1976-2020. JAMA. 2021;326:420–432.
- Kaltenboeck A, Mehlman A, Pearson SD. Potential policy reforms to strengthen the accelerated approval pathway. J Comp Eff Res. 2021;10:1177–1186.
- Naci H, Salcher-Konrad M, Kesselheim AS, et al. Generating comparative evidence on new drugs and devices before approval. Lancet. 2020;395:986–997.
- Kadakia KT, Beckman AL, Ross JS, et al. Renewing the call for reforms to medical device safety-the case of penumbra. JAMA Intern Med. 2022;182:59–65.
- Ciani O, Buyse M, Drummond M, et al. Time to review the role of surrogate end points in health policy: state of the art and the way forward. Value Health. 2017;20:487–495.
- Ciani O, Grigore B, Blommestein H, et al. Validity of surrogate endpoints and their impact on coverage recommendations: a retrospective analysis across international health technology assessment agencies. Med Decis Making. 2021;41:439–452.
- The EU med device regulation timeline: what you should know in [Internet] 2020. [cited 2022 Jan 15]. Available from: https://www.qualio.com/blog/eu-medical-device-regulation-timeline.
- How do you get medical device CE marking approval? [Internet]. Medical device academy. [cited 2022 Jan 15]. Available from: https://medicaldeviceacademy.com/medical-device-ce-marking/.
- Medcert.de. MDR - Medical Device Regulation 2017/745. Initial conformity assessment process [Internet]. [cited 2022 Jan 15]. Available from: https://www.medcert.de/wp-content/uploads/MDR-Zertifizierungsablauf-15042021.pdf.
- Pease AM, Krumholz HM, Downing NS, et al. Postapproval studies of drugs initially approved by the FDA on the basis of limited evidence: systematic review. BMJ. 2017;357:j1680.
- Olavarria OA, Shah P, Bernardi K, et al. The evidence behind products cleared by the 510(k) Process. J Am Coll Surg. 2020;231:S85–S86.
- Darrow JJ, Avorn J, Kesselheim AS, et al. Regulation of Pharmaceuticals, 1983-2018. JAMA. 2020;323:164–176.
- MedTech Europe. MedTech Europe’s Facts and Figures 2021 [Internet]. MedTech Eur. cited 2022 Sep 1; Available from. https://www.medtecheurope.org/resource-library/medtech-europes-facts-and-figures-2021/
- Kanavos P, Angelis A, An DM. EU-wide approach to HTA: an irrelevant development or an opportunity not to be missed? Eur J Health Econ. 2019;20:329–332.
- Campbell B, Wilkinson J, Marlow M, et al. Generating evidence for new high-risk medical devices. BMJ Surg Interv Health Technol. 2019;1:e000022.
- Paez A, Rovers M, Hutchinson K, et al. Beyond the RCT: when are randomised trials unnecessary for new implantable therapeutic devices, and what should we do instead? Annals of surgery (under review). 2021;
- Drummond M, Griffin A, Tarricone R. Economic evaluation for devices and drugs—same or different? - Drummond - 2009 - value in health - Wiley online library. Value Health. 2009;12:402–406.
- Holmes DR, Califf R, Farb A, et al. Overcoming the challenges of conducting early feasibility studies of medical devices in the United States. J Am Coll Cardiol. 2016;68:1908–1915.
- Callea G, Federici C, Freddi R, et al. Recommendations for the design and implementation of an Early Feasibility Studies program for medical devices in the European Union. Expert Rev Med Devices. 2022;19:315–325.
- Bhatt DL, Mehta C. Adaptive designs for clinical trials. Drazen JM, Harrington DP, McMurray JJV, et al., editors. N Engl J Med. 2016;375:65–74.
- Fleetcroft C, McCulloch P, Campbell B. IDEAL as a guide to designing clinical device studies consistent with the new European medical device regulation. BMJ Surg Interv Health Technol. 2021;3:e000066.
- Bolislis WR, Fay M, Kühler TC. Use of real-world data for new drug applications and line extensions. Clin Ther. 2020;42:926–938.
- Campbell B, Wilkinson J, Marlow M, et al. Long-term evidence for new high-risk medical devices. Lancet. 2018;391:2194–2195.
- In: Registries for evaluating patient outcomes: a user’s guide [Internet] 3rd. Gliklich RE, Dreyer NA, Leavy MB, (editors.) Rockville (MD): Agency for Healthcare Research and Quality (US); 2014. cited 2021 Nov 29]. Available from. http://www.ncbi.nlm.nih.gov/books/NBK208616/
- Mahase E. FDA allows drugs without proven clinical benefit to languish for years on accelerated pathway. BMJ. 2021;374:n1898.
- McGettigan P, Alonso Olmo C, Plueschke K, et al. Patient registries: an underused resource for medicines evaluation: operational proposals for increasing the use of patient registries in regulatory assessments. Drug Saf. 2019;42:1343–1351.
- European Commission. Q&A: commission proposal on health technology assessment [Internet]. European commission - European commission. [cited 2021 Nov 15]. Available from: https://ec.europa.eu/commission/presscorner/detail/en/MEMO_18_487.
- Bouttell J, Briggs A, Hawkins N. A different animal? Identifying the features of health technology assessment for developers of medical technologies. Int J Technol Assess Health Care. 2020;36:285–291.
- Basu S, Hassenplug JC. Patient access to medical devices — a comparison of U.S. and European review processes [Internet]. Massachusetts Medical Society; 2012 [cited 2021 Nov 19]. Available from: https://www.nejm.org/doi/10.1056/NEJMp1204170.
- Therapeutic Goods Administration (TGA). In: Australian department of health. consultation: accelerated assessment of medical devices - priority review pathway - implementation [Internet]. Australia: Therapeutic Goods Administration (TGA). Australian Government Department of Health. 2017 cited 2021 Jul 1. Available from. https://www.tga.gov.au/consultation/consultation-accelerated-assessment-medical-devices-priority-review-pathway-implementation
- US Food and Drug Administration. Fast track, breakthrough therapy, accelerated approval, priority review [Internet]. FDA; 2019 [cited 2021 Nov 18]. Available from: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/fast-track-breakthrough-therapy-accelerated-approval-priority-review.
- European Medicines Agency. Adaptive pathways: key learnings and next steps [Internet]. Eur Med Agen. 2018 cited 2021 May 17; Available from. https://www.ema.europa.eu/en/news/adaptive-pathways-key-learnings-next-steps
- Reckers-Droog V, Federici C, Brouwer W, et al. Challenges with coverage with evidence development schemes for medical devices: a systematic review. Health Policy Technol. 2020;9:146–156.
- Federici C, Reckers-Droog V, Ciani O, et al. Coverage with evidence development schemes for medical devices in Europe: characteristics and challenges [Internet]. Eur J Health Econ. 2021 cited 2021 Jul 19; DOI: 10.1007/s10198-021-01334-9
- Martelli N, van den Brink H, Borget I. New French coverage with evidence development for innovative medical devices: improvements and unresolved issues. Value Health. 2016;19:17–19.
- Adenot I, Camus D, A-A ÉDF, et al. Early patient access to health technologies: is innovation needed for early management? Therapies. 2020;75:71–83.
- Boriani G, Vitolo M, Svennberg E, et al. Performance-based risk-sharing arrangements for devices and procedures in cardiac electrophysiology: an innovative perspective. Europace. 2022;24:1541–1547.
- Garrison LP, Towse A, Briggs A, et al. Performance-based risk-sharing arrangements—good practices for design, implementation, and evaluation: report of the ISPOR good practices for performance-based risk-sharing arrangements task force. Value in Health. 2013;16:703–719.
- Bond K, Stiffell R, Ollendorf DA. Principles for deliberative processes in health technology assessment. Int J Technol Assess Health Care. 2020;36:445–452.
- Drummond MF, Schwartz JS, Jönsson B, et al. Key principles for the improved conduct of health technology assessments for resource allocation decisions. Int J Technol Assess Health Care. 2008;24:244–258.
- Fraser AG, Butchart EG, Szymański P, et al. The need for transparency of clinical evidence for medical devices in Europe. Lancet. 2018;392:521–530.
- Zettler M, Nabhan C. Fulfillment of postmarketing requirements to the FDA for therapies granted oncology indications between 2011 and 2016. JAMA Oncol. 2018;4:993–994.
- Oortwijn W, Sampietro-Colom L, editors. The VALIDATE handbook: an approach on the integration of values in doing assessments of health technologies [Internet]. Netherlands: Radboud University Press. 2022. cited 2022 Sep 1. Available from. https://library.oapen.org/handle/20.500.12657/57096
- Gj van der W, Oortwijn W. Health technology assessment: a matter of facts and values. Int J Technol Assess Health Care. 2022;38:e53.
- Marsh K, Ganz ML, Hsu J, et al. Expanding health technology assessments to include effects on the environment. Value Health. 2016;19:249–254.
- Polisena J, Angelis GD, Kaunelis D, et al. ENVIRONMENTAL IMPACT ASSESSMENT OF A HEALTH TECHNOLOGY: a SCOPING REVIEW. Int J Technol Assess Health Care. 2018;34:317–326.
- Sherman JD, MacNeill A, Thiel C. Reducing pollution from the health care industry. JAMA. 2019;322:1043–1044.
- Sørensen BL, Grüttner H. Comparative study on environmental impacts of reusable and single-use bronchoscopes. Am J Environ Protec. 2018;7:55.
- Kinch MS, Haynesworth A, Kinch SL, et al. An overview of FDA-approved new molecular entities: 1827–2013. Drug Discov Today. 2014;19:1033–1039.
- Banzi R, Gerardi C, Bertele’ V, et al. Approvals of drugs with uncertain benefit–risk profiles in Europe. Eur J Intern Med. 2015;26:572–584.
Appendix
Appendix 1. Participants, project to explore accelerated pathways to approval, patient access, and evidence generation for innovative, high-risk, implantable medical devices (in reverse alphabetical order).