
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Carbon nanoparticles (CNP) are generated by incomplete combustion of diesel engines. Several epidemiological studies associated higher susceptibility to particulate matter related adverse respiratory outcomes with preexisting conditions like chronic bronchitis (CB). Therefore, we compared the effect of CNP exposure on primary bronchial epithelial cells (PBEC) developed in air-liquid interface (ALI) models of normal versus CB-like-mucosa.
PBEC cultured at ALI represented normal mucosa (PBEC-ALI). To develop CB-like-mucosa (PBEC-ALI/CB), 1 ng/ml interleukin-13 was added to the basal media of PBEC-ALI culturing. PBEC-ALI and PBEC-ALI/CB were exposed to sham or to aerosolized CNP using XposeALI® system. Protein levels of CXCL-8 and MMP-9 were measured in the basal media using ELISA. Transcript expression of pro-inflammatory (CXCL8, IL6, TNF, NFKB), oxidative stress (HMOX1, SOD3, GSTA1, GPx), tissue injury/repair (MMP9/TIMP1) and bronchial cell type markers (MUC5AC, CC10) were assessed using qRT-PCR.
Increased secretion of CXCL-8 and MMP-9 markers was detected 24 h post-exposure in both PBEC-ALI and PBEC-ALI/CB with more pronounced effect in the later. Pro-inflammatory and tissue injury markers were increased at both 6 h and 24 h post-exposure in PBEC-ALI/CB. Oxidative stress markers exhibited similar responses at 6 h and 24 h post-exposure in PBEC-ALI/CB. The club cell specific marker CC10 was increased by 300 fold in PBEC-ALI/CB and 20 fold in PBEC-ALI following CNP exposure.
Our data indicates an earlier and stronger reaction of pro-inflammatory, oxidative stress and tissue injury markers in PBEC-ALI/CB models compared to PBEC-ALI models following CNP exposure. The findings may provide insight into the plausible mechanisms of higher susceptibility among predisposed individuals to nanoparticle exposure.
Introduction
Particulate matter (PM) has been defined as a ‘criteria air pollutant’ by the US-Environmental protection agency (EPA Citation2018). Several epidemiological studies reported association between exposure to ambient PM and cardio-pulmonary impairments, one of the major public health concerns in industrialized cities throughout the world (Rothen-Rutishauser, Kiama, & Gehr Citation2005; Rich et al. Citation2012; Hoek et al. Citation2013; WHO Citation2018). Both short-term and long-term PM exposures lead to increased morbidity and mortality (Garshick Citation2014; Kloog et al. Citation2013; Pope et al. Citation2002; Tian et al. Citation2018). World Health Organization (WHO) estimated that over 7 million premature deaths worldwide per year can be attributed to PM air pollution in 2012 (WHO Citation2014). Exposure to ultrafine particles (UFP; diameter <100 nm) in the PM are considered as a major risk factor for pulmonary impairments (Nemmar et al. Citation2013; Autengruber et al. Citation2014). The majority of these effects occurred among pre-disposed individuals with preexisting disorders like chronic bronchitis (CB), chronic obstructive pulmonary diseases (COPD), asthma and cardiovascular diseases (Inoue and Takano Citation2011; Nichols et al. Citation2013). It has been shown that individuals with impaired lung physiology are at higher risk to various chronic respiratory diseases like COPD which are exacerbated due to exposure to environmental stressors like UFP and/or nano-sized particles (Burrows, Knudson, and Lebowitz Citation1977; Samet, Tager, and Speizer Citation1983; Layachi et al. Citation2013). Chronic bronchitis is often associated with cigarette smoking as well as long term exposure to highly polluted ambient and/or occupational air quality environment (Axelsson et al. Citation2016). It is characterized by chronic productive cough due to mucus hypersecretion and proliferation of mucus-secreting cells, recurrent or persistent airway inflammation, airflow obstruction, and often airway hyper-responsiveness (Jeffery Citation1991; O’Byrne and Postma Citation1999). The experts of the European Agency for Safety and Health at Work identified inhalation exposure to nanoparticles (NP; diameter <100 nm) as the strongest emerging risk at work (EU-OSHA Citation2009), and chronic bronchitis as the most common respiratory disease among workers exposed to work place fumes and dusts .
Carbon-based nanomaterials (black nanoparticles, fullerenes, carbon nanotubes, fibers, etc.) are considered as the most promising products of nanotechnology with applications ranging from drug delivery to electronics (Yuan et al. Citation2019). Carbon black nanoparticles are the traditional nano-sized carbonaceous nanomaterials. Exposure to ultrafine carbon particles, the major constituent of urban UFP, has increased dramatically due to increased emissions of combustion derived ultrafine particles (soot) from traffic, industry and heating activities (Yuan et al. Citation2019). The major constituent of urban ultrafine particles is soot, i.e. carbonaceous material consisting of an elemental carbon core surrounded by various types of organic carbon and small fractions of metal, sulfate, and nitrate compounds. Moreover, CNPs with primary particle diameter ∼25 nm constitute the core of combustion derived particles and represent a relevant toxicological surrogate for exhaust particles from diesel engines (Su et al. Citation2004; BeruBe et al. Citation2007; Stoeger et al. Citation2009) Motor-vehicle emissions consist of a complex mixture of particulate, chemical and gaseous pollutants such as fine particulate matter (PM2.5; diameter <2.5 μm), UFP, metals, volatile organic material, black carbon, ozone, etc. A major component of urban PM air pollution is CNPs. They constitute the core of combustion derived particles and represents relevant surrogates for exhaust particles from modern diesel engines (Müller et al. Citation2006; Ning et al. Citation2007; Ganguly et al. Citation2017). CNPs refers to industrially produced, combustion derived carbonaceous nanoparticles with low organic carbon content, but is also an ingredient in many rubber products, plastics, inks and paints with an annual production of 10 million tons (Jacobsen et al. Citation2009; Jacobsen et al. Citation2011) indicating its wide usage and potentially massive exposure in our regular life. The concern for the toxicity of CNP arises because of the large surface area, extensive respiratory tract deposition, high reactivity, and slow clearance (Jacobsen et al. Citation2009; Schmid and Stoeger Citation2016).
Both in vitro and in vivo studies on health effects of ambient PM have identified the generation of oxidative stress as one of the major mechanisms by which air pollution particles exert adverse biological effects (Li et al. Citation1999; Li, Xia, and Nel Citation2008). Animal experiments, exposing healthy mice to high levels of laboratory-generated CNP by inhalation and instillation have shown local acute inflammatory effects in the lungs (Stoeger et al. Citation2009; Andre Citation2006). Many studies show that nanoparticles by virtue of their small size can easily penetrate into the deeper part of lung (alveolar compartment) by evading airway mucocillary as well as clearance by macrophages. However, NP do not evade deposition on the central airway mucosa. Per surface area, deposition in the central airway mucosa will be about the same or higher than in the alveolar region. The higher deposited fraction in the peripheral lung primarily reflects the much larger surface area of the alveolar region (Oberdörster et al. Citation2005). This warrants the careful study of NP effects in both regions. Additionally, prolonged airway inflammation associated with NP exposure may lead to systemic inflammation, which may increase the susceptibility to other chronic pulmonary diseases (Daigle et al. Citation2003; Chalupa et al. Citation2004). There are ample evidence available from epidemiological, in vivo and in vitro studies suggesting that exposure to CNP or other NP may contribute to pulmonary morbidity by eliciting pro-inflammatory and oxidative stress response effects in the lung, and/or by acting as inducing factors for susceptible individuals (Paulin and Hansel Citation2016; Upadhyay et al. Citation2014; Ganguly et al. Citation2011; Ovrevik et al. Citation2015). However, the mechanisms causing exacerbation of the response in pre-disposed individuals following exposure to CNP compare to healthy individuals are still not well elucidated.
Thus, in this study, we aimed at investigating the inflammatory and oxidative stress response of normal (PBEC-ALI) versus chronic bronchitis-like mucosa (PBEC-ALI/CB) models using physiologically relevant in vitro (ALI) models of PBEC, following exposure to aerosolized CNP. Printex 90 (Degussa, Frankfurt, Germany) were used as the representative of CNP in this study. Printex 90 is a well-characterized carbonaceous core particle that has been used extensively as a benchmark and as a model for diesel emission particles without adhered chemicals and metals (Jackson et al. Citation2012). Moreover, these CNP (Printex 90) has high volume of industrial application (Kyjovska et al. Citation2015). The Danish Working Environment Authority, 2007 have shown occupational exposures limit to carbon black of all particle size is 3.5 mg/m3 for an 8-h working day (Kyjovska et al. Citation2015). Printex 90 is considered as a low-toxicity insoluble material, but on the other hand, it is a potent generator of reactive oxygen species (Jacobsen et al. Citation2009) and it induces oxidative stress generated DNA damage. Several studies with human and rat lung epithelial cells have reported that pure CNP induced adverse endpoints like the upregulation of pro-inflammatory cytokines, apoptosis, and proliferation (Blanchet et al. Citation2004; Sydlik et al. Citation2006). Moreover, it is already evident that pulmonary exposure to carbon black by instillation or inhalation induces an inflammatory response in rats and mice (Jacobsen et al. Citation2009; Sager et al. Citation2013; Hougaard et al. Citation2010). Printex 90 CNP is one of the most studied materials in particle toxicology, due to its wide industrial application as well chance to exposure through combustion derived carbon black particles in the environment. However, the dose and time dependent-response in normal versus susceptible group following exposure to CNP has not been studied in detail. Therefore, we compared at two different time-points the pro-inflammatory and oxidative stress response between normal mucosa (PBEC-ALI) versus chronic bronchitis like-mucosa (PBEC-ALI/CB) models, which is comparable to healthy versus predisposed individuals to identify the potentially susceptibility attributes (preexisting complications) that may lead to increased risk of CNP mediated health effects.
Methods
Primary bronchial epithelial cells (PBEC)
In the current study, the bronchial mucosa model was developed separately using PBEC from three different (N = 3) donors cultured at ALI. The cells used in this study are well characterized and have been used in several other studies (von Scheele, Larsson, and Palmberg Citation2010; Ji et al. Citation2017, Citation2018; Strandberg, Palmberg, and Larsson Citation2008). The PBEC were harvested from healthy bronchial tissues obtained from donors in connection with lobectomy following their informed and written consent. All procedures performed in this study were in accordance with the approval of the Ethics Committee of Karolinska Institutet, Stockholm.
Development of air Liquid-Interface (ALI) models using PBEC: normal models (PBEC-ALI) and IL-13 induced chronic bronchitis models (PBEC-ALI/CB)
The air-lifted PBEC models were developed as previously described (Ji et al. Citation2017). Briefly, the PBEC were seeded (1 × 105 cell/cm2) and cultured on transwell inserts (0.4 µm pore size, Falcon™, USA) in twelve-well plates under standard conditions (5% CO2 at 37 °C). Transwell inserts were pre-coated with coating buffer (fibronectin (1 mg/ml, Gibco, Life Technologies, Paisley, UK), bovine serum albumin fraction V (BSA; 1 mg/ml, Sigma, Germany), vitrogen 100 collagen (3.1 mg/ml, Cohesion Technologies, Palo Alto, CA)). One ml PneumaCult™-Ex expand medium (Stemcell Technologies, Cambridge, UK) supplemented with 96 µg/ml hydrocortisone (Stemcell technologies, Cambridge, UK) and penicillin streptomycin antibiotics (PEST, 1%, BioWhittaker, Lonza, Basel, Switzerland) was added to the basal and apical chamber of the insert. Cells were maintained in the above-mentioned condition for about 7–10 days and expand medium was replaced every second day. At confluence (95%), the models were air-lifted by aspirating all the PneumaCult™-Ex expand medium and adding 1 ml PneumaCult™-ALI maintenance medium (Stemcell Technologies, Cambridge, UK) supplemented with 96 µg/ml hydrocortisone, 2 mg/ml heparin (Stemcell technologies, Cambridge, UK), and 1% PEST to the basal chamber only. Maintenance medium was changed every second day. After 3 weeks of airlifting, the PBEC count reached 1.5 × 106 cells/insert and the cells were observed in a well-differentiated state including ciliated cells and mucus-producing cells.
To develop chronic bronchitis-like mucosa, 1 ng/ml IL-13 was added to the basal medium of each inserts from first day of PBEC in ALI condition. The details cellular differentiation (club cells, goblet cells, basal cells, ciliated cells, etc.) of the PBEC-ALI and PBEC-ALI/CB model development have been described previously (Ji et al. Citation2017).
Exposure of PBEC-ALI and PBEC-ALI/CB models to CNP
In order to mimic the in vivo exposure situation of the lung, both PBEC-ALI and PBEC-ALI/CB models were exposed to clean air (sham) or to aerosolized CNP (exposed) using the XposeALI® (Inhalation Sciences, Sweden) exposure system as previously described (Ji et al. Citation2017). In brief, during each exposure, 3 inserts were placed inside the exposure module. Exposure to aerosolized CNP was performed for 3 different durations (1, 2, and 3 min) to achieve low, medium and high CNP concentrations. The inside of the aerosol holding chamber was covered with wet filter papers, which maintained the humidity in order to maintain the cell viability. Additionally, in the exposure module, the inserts including bronchial models were always in contact with the basal medium during the exposures to avoid any physiological stress to the cells. Compressed air of 100 bars was used to aerosolize CNP into the 300 ml holding chamber. The CNP aerosol was pulled from the holding chamber with a constant flow rate (120 ml/min) and diverted into triplicate branch exposures at a flow rate of 10 ml/min per branch. Supplementary Figures 1 and 2 represent positive control experiments using lipopolysaccharide (LPS; 100 ng/ml) and hydrogen peroxide (H2O2; 50 µm) exposure to PBEC-ALI and PBEC-ALI/CB models compared to sham exposed and incubator control. Supplementary Figure 3 shows the transepithelial electrical resistance (TEER) as a measure of transepithelial permeability following low, medium and high CNP exposure compared to sham exposed and incubator control PBEC-ALI and PBEC-ALI/CB models. The corresponding methods are explained in the Supplementary material section.
CNP characterization
Endotoxin-free Printex 90 particles used in this study have been previously characterized (Ganguly et al. Citation2011). Briefly, the primary particle size of Printex 90 is 14 nm, the specific surface area about 300 m2/g, and the organic content was low 1–2% (Matuschek et al. Citation2007). The zeta potential and intensity weighted median dynamic light scattering diameter for Printex 90 particles in a pyrogen-free distilled water suspension at a concentration of 20 μg/50 μL are 33 mV and 0.17 μm, respectively (Ganguly et al. Citation2009, Citation2011).
CNP dose measurements
To measure the CNP exposure doses the ALI models (n = 3 in each time point) were exposed to CNP for 1, 2 and 3 min. Immediately after each exposure, CNP were collected from all 3 inserts by suspending the deposited CNP from each insert in 0.5 mL 99% ethanol. The deposited CNP dose in each insert was quantified by measuring the absorbance using spectrophotometric technique (Cary 60 UV-Vis, Agilent Technologies, Palo Alto, CA, United States). The absorbance was converted to CNP concentration using a standard calibration curve. Finally, the actual exposure dose of CNP in each insert (CI) was calculated using the following formula:
Cell viability assay
The cell viability assay of each model was determined 24 h following exposure to clean air (sham) and all three different concentrations of CNP using two different methods described below.
Alamar blue assay
In order to establish the cell viability, we performed the Alamar Blue Assay. The assay is based on the detection of metabolically active cells through changes of the fluorescence of resazurin (blue, non-fluorescent) to the reduced form resorufin (red, highly fluorescent). The 400 μl of 10% Alamar Blue (Invitrogen™, US) solution was added for 2 h to 8 inserts (sham and 2 inserts for each CNP concentration). Following 2 h of incubation, the absorbance was measured using spectrophotometer at an emission of 570 nm with 600 nm as a reference wavelength. To perform the positive control test 2 inserts were treated with 1% TritonX for 2 h. After 2 h of incubation, TritonX was removed and 400 µl of 10% Alamar Blue was added to detect the cell viability as mentioned above.
Lactate dehydrogenase release (LDH) assay
Cytotoxicity of both clean air exposed (sham) and CNP exposed (three doses) PBEC-ALI as well PBEC-ALI/CB models were determined by measuring the level of released LDH in the apical and basal media (BM). The assay was carried out according to the manufacturer’s instruction (Thermo Fisher Scientific, US). Cytotoxicity of both sham and CNP exposed lung mucosa models of both types were compared with cytotoxicity of positive control (manufacturer provided) samples.
ELISA
In order to assess the concentration of inflammatory and tissue injury markers, CXCL-8 and MMP-9 were measured in BM from both sham and CNP exposed models using the in-house ELISA method described previously (Ji et al. Citation2014; Ji et al. Citation2016). Commercially available antibody pairs MAB208 and MAF208 (R&D SYSTEMS®, UK) were used to measure CXCL-8. The detection limit was 12.5 pg/ml for CXCL-8. Concentrations of MMP-9 were measured using DouSet MMP-9 ELISA Kit (R&D SYSTEMS, UK). The detection limit of the MMP-9 was 31.2 pg/ml.
Quantitative real time polymerase chain reaction (qRT-PCR)
Transcript expression of genes involved in oxidative stress, pro-inflammation tissue injury/repair, and cell-specific markers were assayed using the qRT-PCR technique. The list of genes assessed, and corresponding primer pairs are provided in . Total RNA was isolated from cells (PBEC-ALI and PBEC-ALI/CB) following 6 h and 24 h exposure (sham and CNP at three different doses) using the RNeasy Mini Kit (Qiagen; n = 6) as described previously (Dwivedi et al. Citation2018). The concentration of RNA was measured using the Nanodrop (ND1000 Technology). One μg mRNA was reverse transcribed to generate complementary DNA (cDNA) using the high capacity RNA to cDNA kit (Life Technologies, Paisley, UK) using and a thermal cycler (MycyclerTM, Biorad). The qRT-PCR was performed using the AB 7500 System. The 20 μl qRT-PCR reaction mix consisted of 10 μl Fast SYBR® Green Master Mix (Life Technologies, Paisley, UK), 200 nmol of each primer, 5 ng cDNA, and nuclease-free water. Beta actin (ACTB) was used as the reference control. Expression of each target gene was quantified as a fold change following normalization with ACTB and sham in PBEC-ALI. The results were calculated as 2−ΔCt (ΔCt = Ct (gene of interest) − Ct (beta actin)).
Table 1. Primer pairs used for Quantitative Real-Time PCR (qRT-PCR).
Experimental strategy and sample collection
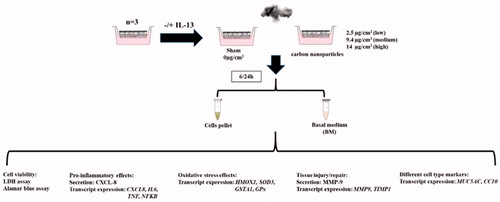
The experimental strategy is shown in . PBEC-ALI and PBEC-ALI/CB models were developed from same donor in parallel and exposed for 1, 2, and 3 min which corresponded to low (2.5 μg/cm2), medium (9.4 μg/cm2), and high (14 μg/cm2) doses of CNP. PBEC-ALI and PBEC-ALI/CB models from 3 different donors (N = 3) were developed separately and corresponding exposure to clean air (sham) or to CNP were performed using 5 technical replicates (n = 5 inserts form each donor). Both PBEC-ALI and PBEC-ALI/CB models are well-differentiated tissue-like models, which contained multiple layers of cells including different cell types of unique distribution. Hence, in this study, every model can be recognized as a unique in vivo-like in vitro model with its own distribution of different cell types. Assessment of transcript and protein expression of pro-inflammatory, oxidative stress and tissue injury/repair markers were performed following 6 h and 24 h post-CNP exposure. Effects of CNP exposure at all 3 concentrations were compared to the corresponding sham-exposed samples within each group (normal and CB). Additionally, comparisons were also performed between normal and chronic bronchitis-like mucosa (normal versus CB). BM was collected immediately at the end of each incubation period (6 h and 24 h) following exposure. Inserts with cells were stored at -80 °C after collection of the BM and used for RNA extraction. Five lung mucosa models (‘technical replicates’: n = 5) were collected from each of the three exposure concentrations and sampling times, resulting in a total of 90 samples (3 donors × 3 concentrations × 5 replicates × 2 time points) of BM and inserts with cells. Out of five, 2 inserts from each of the three exposure concentrations and sampling times were used for Alamar blue assay and rest 3 inserts with cells were used for RNA extraction. LDH assay and secreted protein concentration were measured from BM of 3 inserts of each donor after exposure to three exposure concentrations and sampling times. Additionally, in order to measure the actual deposited CNP on the inserts, 3 extra inserts per dose were exposed to CNP under identical exposure duration and used for dose measurements.
Figure 1. The flow chart of experimental strategy.
Statistics
The results are expressed as median and interquartile ranges (25th–75th percentiles) and normalized to sham exposure in PBEC-ALI. However, all statistics were performed to its own sham control. Within each group (PBEC-ALI or PBEC-ALI/CB models), the comparisons between sham and different CNP exposure concentrations were assessed by Friedman test and followed by Wilcoxon signed-rank t-test as a post hoc test. The comparisons between PBEC-ALI and PBEC-ALI/CB models were performed by Wilcoxon signed-rank t-test both regarding sham exposure and the different concentrations of CNP. A p-value <0.05 was considered as significant. All the data were analyzed using the STATISTICA9 software (StatSoft, Inc. Uppsala, Sweden).
Results
CNP dose calculation
From spectrophotometric analysis the final calculated dose of CNP following 1, 2, and 3 min of exposure were 2.5, 9.4, and 14 μg/cm2, respectively, corresponding to low, medium, and high CNP dose in our experimental setup.
Effects of CNP on cell viability
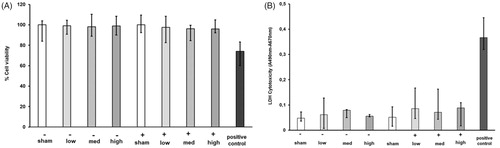
Alamar blue and LDH assay did not exhibit any cytotoxic effects after sham and CNP exposure at all tested doses (; Supplementary Figure 4). None of the CNP doses used in this study were therefore cytotoxic. Additionally, sham exposure did not cause any inflammatory and oxidative stress response, as well as transepithelial permeability, remained unaltered compared to incubator control (Supplementary Figures 1–3).
Figure 2. Cell viability and Cytotoxicity assays to analyze the effect of carbon nanoparticles (CNP) exposure in air-liquid interface-cultured models using Alamar Blue (A) and Lactate Dehydrogenase Assay (LDH: B). (A) The percentage of cell viability was assayed using colorimetric Alamar blue assay of PBEC-ALI (−) and PBEC-ALI/CB (+) 24 h after exposure to Sham (clean air) or aerosolized CNP at three doses compared to a positive control (1% Titron X). Data presented as median and 25th–75th percentiles (n = 6). (B) The colorimetric LDH assay to measure the cytotoxic effect in PBEC-ALI (−) and PBEC-ALI/CB (+) 24 h post-exposure to sham (clean air) and aerosolized CNP at three concentrations compare to a positive control (manufacturer provided). Data presented as median and 25th–75th percentiles (n = 9).
CNP exposure induced inflammatory effect on PBEC-ALI versus PBEC-ALI/CB models
Pro-inflammatory effects
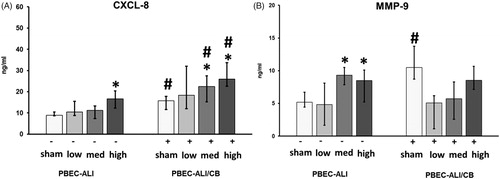
In PBEC-ALI, CXCL-8 secretion was increased at 24 h post-exposure to CNP (high dose) compared to sham exposure (, left panel). In PBEC-ALI/CB, 24 h post-exposure to the two highest doses of CNP resulted in increased CXCL-8 secretion compared to sham exposure (, right panel). Additionally, CXCL-8 secretion was increased after exposure to sham and the two highest doses of CNP in PBEC-ALI/CB model compared to corresponding PBEC-ALI model (. After 6 h following CNP exposure at all doses, CXCL-8 levels remained unchanged in both PBEC-ALI and PBEC-ALI/CB (data not shown).
Figure 3. Carbon nanoparticle (CNP) induced protein secretion in basal media (BM) of normal (PBEC-ALI: −) and interleukin-13 (IL-13)-induced chronic bronchitis-like mucosa (PBEC-ALI/CB: +). Concentration of CXCL-8 (A) and MMP-9 (B) was measured using ELISA from BM, collected from both PBEC-ALI (−) and PBEC-ALI/CB (+) after incubation for 24 h following exposure to sham (clean air), low (2.5 μg/cm2), medium (9.4 μg/cm2), and high (14 μg/cm2) doses of CNP. Data presented as median and 25th–75th percentiles (n = 9). *: p < 0.05 within-group comparison of effects; effects of CNP exposure at all three concentrations vs corresponding sham (PBEC-ALI or PBEC-ALI/CB), #: p < 0.05 between-group comparisons (PBEC-ALI/CB vs PBEC-ALI) between sham or corresponding dose of CNP.
Transcript expression of pro-inflammatory markers (CXCL8, IL6, TNFα, and NFKB) were assayed from both PBEC-ALI and PBEC-ALI/CB model at 6 h and 24 h post-exposure to sham and aerosolized CNP at 3 different doses. Compare with sham exposure, transcript expression of CXCL8 was increased in PBEC-ALI following 6 h post-CNP exposure (medium dose; , left panel). However, IL6, TNFα, and NFKB remained unchanged 6 h after exposure to all three doses in PBEC-ALI (, left panel). In contrast, exposure to aerosolized CNP resulted in increased transcript expression of CXCL8, IL6, and TNFα after 6 h in PBEC-ALI/CB (highest dose vs its own sham; , right panel). Interestingly, 24 h following exposure to CNP in the PBEC-ALI, the mRNA expressions of IL6 and TNFα were increased (low and medium doses vs sham; , left panel) while expressions of CXCL8 and NFKB remained unaltered after exposure to any CNP dose (, left panel). Similarly, 24 h following exposure in the PBEC-ALI/CB, transcript expression of CXCL8 and NFKB were enhanced by CNP exposure compare with its own sham exposure (high and low dose vs sham; , right panel).
Figure 4. Transcript expression of pro-inflammatory markers in normal (PBEC-ALI: −) and interleukin-13 (IL-13) induced chronic bronchitis-like mucosa models (PBEC-ALI/CB: +) after 6 h (A–D) and 24 h (E–H) incubation following exposure to carbon nanoparticles (CNP). Fold change of CXCL8 (A and E), IL6 (B and F), TNFα (C and G), and NFKB (D and H) 6 h and 24 h post-exposure to sham (clean air), low (2.5 μg/cm2), medium (9.4 μg/cm2), and high (14 μg/cm2) CNP doses. Data presented as median and 25th–75th percentiles (n = 9) and normalized to sham exposure of PBEC-ALI. *: p < 0.05 within-group comparison of effects; effects of CNP exposure at all three concentrations vs corresponding sham (PBEC-ALI or PBEC-ALI/CB), #: p < 0.05 between-group comparisons (PBEC-ALI/CB vs PBEC-ALI) between sham or corresponding dose of CNP.
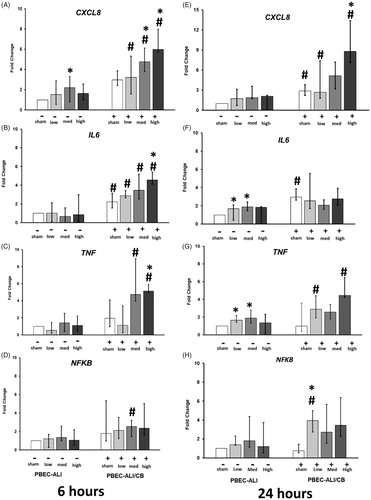
After both 6 h and 24 h following CNP exposure, CXCL8 expression increased more in PBEC-ALI/CB compared to PBEC-ALI (sham, medium, and high doses, ). Also, IL6 mRNA expression was higher in PBEC-ALI/CB compared to PBEC-ALI 6 h after exposure to both sham and CNP at all 3 doses (. Besides, CNP exposure induced a higher expression of TNFα in PBEC-ALI/CB than in PBEC-ALI (6 h, medium, and high dose; 24 h, low and high dose; ).
Oxidative stress
Transcript expression analysis of oxidative stress genes (HMOX1, SOD3, GSTA1, and GPx) was performed from both PBEC-ALI and PBEC-ALI/CB at 6 h and 24 h post-exposure to sham and aerosolized CNP at 3 different doses. In the PBEC-ALI/CB, HMOX1 transcript expression was increased 6 h post CNP exposure (low and high doses vs its own sham; , right panel) while the other oxidative stress genes (SOD3, GSTA1, and GPx) remained unchanged 6 h after CNP exposure at all tested doses in both normal and chronic bronchitis-like models (). After 24 h, expression of HMOX1 was induced by CNP exposure in PBEC-ALI (low dose vs its own sham; , left panel). Additionally, in the PBEC-ALI, transcript expressions of SOD3 and GSTA1 were increased 24 h post-exposure to all three doses of CNP compare to its own sham exposure (, left panel).
Figure 5. Transcript expression of oxidative stress markers in normal (PBEC-ALI: −) and interleukin-13 (IL-13) induced chronic bronchitis-like mucosa models (PBEC-ALI/CB: +) after 6 h (A–D) and 24 h (E–H) incubation following exposure to carbon nanoparticles (CNP). Fold change of HMOX1 (A and E), SOD3 (B and F), GSTA1 (C and G), and GPx (D and H) 6 h and 24 h post-exposure to sham (clean air), low (2.5 μg/cm2), med (9.4 μg/cm2), and high (14 μg/cm2) CNP doses. Data presented as median and 25th–75th percentiles (n = 9) and normalized to sham exposure of PBEC-ALI. *: p < 0.05 within-group comparison of effects; effects of CNP exposure at all three concentrations vs corresponding sham (PBEC-ALI or PBEC-ALI/CB), #: p < 0.05 between-group comparisons (PBEC-ALI/CB vs PBEC-ALI) between sham or corresponding dose of CNP.
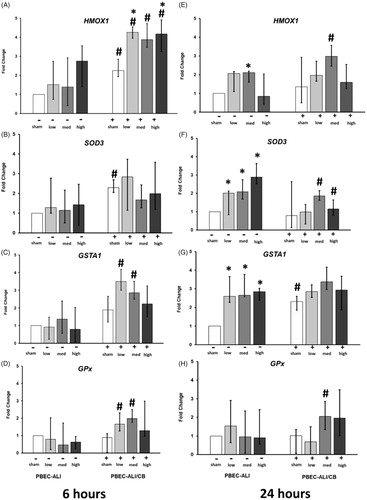
The mRNA expression of HMOX1 was higher in PBEC-ALI/CB compared to PBEC-ALI after exposure to both sham and CNP at all 3 doses (. As for SOD3, an enhanced mRNA expression in PBEC-ALI/CB compared to PBEC-ALI could only be found 6 h post sham exposure () while a decreased expression 24 h post-CNP exposure (medium and high doses; ) where observed in PBEC-ALI/CB compared with PBEC-ALI. Both GSTA1 and GPx were increased 6 h after CNP exposure at the two lowest doses in PBEC-ALI/CB compared with PBEC-ALI (). Furthermore, significant increase of reactive oxygen species (ROS) was measured in PBEC-ALI/CB following 3 h of exposure to high CNP dose (14 μg/cm2) using the flow cytometer (LSRFortessa™, BD Bioscienc, Supplementary Figure 5).
Tissue injury/repair
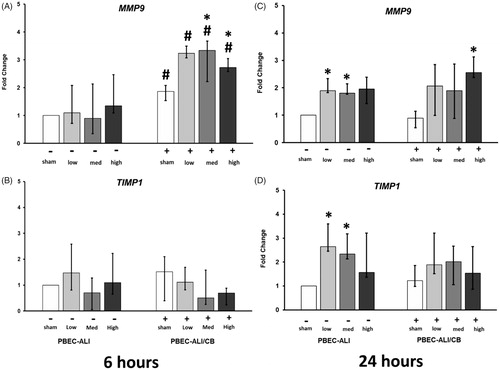
In PBEC-ALI, secretion of MMP-9 in BM was increased at 24 h following exposure to the two highest doses of CNP (, left panel) while 6 h after exposure to CNP, MMP-9 level in BM remained unchanged in normal model (data not shown). In PBEC-ALI/CB, exposure to sham induced a higher release of MMP-9 levels compared to sham exposed PBEC-ALI 24 h post-exposure (. CNP exposure did not result in any altered MMP-9 levels in the BM of PBEC-ALI/CB following CNP exposure at either 6 h or 24 h.
Six hours post-exposure to the two highest doses of CNP the expression of MMP9 were upregulated in PBEC-ALI/CB while corresponding TIMP1 expression remained unchanged (, right panel). After 24 h, the expression of both MMP9 and TIMP1 increased after exposure to the two lowest doses of CNP in PBEC-ALI (, left panel). At 6 h post-exposure to both sham and all three concentrations of CNP exposure, there was a higher MMP9 expression in PBEC-ALI/CB compared to PBEC-ALI (.
Figure 6. Transcript expression of tissue injure/repair markers in normal (PBEC-ALI: −) and Interleukin-13 (IL-13) induced chronic bronchitis-like mucosa (PBEC-ALI/CB: +) after 6 h (A and B) and 24 h (C and D) incubation following exposure to Carbon nanoparticles (CNP). Fold change of MMP9 (A and C), and TIMP1 (B and D) 6 h and 24 h post-exposure to sham (clean air), low (2.5 μg/cm2), med (9.4 μg/cm2), and high (14 μg/cm2) CNP doses. Data presented as median and 25th–75th percentiles (n = 9) and normalized to sham exposure of PBEC-ALI). *p < 0.05 within-group comparison of effects; effects of CNP exposure at all three concentrations vs corresponding sham (PBEC-ALI or PBEC-ALI/CB), #p < 0.05 between-group comparisons (PBEC-ALI/CB vs PBEC-ALI) between sham or corresponding dose of CNP.
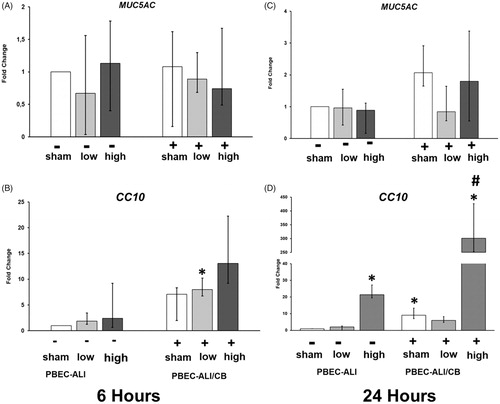
Club cell and mucus-producing cell markers
To assess the plausible interaction of CNP (low and high doses) with mucus-producing cells and club cells in PBEC-ALI and PBEC-ALI/CB models, transcript expression of mucin 5AC, oligomeric mucus/gel-forming (MUC5AC) and secretoglobin family 1 A member 1 (SCGB1A1, also known as CC10) were measured. Expression of MUC5AC remained unaltered (), whereas CC10 levels were increased 24 h after in the high CNP dose exposed PBEC-ALI (>20 fold,) and PBEC-ALI/CB (>300 fold) models (.
Figure 7. mRNA expression of Mucin 5AC (MUC5AC) and Club cell 10 kD protein (CC10: C and D) in normal (PBEC-ALI: −) and interleukin-13 (IL-13) induced chronic bronchitis-like mucosa models (PBEC-ALI/CB: +) after 6 h and 24 h (incubation following exposure to sham (clean air), and carbon nanoparticles (CNP). Fold change of MUC5AC (A and B), CC10 (C and D) 6 h and 24 h post-exposure to sham (clean air), low (2.5 μg/cm2), medium (9.4 μg/cm2), and high (14 μg/cm2) CNP doses. Data presented as median and 25th–75th percentiles (n = 9) and normalized to sham exposure of PBEC-ALI). *p < 0.05 within-group comparison of effects; effects of CNP exposure at all three concentrations vs corresponding sham (PBEC-ALI or PBEC-ALI/CB), #p < 0.05 between-group comparisons (PBEC-ALI/CB vs PBEC-ALI) between sham or corresponding dose of CNP.
Discussion
This study analyzed the pro-inflammatory, oxidative stress and tissue injury/repair responses of PBEC-ALI and PBEC-ALI/CB models at 6 h and 24 h post-exposure to aerosolized CNP. The results showed an earlier onset of CNP-mediated detrimental effects in PBEC-ALI/CB compared with PBEC-ALI, as indicated by significant up-regulation of pro-inflammatory, oxidative stress and tissue injury markers already at 6 h and still to some extent present at 24 h. However, pro-inflammatory, oxidative stress, and tissue injury responses started at 24 h post-CNP exposure in PBEC-ALI models. Furthermore, at both time points, exposure to sham or CNP induced a higher expression of pro-inflammatory-, oxidative stress- and tissue injury markers in PBEC-ALI/CB compare to PBEC-ALI. Finally, exposure to CNP in PBEC-ALI induced both tissue injury and repair markers (MMP9 and TIMP1) while in PBEC-ALI/CB only the tissue injury marker MMP9 increased with an earlier onset and significantly more compared with normal mucosa. Previously (Ji et al. Citation2017), we demonstrated the differentiation of ciliated cells, basal cells, and goblet cells in the bronchial mucosa models cultured at ALI using PBEC. Light/confocal microscopy, scanning and transmission electron microscopy, cell-specific gene and protein expression were performed for this purpose (Ji et al. Citation2017). Presence of the aforementioned cell types are consistent with in vivo situation where human bronchial epithelium consists of 50–70% ciliated, up to 30% basal cells, up to 25% goblet cells and 11% club (clara) cells (Schamberger et al. Citation2015). Further, PAS stained cross sections of the chronic-bronchitis-like model exhibited more mucus-producing cells along with other cell types compared to the healthy bronchial mucosa model (Ji et al. Citation2017), again showing the physiological relevance of the PBEC-ALI/CB model to in vivo situation.
In PBEC-ALI/CB, a higher secretion and expression of pro-inflammatory mediators already at 6 h were observed compared with PBEC-ALI. In PBEC-ALI, an increased release of CXCL-8 at protein level was observed only after 24 h exposure to high CNP dose compared to sham. Whereas in the PBEC-ALI/CB, an increased CXCL-8 secretion in the BM was detected at 24 h post the two highest CNP exposures compared to sham exposure. These data illustrated an early pro-inflammatory effect following CNP exposure in the PBEC-ALI/CB compared to PBEC-ALI. The increased CXCL-8 secretion and expression already after sham exposure in PBEC-ALI/CB compared to PBEC-ALI models indicated an ongoing preexisting condition similar to the in vivo situation observed in COPD patients suffering from chronic bronchitis (Ji et al. Citation2014). These findings were also observed in our previous study (Ji et al. Citation2017), where exposure to palladium nanoparticles resulted in higher releases of CXCL-8 in PBEC-ALI/CB compare to PBEC-ALI. Loxham et al. (Citation2015) have compared the release of CXCL-8 from healthy and severely asthmatic donor differentiated PBEC cultures at ALI after 24 h exposure to varying concentrations of coarse fine, or ultrafine underground railway PM. In line with our previous and present results, they reported higher induction of CXCL8 release from severely asthmatic donor differentiated PBEC cultures at ALI after exposure to 5.6 μg/cm2 coarse underground PM and 2.2 μg/cm2 ultrafine PM compared to models with cells from healthy donors (Loxham et al. Citation2015). We also found increased IL6 and TNFα expression at 6 h following CNP exposure in the PBEC-ALI/CB and significantly higher expression in PBEC-ALI/CB compared to PBEC-ALI. In PBEC-ALI, CNP exposure, increased IL6 and TNFα expressions at 24 h indicating a later onset of the inflammatory response in normal compared to chronic bronchitis-like mucosa. Taken together, these findings may indicate an earlier and more intense inflammatory reaction in patients with preexisting complication like chronic bronchitis.
Moreover, the transcription levels of oxidative stress markers at two different time points (6 h and 24 h) following exposure to CNP were assessed to determine whether the response patterns were time-dependent and if there was any difference between normal and chronic bronchitis-like models. We found an increased expression of HMOX1 in PBEC-ALI/CB already after 6 h post-CNP exposure and significantly higher expression of HMOX1, GSTA1, and GPx in PBEC-ALI/CB compared to PBEC-ALI. On the other hand, in PBEC-ALI, HMOX1, SOD3, and GSTA1 increased 24 h post-CNP exposure. This altered expression of oxidative stress marker resembles that of the pro-inflammatory reaction with an earlier onset and stronger reaction in PBEC-ALI/CB compared to the PBEC-ALI. Accordingly, Kodavanti (Citation2000) reported that the exposure of bronchitis rat models (SO2 induced bronchitis rat model) to concentrated ambient particles exhibits increased pulmonary injury than the normal healthy rats. In line with this study, other studies (Upadhyay et al. Citation2014; Upadhyay et al. Citation2008) demonstrated that, that exposure of aged spontaneously hypertensive rats (SHRs) to ultrafine carbon particles results in early (24 h post-exposure) detection of increased cardiovascular parameters (heart rate, blood pressure) in association with pulmonary and systemic inflammation compare to adult healthy rats. The authors concluded that aged SHRs which suffers from preexisting disease likely to be more susceptible than healthy controls to CNP-mediated pulmonary impairment in respect to particle-induced physiological and immune responses (Upadhyay et al. Citation2014). Therefore, the findings of these physiologically relevant in vitro models (PBEC-ALI and PBEC-ALI/CB), which are comparable with healthy versus pre-disposed individuals, are consistent with results from in vivo studies.
Additionally, at 6 h post-CNP exposure, MMP9 expression was increased compared to sham and significantly higher in the PBEC-ALI/CB compared to PBEC-ALI. However, TIMP1 expression was not altered. This suggested that the chronic bronchitis-like model could not counteract the increased expression of the matrix-degradation enzymes after exposure to CNP. Besides, no effects on the tissue injury/repair markers (MMP9/TIMP1) were detected in PBEC-ALI at 6 h post CNP exposures. This again indicated an early effect of CNP in the chronic bronchitis-like model. At 24 h post-exposure, both MMP9 and TIMP1 were increased after CNP exposure in PBEC-ALI, which suggested an initiation of matrix degradation protection in normal mucosa. Similarly, increased MMP-9 secretion was detected at 24 h after exposure to CNP in PBEC-ALI. These findings regarding tissue injury/repair markers were again consistent with the pro-inflammatory reaction and oxidative stress response exhibiting early response and more susceptibility in the chronic bronchitis-like mucosa than in normal mucosa.
Interestingly, the 20 fold (PBEC-ALI) to 300 fold (PBEC-ALI/CB) increased CC10 transcript levels following exposure to the high dose CNP may represent a protective phenomenon that warrants detailed mechanistic studies. CC10 has been implicated in anti-inflammatory actions and defects in this gene are associated with a susceptibility to asthma. Low serum CC10 levels were detected among patients with asthma-COPD overlap syndrome (Oh et al. Citation2018). Further, serum CC10 levels were positively correlated with forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), forced expiratory flow at 25–75% of FVC, FEV1/FVC, vital capacity, and diffusing capacity of the lung for carbon monoxide (Oh et al. Citation2018). Serum CC10 levels were negatively correlated with smoking amount (pack-years), bronchodilator response, fractional residual capacity, residual volume, and number of exacerbations per year. FEV1 and serum CC10 levels were also significantly lower in patients with frequent exacerbations (Oh et al. Citation2018). Protective role of CC10 in the development of COPD has been described by Laucho-Contreras et al. (Citation2015). The authors reported reduced immunoreactive CC10 in the airways of among smokers and COPD patients that decreased with increasing COPD severity. Mice lacking CC10 exhibited greater cigarette smoke-induced emphysema, airway remodeling, pulmonary inflammation, alveolar cell apoptosis, airway MUC5AC expression, and more compliant lungs compared to wild type mice (Laucho-Contreras et al. Citation2015).
In the present study, we have illustrated that our physiologically relevant normal (PBEC-ALI) and chronic bronchitis-like models (PBEC-ALI/CB) mimic the in vivo situation regarding inflammatory response after exposure to PM present in air pollution. This may indicate that such models could be used to study important mechanisms involved during development of lung diseases. They might also be a good alternative to study mechanisms behind health effects of air pollution exposures and how this varies between healthy and diseased individuals.
By stimulating our normal mucosa models with IL-13 the number of mucus-producing cells increased, which is a characteristic feature of chronic bronchitis (Ji et al. Citation2017). Also, IL-13 is increased in smokers with chronic bronchitis (Miotto et al. Citation2003). Based on our previous study (Ji et al. Citation2017), we used very low IL-13 concertation to stimulate the mucus hyper-secretion. According to this study, it was shown by PAS stained cross sections that models treated with moderate concertation of IL-13 (1 ng/ml) showed increased numbers of mucus producing cells but different cell types still existed. However, IL-13 can also induce ROS production and oxidative stress via activation of NADPH oxidase in respiratory tract epithelium (Harper et al. Citation2005). This could explain the difference regarding oxidative stress between PBEC-ALI and PBEC-ALI/CB models upon CNP exposure in such a way that the signal pathway was pre-activated by IL-13 treatment. Since chronic bronchitis patients have higher levels of IL-13 in central airways (Miotto et al. Citation2003), and COPD patients showed evidence of increased oxidative stress (Repine, Bast, and Lankhorst Citation1997), we think the model treated with IL-13 rather represents chronic bronchitis conditions. In this study, we performed acute exposure of both normal and chronic bronchitis-like models to three different doses of CNP. However, further studies are needed both on the repeated exposure to CNP as well as combining exposure to both particles and ambient gaseous agents (sulfur dioxide) (Kodavanti Citation2000), which are likely responsible for the development of chronic lung diseases.
Finally, some short notes on the difficult task of linking air pollutant dosimetry of short-term cell culture experiments with in vivo animal exposures as well as with human acute- or chronic exposures in the polluted environment. While much progress can be made in cell culture work in understanding the various mechanisms of action of toxicity in humans, more research will be needed to link the dose metrics between these vastly disparate dosing scenarios (Donaldson et al. Citation2008). Several researchers have shown the mass concentration of black carbon (BC) in the ambient air in different part of the world. For example, Sattar et al. (Citation2014) have shown the concentration of black carbon in the ambient air of urban area in Indonesia is varying between 2.6 and 3.8 μg/m3, which constitute 6.1% of the particulate mass during the dry season. Similarly, Islam et al. (Citation2014) reported that average concentration of BC in PM10 and PM2.5 was 29.1 µg/m³ and 31.6 µg/m3 in Dhaka, Bangladesh.
To conclude, our study indicates an earlier onset and stronger reaction of pro-inflammatory, oxidative stress and tissue injury response in chronic bronchitis-like models compared to normal models following exposure to aerosolized CNP. Further, the chronic bronchitis-like models increased the expression of a matrix degradation enzyme and not the counteract tissue repairing enzyme. This was in contrast to the normal mucosa model, where both matrix degradation and repairing enzymes increased after exposure to CNP. Greater increase of the protective marker CC10 following CNP exposure in chronic-bronchitis-like compared to normal mucosa model is consistent with the overall findings of the study suggesting higher susceptibility in pre-disposed condition. These findings are in agreement with epidemiological observations showing higher susceptibility to air pollution episodes among individuals with chronic bronchitis (Kloog et al. Citation2013; Pope et al. Citation2002; Sager et al. Citation2013). Therefore, our chronic bronchitis-like models using PBEC cultured at ALI conditions serve as physiologically relevant models for pursuing in vitro studies. Exposure experiments with in vitro multi-cellular airway mucosa ALI-models that include different cell types (epithelial cells, macrophages, endothelial cells, and/or dendritic cells) may provide even higher physiological relevance (Klein et al. Citation2013; Upadhyay and Palmberg Citation2018). We also confirmed that a preexisting condition like chronic bronchitis might lead to an increased risk of nanoparticle-mediated healthy effects. This is in consistence with the clinical findings of an increased susceptibility of individuals with chronic lung diseases compared to healthy people to air pollution exposure.
TNAN-2018-OR-0286-File009.docx
Download MS Word (621.5 KB)Acknowledgments
The authors sincerely acknowledge the technical assistance received from Tania A Thimraj and Mizanur Rahman, Institute of Environmental Medicine (IMM), Karolinska Institutet.
Disclosure statement
All authors have read and approved the manuscript. The Coauthors Dr. Per Gerde has stocks in Inhalation Sciences, Sweden. All other authors have no competing financial interests to disclose.
Additional information
Funding
Related Research Data
References
- Andre, E. 2006. “Inhalation of Ultrafine Carbon Particles Triggers Biphasic Pro-Inflammatory Response in the Mouse Lung.” European Respiratory Journal 28(2): 275–285. doi:10.1183/09031936.06.00071205.
- Autengruber, Andrea, Ulrich Sydlik, Matthias Kroker, Tamara Hornstein, Niloofar Ale-Agha, Daniel Stöckmann, Andreas Bilstein, et al. 2014. “Signalling-Dependent Adverse Health Effects of Carbon Nanoparticles Are Prevented by the Compatible Solute Mannosylglycerate (Firoin) in Vitro and in Vivo.” PLoS One 9(11): E111485. doi:10.1371/journal.pone.0111485.
- Axelsson, Malin, Linda Ekerljung, Jonas Eriksson, Stig Hagstad, Eva Rönmark, Jan Lötvall, and Bo Lundbäck. 2016. “Chronic Bronchitis in West Sweden – A Matter of Smoking and Social Class.” European Clinical Respiratory Journal 3: 30319doi:10.3402/ecrj.v3.30319.
- Berube, K., D. Balharry, K. Sexton, L. Koshy, and T. Jones. 2007. “Combustion-Derived Nanoparticles: Mechanisms of Pulmonary Toxicity.” Clinical and Experimental Pharmacology and Physiology 34: 1044–1050. doi:10.1111/j.1440-1681.2007.04733.x.
- Blanchet, Sophie, Kiran Ramgolam, Augustin Baulig, Francelyne Marano, and Armelle Baeza-Squiban. 2004. “Fine Particulate Matter Induces Amphiregulin Secretion by Bronchial Epithelial Cells.” American Journal of Respiratory Cell and Molecular Biology 30(4): 421–427.
- Burrows, B., R. J. Knudson, and M. D. Lebowitz. 1977. “The Relationship of Childhood Respiratory Illness to Adult Obstructive Airway disease.” The American Review of Respiratory Disease 115(5): 751–760. doi:10.1164/arrd.1977.115.5.751.
- Chalupa, David C., Paul E. Morrow, Günter Oberdörster, Mark J. Utell, and Mark W. Frampton. 2004. “Ultrafine Particle Deposition in Subjects with Asthma.” Environmental Health Perspectives 112(8): 879–882. doi:10.1289/ehp.6851.
- Daigle, Christopher C., David C. Chalupa, F Raymond Gibb, Paul E. Morrow, Günter Oberdörster, Mark J. Utell, and Mark W. Frampton. 2003. “Ultrafine Particle Deposition in Humans during Rest and Exercise.” Inhalation Toxicology 15(6): 539–552. doi:10.1080/08958370304468.
- Donaldson, K., P J A. Borm, G. Oberdorster, K E. Pinkerton, V. Stone, and C L. Tran. 2008. “Concordance between in Vitro and in Vivo Dosimetry in the Proinflammatory Effects of Low-Toxicity, Low-Solubility Particles: The Key Role of the Proximal Alveolar Region.” Inhalation Toxicology 20(1): 53–62. doi:10.1080/08958370701758742.
- Dwivedi, Aishwarya M., Swapna Upadhyay, Gunnar Johanson, Lena Ernstgård, and Lena Palmberg. 2018. “Inflammatory Effects of Acrolein, Crotonaldehyde and Hexanal Vapors on Human Primary Bronchial Epithelial Cells Cultured at Air-Liquid Interface.” Toxicology in Vitro 46: 219–228. doi:10.1016/j.tiv.2017.09.016.
- EPA. 2018. “Criteria Air Pollutants”. https://www.epa.gov/criteria-air-pollutants.
- EU-OSHA. 2009. “EU-OSHA Strategy 2009–13”. https://osha.europa.eu/en/publications/work_programmes/strategy2009-2013/view.
- Ganguly, Koustav, Swapna Upadhyay, Martin Irmler, Shinji Takenaka, Katrin Pukelsheim, Johannes Beckers, Martin Hrabé De Angelis, et al. 2011. “Impaired Resolution of Inflammatory Response in the Lungs of JF1/Msf Mice following Carbon Nanoparticle Instillation.” Respiratory Research 12: 94. doi:10.1186/1465-9921-12-94.
- Ganguly, Koustav, Swapna Upadhyay, Martin Irmler, Shinji Takenaka, Katrin Pukelsheim, Johannes Beckers, Eckard Hamelmann, Holger Schulz, and Tobias Stoeger. 2009. “Pathway Focused Protein Profiling Indicates Differential Function for IL-1B, -18 and VEGF during Initiation and Resolution of Lung Inflammation Evoked by Carbon Nanoparticle Exposure in mice.” Particle and Fibre Toxicology 6: 31. doi:10.1186/1743-8977-6-31.
- Ganguly, Koustav, Leema George, Tania A. Thimraj, Swapna Upadhyay, Holger Schulz, and Tobias Stoeger. 2017. “Skewed Alveolar Macrophage Polarization Signature in Mice with Impaired Pulmonary Function following Carbon Nanoparticle Exposure.” American Journal of Respiratory and Critical Care Medicine 195: A6839
- Garshick, E. 2014. “Effects of Short- and Long-Term Exposures to Ambient Air Pollution on COPD.” The European Respiratory Journal 44(3): 558–561. doi:10.1183/09031936.00108814.
- Harper, Richart W., Changhong Xu, Jason P. Eiserich, Yin Chen, Cheng-Yuan Kao, Philip Thai, Henny Setiadi, and Reen Wu. 2005. “Differential Regulation of Dual NADPH Oxidases/Peroxidases, Duox1 and Duox2, by Th1 and Th2 Cytokines in Respiratory Tract Epithelium.” FEBS Letters 579(21): 4911–4917. doi:10.1016/j.febslet.2005.08.002.
- Hoek, Gerard, Ranjini M. Krishnan, Rob Beelen, Annette Peters, Bart Ostro, Bert Brunekreef, and Joel D. Kaufman. 2013. “Long-Term Air Pollution Exposure and Cardio- Respiratory Mortality: A Review.” Environmental Health 12(1): 43. doi:10.1186/1476-069X-12-43.
- Hougaard, Karin S., Petra Jackson, Keld A. Jensen, Jens J. Sloth, Katrin Löschner, Erik H. Larsen, Renie K. Birkedal, et al. 2010. “Effects of Prenatal Exposure to Surface-Coated Nanosized Titanium Dioxide (UV-Titan). A Study in Mice.” Particle and Fibre Toxicology 7: 16. doi:10.1186/1743-8977-7-16.
- Inoue, K., and H. Takano. 2011. “Particulate Matter-Induced Health Effects: Who Is Susceptible?” Environmental Health Perspectives 119(7): A285. doi:10.1289/ehp.1103846.
- Islam, J., A. K. M. Rahman, A. Sarkar, K. Ahmed, and B. Begum, 2014. “Particulate Matter and Black Carbon Concentration in Ambient Air of an Urban-Traffic Influenced Site at Farm Gate, Dhaka, Bangladesh.” Jagannath University Journal of Science 3(I): 87–96.
- Jackson, Petra, Karin Sørig Hougaard, Anne Mette Z. Boisen, Nicklas Raun Jacobsen, Keld Alstrup Jensen, Peter Møller, Gunnar Brunborg, Kristine Bjerve Gutzkow, et al. 2012. “Pulmonary Exposure to Carbon Black by Inhalation or Instillation in Pregnant Mice: Effects on Liver DNA Strand Breaks in Dams and Offspring.” Nanotoxicology 6(5): 486–500. doi:10.3109/17435390.2011.587902.
- Jacobsen, Nicklas, Peter Møller, Keld Jensen, Ulla Vogel, Ole Ladefoged, Steffen Loft, and Håkan Wallin. 2009. “Lung Inflammation and Genotoxicity following Pulmonary Exposure to Nanoparticles in ApoE-/- Mice.” Particle and Fibre Toxicology 6(1): 2. doi:10.1186/1743-8977-6-2.
- Jacobsen, Nicklas Raun, Paul A. White, John Gingerich, Peter Møller, Anne Thoustrup Saber, George R. Douglas, Ulla Vogel, and Håkan Wallin. 2011. “Mutation Spectrum in FE1-MUTA(TM) Mouse Lung Epithelial Cells Exposed to Nanoparticulate Carbon Black.” Environmental and Molecular Mutagenesis 52(4): 331–337. doi:10.1002/em.20629.
- Jeffery, P. K. 1991. “Morphology of the Airway Wall in Asthma and in Chronic Obstructive Pulmonary Disease.” American Review of Respiratory Disease 143(5_pt_1): 1152–1158. discussion 1161. doi:10.1164/ajrccm/143.5_Pt_1.1152.
- Ji, Jie, Ida von Schéele, Jan Bergström, Bo Billing, Barbro Dahlén, Ann-Sofie Lantz, Kjell Larsson, and Lena Palmberg. 2014. “Compartment Differences of Inflammatory Activity in Chronic Obstructive Pulmonary Disease.” Respiratory Research 15: 104. doi:10.1186/s12931-014-0104-3.
- Ji, Jie, Ida von Schéele, Bo Billing, Barbro Dahlén, Ann-Sofie Lantz, Kjell Larsson, and Lena Palmberg. 2016. “Effects of Budesonide on Toll-like Receptor Expression in Alveolar Macrophages from Smokers with and without COPD.” International Journal of Chronic Obstructive Pulmonary Disease 11: 1035–1043. doi:10.2147/COPD.S102668.
- Ji, Jie, Anna Hedelin, Maria Malmlöf, Vadim Kessler, Gulaim Seisenbaeva, Per Gerde, and Lena Palmberg. 2017. “Development of Combining of Human Bronchial Mucosa Models with XposeALI® for Exposure of Air Pollution Nanoparticles.” PLoS One 12(1): e0170428. doi:10.1371/journal.pone.0170428.
- Ji, Jie, Swapna Upadhyay, Xiaomiao Xiong, Maria Malmlöf, Thomas Sandström, Per Gerde, and Lena Palmberg. 2018. “Multi-Cellular Human Bronchial Models Exposed to Diesel Exhaust Particles: Assessment of Inflammation, Oxidative Stress and Macrophage Polarization.” Particle and Fibre Toxicology 15(1): 19
- Klein, Sebastian G., Tommaso Serchi, Lucien Hoffmann, Brunhilde Blömeke, and Arno C. Gutleb. 2013. “An Improved 3D Tetraculture System Mimicking the Cellular Organisation at the Alveolar Barrier to Study the Potential Toxic Effects of Particles on the Lung.” Particle and Fibre Toxicology 10(1): 31. doi:10.1186/1743-8977-10-31.
- Kloog, Itai, Bill Ridgway, Petros Koutrakis, Brent A. Coull, and Joel D. Schwartz. 2013. “Long- and short-term exposure to PM2.5 and mortality: using novel exposure models.” Epidemiology (Cambridge, Mass.) 24(4): 555–561. doi:10.1097/EDE.0b013e318294beaa.
- Kodavanti, U. P. 2000. “Variable Pulmonary Responses from Exposure to Concentrated Ambient Air Particles in a Rat Model of Bronchitis.” Toxicological Sciences 54(2): 441–451. doi:10.1093/toxsci/54.2.441.
- Kyjovska, Zdenka O., Nicklas R. Jacobsen, Anne T. Saber, Stefan Bengtson, Petra Jackson, Håkan Wallin, and Ulla Vogel. 2015. “ArticleDNA Damage following Pulmonary Exposure byInstillation to Low Doses of Carbon Black (Printex 90)N Anoparticles in MiceZdenka.” Environmental and Molecular Mutagenesis 56(1): 41–49. doi:10.1002/em.21888.
- Laucho-Contreras, Maria E., Francesca Polverino, Kushagra Gupta, Katherine L. Taylor, Emer Kelly, Victor Pinto-Plata, Miguel Divo, et al. 2015. “Protective Role for Club Cell Secretory Protein-16 (CC16) in the Development of COPD.” The European Respiratory Journal 45(6): 1544–1556. doi:10.1183/09031936.00134214.
- Layachi, Skander, Françoise Rogerieux, Franck Robidel, Ghislaine Lacroix, and Sam Bayat. 2013. “Correction: Effect of Combined Nitrogen Dioxide and Carbon Nanoparticle Exposure on Lung Function during Ovalbumin Sensitization in Brown Norway Rat.” PLoS One 7(9): e45687. doi:10.1371/annotation/d271d9c1-5588-4b43-85c3-d3de58ab61a4.
- Li, X. Y., D. Brown, S. Smith, W. MacNee, and K. Donaldson. 1999. “Short-Term Inflammatory Responses following Intratracheal Instillation of Fine and Ultrafine Carbon Black in Rats.” Inhalation Toxicology 11(8): 709–731.
- Li, N., T. Xia, and A. E. Nel. 2008. “The Role of Oxidative Stress in Ambient Particulate Matter-Induced Lung Diseases and Its Implications in the Toxicity of Engineered Nanoparticles.” Free Radical Biology and Medicine 1(44): 1689–1699. doi:10.1016/j.freeradbiomed.2008.01.028.
- Loxham, Matthew, Rebecca J. Morgan-Walsh, Matthew J. Cooper, Cornelia Blume, Emily J. Swindle, Patrick W. Dennison, Peter H. Howarth, et al. 2015. “The Effects on Bronchial Epithelial Mucociliary Cultures of Coarse, Fine, and Ultrafine Particulate Matter from an Underground Railway Station.” Toxicological Sciences 145(1): 98–107. doi:10.1093/toxsci/kfv034.
- Matuschek, Georg, Erwin Karg, Andreas Schröppel, Holger Schulz, and Otmar Schmid. 2007. “Chemical Investigation of Eight Different Types of Carbonaceous Particles Using Thermoanalytical Techniques.” Environmental Science & Technology 41(24): 8406–8411. doi:10.1021/es062660v.
- Miotto, D., M. P. Ruggieri, P. Boschetto, G. Cavallesco, A. Papi, I. Bononi, C. Piola, et al. 2003. “Interleukin-13 and -4 Expression in the Central Airways of Smokers with Chronic Bronchitis.” European Respiratory Journal 22(4): 602–608.
- Müller, J.-O., D. S. Su, R. E. Jentoft, U. Wild, and R. Schlögl. 2006. “Diesel Engine Exhaust Emission: Oxidative Behavior and Microstructure of Black Smoke Soot Particulate.” Environmental Science & Technology 40(4): 1231–1236. doi:10.1021/es0512069.
- Nemmar, Abderrahim, Jørn A. Holme, Irma Rosas, Per E. Schwarze, and Ernesto Alfaro-Moreno. 2013. “Recent Advances in Particulate Matter and Nanoparticle Toxicology: A Review of the in Vivo and in Vitro Studies.” Biomed Research International 2013: 279371. doi:10.1155/2013/279371.
- Nichols, Jennifer L., Elizabeth Oesterling Owens, Steven J. Dutton, and Thomas J. Luben. 2013. “Systematic Review of the Effects of Black Carbon on Cardiovascular Disease among Individuals with pre-existing disease.” International Journal of Public Health 58(5): 707–724. doi:10.1007/s00038-013-0492-z.
- Ning, Zhi, Michael D. Geller, Katharine F. Moore, Rebecca Sheesley, James J. Schauer, and Constantinos Sioutas. 2007. “Daily Variation in Chemical Characteristics of Urban Ultrafine Aerosols and Inference of Their Sources.” Environmental Science & Technology 41(17): 6000–6006. doi:10.1021/es070653g.
- Oberdörster, Günter, Andrew Maynard, Ken Donaldson, Vincent Castranova, Julie Fitzpatrick, Kevin Ausman, Janet Carter, et al. 2005. “Principles for Characterizing the Potential Human Health Effects from Exposure to Nanomaterials: elements of a Screening Strategy.” Particle and Fibre Toxicology 6(2): 8.
- O'Byrne, P. M., and D. S. Postma. 1999. “The Many Faces of Airway Inflammation. Asthma and Chronic Obstructive Pulmonary Disease. Asthma Research Group.” American Journal of Respiratory and Critical Care Medicine 159: S41–S63. doi:10.1164/ajrccm.159.supplement_2.mfa-1.
- Oh, Jee Youn, Young Seok Lee, Kyung Hoon Min, Gyu Young Hur, Sung Yong Lee, Kyung Ho Kang, Chin Kook Rhee, Seoung Ju Park, and Jae Jeong Shim. 2018. “Decreased Serum Club Cell Secretory Protein in Asthma and Chronic Obstructive Pulmonary Disease Overlap: A Pilot Study.” International Journal of Chronic Obstructive Pulmonary Disease 13: 3411–3417. doi:10.2147/COPD.S174545.
- Ovrevik, Johan, Magne Refsnes, Marit Låg, Jørn A. Holme, and Per E. Schwarze. 2015. “Activation of Proinflammatory Responses in Cells of the Airway Mucosa by Particulate Matter: Oxidant- and Non-Oxidant-Mediated Triggering Mechanisms.” Biomolecules 5: 1399–1440.
- Paulin, L., and N. Hansel. 2016. “Particulate Air Pollution and Impaired Lung Function.” F1000Res 5: 201.
- Pope, C. A., R. T. Burnett, M. J. Thun, E. E. Calle, D. Krewski, K. Ito, and G. D. Thurston. 2002. “Lung Cancer, Cardiopulmonary Mortality, and Long-Term Exposure to Fine Particulate Air Pollution.” JAMA 287: 1132–1141.
- Repine, John E., Aalt Bast, and Ida Lankhorst. 1997. “Oxidative Stress in Chronic Obstructive Pulmonary Disease. Oxidative Stress Study Group.” American Journal of Respiratory and Critical Care Medicine 156(2): 341–357. doi:10.1164/ajrccm.156.2.9611013.
- Rich, K., H. M. Huang, W. Wang, G. Wang, Y. Zhu, P. Ohman-Strickland, P. Hu, et al. 2012. “Association between Changes in Air Pollution Levels during the Beijing Olympics and Biomarkers of Inflammation and Thrombosis in Healthy Young Adults.” JAMA 307(19): 2068–2078. doi:10.1001/jama.2012.3488.
- Rothen-Rutishauser, Barbara M., Stephen G. Kiama, and Peter Gehr. 2005. “A Three-Dimensional Cellular Model of the Human Respiratory Tract to Study the Interaction with Particles.” American Journal of Respiratory Cell and Molecular Biology 32(4): 281–289. doi:10.1165/rcmb.2004-0187OC.
- Sager, Tina M., Michael W. Wolfarth, Lori A. Battelli, Stephen S. Leonard, Michael Andrew, Thomas Steinbach, Morinobu Endo, et al. 2013. “Investigation of the Pulmonary Bioactivity of Double-Walled Carbon Nanotubes.” Journal of Toxicology and Environmental Health. Part A 76(15): 922–936. doi:10.1080/15287394.2013.825571.
- Samet, Jonathan M., Ira B. Tager, and Frank E. Speizer. 1983. “The Relationship between Respiratory Illness in Childhood and Chronic Air-Flow Obstruction in Adulthood.” American Review of Respiratory Disease 127(4): 508–523. doi:10.1164/arrd.1983.127.4.508.
- Sattar, Y., M. Rashid, M. Ramli1, and B. Sabariah 2014. “Black Carbon and Elemental Concentration of Ambient Particulate Matter in Makassar Indonesia.” IOP Conference Series: Earth and Environmental Science 18(1): 012099 doi:10.1088/1755-1315/18/1/012099.
- Schamberger, Andrea C., Claudia A. Staab-Weijnitz, Nikica Mise-Racek, and Oliver Eickelberg. 2015. “Cigarette Smoke Alters Primary Human Bronchial Epithelial Cell Differentiation at the Air-Liquid Interface.” Scientific Reports 5, 8163.
- Schmid, O., and T. Stoeger. 2016. “Surface Area Is the Biologically Most Effective Dose Metric for Acute Nanoparticle Toxicity in the Lung.” Journal of Aerosol Science 99: 133–143. doi:10.1016/j.jaerosci.2015.12.006.
- Stoeger, Tobias, Shinji Takenaka, Birgit Frankenberger, Baerbel Ritter, Erwin Karg, Konrad Maier, Holger Schulz, and Otmar Schmid. 2009. “Deducing in Vivo Toxicity of Combustion-Derived Nanoparticles from a Cell-Free Oxidative Potency Assay and Metabolic Activation of Organic Compounds.” Environmental Health Perspectives 117(1): 54–60. doi:10.1289/ehp.11370.
- Strandberg, Karin, Lena Palmberg, and Kjell Larsson. 2008. “Effect of Budesonide and Formoterol on IL-6 and IL-8 Release from Primary Bronchial Epithelial Cells.” The Journal of Asthma 45(3): 201–203. doi:10.1080/02770900801890372.
- Su, D.S., R.E. Jentoft, J.-O. Müller, D. Rothe, E. Jacob, C.D. Simpson, Ž. Tomović, K. Müllen, A. Messerer, U. Pöschl., et al. 2004. “Microstructure and Oxidation Behaviour of Euro IV Diesel Engine Soot: A Comparative Study with Synthetic Model Soot Substances.” Catalysis Today 90(1–2): 127–132. doi:10.1016/j.cattod.2004.04.017.
- Sydlik, Ulrich, Katrin Bierhals, Maria Soufi, Josef Abel, Roel P. F. Schins, and Klaus Unfried. 2006. “Ultrafine Carbon Particles Induce Apoptosis and Proliferation in Rat Lung Epithelial Cells via Specific Signaling Pathways Both Using EGF-R.” American Journal of Physiology-Lung Cellular and Molecular Physiology 291(4): L725–733.
- Tian, Yaohua, Xiao Xiang, Juan Juan, Jing Song, Yaying Cao, Chao Huang, Man Li, and Yonghua Hu. 2018. “Short-Term Effects of Ambient Fine Particulate Matter Pollution on Hospital Visits for Chronic Obstructive Pulmonary Disease in Beijing, China.” Environmental Health 17(1): 21.doi:10.1186/s12940-018-0369-y.
- Upadhyay, Swapna, Tobias Stoeger, Volkar Harder, Ronald F. Thomas, Mette C. Schladweiler, Manuela Semmler-Behnke, Shinji Takenaka, Erwin Karg, Peter Reitmeir, Michael Bader., et al. 2008. “Exposure to Ultrafine Carbon Particles at Levels below Detectable Pulmonary Inflammation Affects Cardiovascular Performance in Spontaneously Hypertensive Rats.” Particle and Fibre Toxicology 5(1): 19. doi:10.1186/1743-8977-5-19.
- Upadhyay, Swapna, Tobias Stoeger, Leema George, Mette C. Schladweiler, Urmila Kodavanti, Koustav Ganguly, and Holger Schulz. 2014. “Ultrafine Carbon Particle Mediated Cardiovascular Impairment of Aged Spontaneously Hypertensive Rats.” Particle and Fibre Toxicology 11: 36. doi:10.1186/s12989-014-0036-6.
- Upadhyay, S., and L. Palmberg. 2018. “Air-Liquid Interface: Relevant in Vitro Models for Investigating Air Pollutant-Induced Pulmonary Toxicity.” Toxicological Sciences 164(1): 21–30.
- von Scheele, Ida, Kjell Larsson, and Lena Palmberg. 2010. “Budesonide Enhances Toll-like Receptor 2 Expression in Activated Bronchial Epithelial cells.” Inhalation Toxicology 22(6): 493–499. doi:10.3109/08958370903521216.
- WHO. 2014. “7 Million Premature Deaths Annually Linked to Air Pollution.” https://www.who.int/mediacentre/news/releases/2014/air-pollution/en/
- WHO. 2018. “Ambient (Outdoor) Air Quality and Health.” http://www.who.int/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health.
- Yuan, Xia, Xiangxian Zhang, Lu Sun, Yuquan Wei, and Xiawei Wei. 2019. “Cellular Toxicity and Immunological Effects of Carbon-Based Nanomaterials.” Particle and Fibre Toxicology 16(1): 18. doi:10.1186/s12989-019-0299-z.