
ABSTRACT
Introduction
Congenital hypogonadotropic hypogonadism (CHH) is a genetic disorder of reproduction and development, characterized by deficient gonadotropin-releasing hormone (GnRH) secretion or action, affecting 1-in-4,000–15,000 males. Micropenis and undescended testes are cardinal features of antenatal GnRH deficiency and could indicate absent minipuberty in the first postnatal months. In this review, we outline the pathophysiology and clinical consequences of absent minipuberty and its implications for optimal approaches to the endocrine management of affected boys.
Areas covered
Deficient GnRH activity during fetal development and neonatal-infancy phase of minipuberty accounts for the diminished mass of Sertoli cells and seminiferous tubules among CHH males, enduring impairment of reproductive function even during gonadotropin replacement in adult life. In overcoming this obstacle, several clinical studies of neonatal gonadotropin replacement have consistently shown positive results in inducing testicular development and correcting cryptorchidism.
Expert opinion
A high index of clinical suspicion, combined with hormonal testing undertaken in the postnatal period of 1–4 months, can reliably confirm or refute the diagnosis of CHH. Timely identification of CHH in affected male infants (having characteristic “red flag’ developmental anomalies) opens up the possibility for gonadotropin replacement as a targeted therapy to restore the normal hormonal milieu of minipuberty. Further work is necessary in formulating optimal gonadotropin treatment regimens to be more widely adopted in clinical practice.
1. Introduction
Congenital hypogonadotropic hypogonadism (CHH) is a reproductive disorder characterized by an isolated deficiency in gonadotropin-releasing hormone (GnRH) activity. Disruption in the embryonal development of the hypothalamic GnRH neuronal regulatory network underlies this complex genetic disorder. When this is coupled with defective olfactory axon pathfinding during embryonic development (causing arrested migration of GnRH neurons from the olfactory placode), impaired sense of smell (anosmia) becomes a key defining feature of the approximately 50% of CHH patients with Kallmann syndrome [Citation1,Citation2]. Male predominance is typically described for CHH with a male:female gender ratio of 3.6: 1 [Citation3] although this cannot be satisfactorily explained on the basis of our current knowledge of the genetics.
CHH is widely regarded as a rare medical disorder, but it has not been possible to accurately ascertain the actual prevalence of CHH because of scarcely available literature. Drawing data from two studies – a French study on young military conscripts [Citation4] and a Finnish nationwide study of hospital records [Citation5] – it can be conservatively estimated that the prevalence of CHH in men is at least 1 in 4,000–15,000.
In the context of combined pituitary hormone deficiency (CPHD), or congenital hypopituitarism, patients may also manifest similar CHH-associated features although they tend to present first with complications of adrenocorticotropic hormone (ACTH), growth hormone (GH), thyroid-stimulating hormone (TSH), and/or antidiuretic hormone (ADH) deficiency, including poor feeding and weight gain, hypoglycemia, seizures, and prolonged jaundice in the neonatal period [Citation6]. The estimated incidence of CPHD is between 1 in 4,000–10,000 live births [Citation7].
At present, more than 25 verified disease-causing genes have been linked to CHH, accounting for nearly 50% of cases (an up-to-date analysis of CHH genetics by Cangiano et al. reviewed >60 associated genes) [Citation8]. These genetic variants disrupt the central neuroendocrine regulation of reproduction at key points, including the differentiation of GnRH-secreting neurons – e.g. genes encoding the receptor-ligand pair fibroblast growth factor receptor 1/fibroblast growth factor 8 (FGFR1/FGF8) [Citation9–11] and NMDA receptor synaptonuclear signaling and neuronal migration factor (NMSF) [Citation12], the migration of GnRH neurons – e.g. anosmin-1 (ANOS1) [Citation13] and the ligand-receptor complex prokineticin 2/prokineticin receptor 2 (PROK2/PROKR2) [Citation14,Citation15], or the neuroendocrine control of GnRH secretion – e.g. genes encoding the ligand-receptor complexes kisspeptin/kisspeptin receptor 1 (KISS1/KISS1R) and neurokinin B/neurokinin 3 receptor (TAC3/TACR3) [Citation8]. Consequently, genetic evaluation and counseling remain highly challenging because of the underpinning complex genetic architecture, interplay of pathogenic variants and environmental exposure, and variable penetrance frequently observed within the same family [Citation16,Citation17].
2. Clinical manifestations of CHH across ages
CHH manifestations in male individuals vary at different stages of life [Citation18].
Neonates and infants with CHH may present with micropenis and/or undescended testes in the early postnatal months [Citation18]. When compared to the published prevalence of such congenital anomalies in general population, their prevalence in CHH male infants is at least 20- to 30-fold higher; hence, they are aptly considered as ‘red flag’ features of CHH [Citation19]. This is an important consequence of deficient minipuberty, which not only affects genital development early in life but also has a far-reaching impact on reproductive potential in adulthood.
Besides genital anomalies, several nonreproductive phenotypic features other than anosmia may also be identifiable in patients with CHH (“red flags”), including hearing impairment, synkinesia, midline facial defects, dental and digit anomalies, and renal malformation [Citation20,Citation21]. Clinically and genetically, there is a degree of overlap with the milder end of the CHARGE syndrome spectrum [Citation22,Citation23]. CHARGE syndrome is a rare autosomal dominant disorder due to CHD7 mutations, characterized by iris coloboma, congenital heart disease, choanal atresia, mental and growth retardation, genital hypoplasia, and ear malformations or deafness [Citation24]. In a study by Xu et al, CHD7 rare sequencing variants were found to be significantly enriched in a large cohort of CHH probands versus controls, supporting the implication of CHD7 in CHH [Citation23]. Similarly, the presence of midline developmental abnormalities may be associated with syndromic hypopituitarism e.g. septo-optic dysplasia (HESX1 and FGFR1/FGF8/PROKR2 mutations) [Citation25,Citation26], Hartsfield syndrome (FGFR1 mutation) [Citation25], and Culler-Jones syndrome (GLI2 mutation) [Citation27].
In adolescents, the inactive gonadotropic axis results in failure to initiate or progress through puberty with impaired development of secondary sexual characteristics. Approximately two-thirds of CHH male adolescents do not develop spontaneous puberty at >17 years of age, while rest exhibit features of stalled puberty [Citation21]. Diagnosis tends to be markedly delayed across the board, with meaningful hormonal treatment only being initiated around 18–19 years of age across Europe [Citation28–30], in large part due to the challenge of distinguishing CHH from constitutional delay in growth and puberty (CDGP) [Citation31,Citation32], with the latter being regarded as an extreme of the normal pubertal timing [Citation33]. However, with a history of neonatal cryptorchidism or micropenis, clinicians should have a very high index of suspicion, as the observed prevalence of these in CHH compared to CDGP males is nearly 20-fold elevated [Citation34]. A recent study suggests the possible utility of GnRH-stimulated luteinizing hormone (LH) and inhibin B to differentiate CHH from CDGP, but data remain very limited [Citation35]. Key obstacles to timely diagnosis reported by patients include their own shyness or embarrassment in coming forward, their parents’ assumption that they were simply late developers, and the misapplication by pediatricians of a ‘watch-and-wait’ strategy when there were already very clear “red flag” markers of CHH [Citation36].
Later in adulthood, symptoms of sexual dysfunction and infertility issues are more commonly encountered in clinical practice, and as such, the focus of treatment turns to ensuring adequate long-term testosterone replacement and the induction of spermatogenesis and fertility [Citation18] Much older individuals, who somehow “slipped through the net” when they were younger, occasionally present with osteoporosis, anemia, or sarcopenia.
3. Physiology of fetal genital development, minipuberty, and the impact of their deficiencies
While a majority of CHH males only present clinically from adolescence onward due to pubertal failure, lack of secondary sexual characteristics, sexual dysfunction, and/or infertility, it is important for clinicians to be aware that the disruption in their reproductive tract development occurs much earlier in life due to deficient minipuberty. Indeed, minipuberty plays an integral role in determining long-term testicular function.
Following sexual differentiation at 7–9 weeks of gestation with secretion of testosterone and insulin-like peptide 3 (INSL3) by Leydig cells and anti-Müllerian Hormone (AMH) by Sertoli cells, respectively, masculinization of the fetal external genitalia occurs under the regulation of placental human chorionic gonadotropin (hCG) (first trimester) and fetal pituitary LH (midgestation) [Citation37]. In the 3rd trimester, fetal GnRH-LH take over the control of fetal Leydig tissue in the production of androgens, creating the necessary hormonal milieu to stimulate penile growth and modulate the inguinoscrotal descent of testes via regression of cranial suspensory ligament and their final anchoring in the scrotal position [Citation38–40]. In addition, deficiency in fetal follicle-stimulating hormone (FSH) could possibly affect utero proliferation of Setoli cells and germ cells [Citation41]. This has important implication in CHH, as the lack of antenatal GnRH activity would bring about micropenis, testicular maldescent, as well as microorchidism at birth [Citation42].
After birth, neonatal GnRH activity recovers robustly following an initial dip due to the inhibitory effect of placental estrogens on the GnRH pulse generator and pituitary gonadotropes, marking the start of minipuberty [Citation43]. This represents the next critical milestone in male reproductive tract development that begins shortly after birth and continues until 6 months postnatal [Citation44]. Not unlike adolescence puberty, GnRH axis activation characterizes minipuberty, which is accompanied by increased pituitary gonadotroph secretion of FSH and LH and, in turn, testicular androgens. A peak in the gonadotropin and gonadal sex steroids and peptides occurs between 1 and 4 months postnatally, reaching adult levels, before declining to the prepubertal range by 6–9 months of age [Citation44,Citation45]. During this period, Leydig and Sertoli cells proliferate substantially [Citation46], with the parallel rise in production of androgens playing a key role in driving male genital development. The Sertoli cells do not fully express androgen receptors under age of 5 years [Citation47] and are thus physiologically insensitive to the concurrent high intratesticular concentration of testosterone [Citation48], thereby allowing germ cell proliferation while preventing premature differentiation, as shown by the rising AMH levels during minipuberty. In contrast, as the AMH level falls during male puberty, Sertoli cells undergo differentiation and begin to instead secrete Inhibin B as they support spermatogenesis [Citation49].
Therefore, it can be appreciated that pulsatile GnRH secretion associated with gonadotropin-stimulated secretion of reproductive hormones during the 3rd trimester and neonatal-infancy period is paramount for normal genital growth and gonadal development [Citation50,Citation51]. In addition, postnatal changes in the position of testes and consolidation of the intrascrotal position are strongly correlated with Leydig and Sertoli cell function [Citation39]. As such, any disease process that blunts activation of the fetal hypothalamic-pituitary-testicular may predispose to neonatal microphallus and cryptorchidism, which, in turn, should be regarded as potential cardinal features of severe gonadotropin deficiency.
Furthermore, in the absence of GnRH activity during fetal life and neonatal-infancy, the diminished proliferation of Sertoli cells and seminiferous tubules that would otherwise normally account for 90% of testicular volume [Citation52] resulting from loss of FSH-signaling result in undeveloped testes. Consequently, the Sertoli cell-derived AMH and Inhibin B levels are substantially depressed, reflecting a sparse Sertoli cell mass, which, in turn, has a huge bearing on future spermatogenic potential if not addressed appropriately. In a recent series, severe GnRH deficiency was found to affect two-thirds of CHH men, who manifested with prepubertal testicular volume [Citation53], and this has consistently been linked to diminished spermatogenic response to gonadotropin therapy in other studies. Knowledge gleaned from research in rhesus monkeys showed that transient switching-off of hypothalamic-pituitary-testicular function during the neonatal period led to stunted testicular growth and sperm production later in life [Citation54], underscoring the far-reaching impact of minipuberty on reproductive function.
4. ‘Window of opportunity’ in the diagnosis of CHH in infancy
Minipuberty has also been gaining greater attention among clinicians and investigators in recent years because of the recognition of its integral and fundamental role in male genital development [Citation55].
Bypassing the conundrum of distinguishing between CDGP and CHH that clinicians face in the context of delayed puberty in adolescents, the window of opportunity in the first 6 months of life allows CHH to be diagnosed by establishing deficiency in minipuberty-related hormones [Citation44], so that affected individuals could receive pubertal induction with sex steroids or gonadotropins from 12 years of age and, thereby, experience a normal adolescence, rather than the existing ubiquitous state of uncertainty, delay, and procrastination.
Timely evaluation and intervention of children with hypogonadotropic hypogonadism could modify the course of the disease, which, otherwise, if left inappropriately managed, is associated with poor long-term reproductive health and psychological outcomes. Indeed, the responsiveness of Sertoli cells to gonadotropin in spermatogenesis later in life is critically dependent on the priming process that occurred minipuberty, the lack of which conceivably compromises fertility significantly due to the diminished Sertoli cell population and seminiferous tubular mass.
As it is time-sensitive in nature, a protocolized approach incorporating clinical and biochemical assessment () would therefore be helpful in facilitating timely detection and intervention. Based on the normal physiology observed in male minipuberty, a relatively straightforward and cheap biochemical profiling of basal reproductive hormones during this phase in suspected male infants would provide crucial evidence to support or exclude the possibility of CHH. In one study protocol, the absence of neonatal male minipuberty was confirmed with at least three repeated measurements at 8 hours of undetectable LH, FSH, and testosterone from 2 weeks to 3 months of age [Citation56]. Consistent with this approach, in a cohort of male infants with combined pituitary hormone deficiency (CPHD), 14/15 infants with genitalia anomalies were reliably identified to have CHH based on biochemical profiling (low FSH, LH, and testosterone concentrations), in contrast to the other infants with normal genitalia [Citation57]. This demonstrates the high discriminatory value of reproductive hormone evaluation in the first 6 months of life. For greater diagnostic accuracy, investigations could be performed around age 3–3.5 months when sex hormones tend to be most robust, based on the normative data derived from a large cohort of healthy Danish infants [Citation45]. Besides LH, FSH, and testosterone, the inclusion of AMH and inhibin B levels may be helpful to increase diagnostic confidence. In addition, although non-CHH infants with cryptorchidism may also exhibit hormonal changes, they differ from CHH infants in whom they tend to demonstrate higher FSH, similar testosterone, and slightly depressed inhibin B levels compared to healthy infants [Citation58].
Figure 1. Proposed approach to suspected absent minipuberty in male neonates and infants.
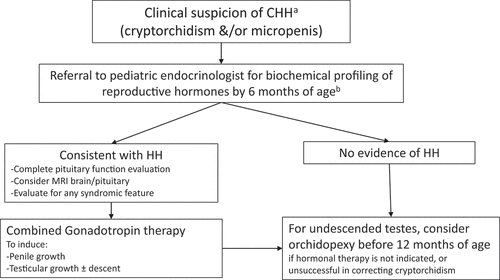
Therefore, it would also be helpful to adopt the targeted strategy that seeks to improve the recognition of infants affected by severe CHH (). Male neonates with a significant degree of congenital GnRH deficiency are at high risk of micropenis and/or cryptorchidism at birth. Indeed, persistent cryptorchidism is found in ~40% of CHH infants, compared to less than 2.5% of infants in the general population [Citation59–62], as cryptorchidism in the absence of underlying pathology including gonadotropin deficiency would tend to resolve spontaneously within the first 3 postnatal months [Citation63]. Hence, the presence of such genital anomalies, regarded as some of the ”red flag” markers of CHH, should trigger referral to pediatric endocrinologists for evaluation of hypothalamic-pituitary-testicular axis, rather than direct referral to a pediatric urological surgeon.
However, opportunities for early diagnosis and appropriate intervention are often missed due to lack of recognition [Citation64,Citation65]. In a large single-center series, only a third of CHH males with history of bilateral orchidopexy in childhood were referred to pediatric endocrinology for evaluation [Citation20]. Moreover, recommendation on hormonal testing in the management of infants born with genital anomalies remains lacking in published guidance [Citation66,Citation67] although data have since emerged to support such testing during the window of opportunity of minipuberty in the workup of a child with disorder of sex development (DSD) [Citation45]. This is similarly applicable to suspected CHH infants.
5. Gonadotropin therapy in neonates and infants
5.1. Correcting congenital anomalies
In male neonates and infants with micropenis (stretched penile length of <2.5 cm in term babies) with or without undescended testes, the goal of treatment is to increase the penile length, ensure normal positioning of testes, and maximize fertility potential [Citation42].
Conventionally, the treatment of congenital micropenis involves low-dose testosterone therapy given as either topical gel or intramuscular injections, which is generally expected to achieve desired phallus development [Citation68]. However, it would not be able to overcome gonadotropin-deficient related testicular maldevelopment. Therefore, an important consideration in the use of gonadotropin early in life for the treatment of CHH-associated genital anomalies lies in the possibility of replicating minipuberty physiology to achieve normal development in infancy, with a longer-term view of optimizing reproductive potential. Growing data on the outcomes of such a treatment strategy have been encouraging, making it an increasingly feasible intervention to be carried out in affected boys ().
Figure 2. Combined gonadotropin therapy regimens and outcomes studied in male neonates and infants with congenital hypogonadotropic hypogonadism.
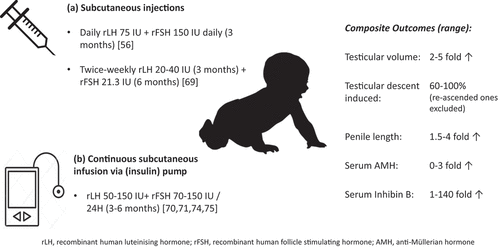
In one of the earliest reported experience, twice-weekly subcutaneous administration of recombinant human LH (rLH) and FSH (rFSH) to a 7.9-month-old child with CHH with micropenis was carried out () [Citation69]. Dose of rLH was increased from initial 20 IU to 40 IU after 2 months, while rFSH was maintained at 2.5 IU/kg throughout. Treatment was effective in increasing the penile length by 50% (1.6 cm → 2.4 cm). The substitution of rLH with testosterone suppositories at age 12.2 months further enhanced phallic growth to 2.7 cm. The six-month hormonal therapy also stimulated testicular volume growth from 31 mm3 to 84 mm3, along with doubling of serum inhibin B level. Treatment was well tolerated by the child without serious adverse effects.
Table 1. Studies of gonadotropin therapy in non-cryptorchid CHH infants
Bougnėres et al. reported their experience with a 6-month continuous subcutaneous infusion of combined gonadotropin therapy in two male infants born with hypogonadotropic hypogonadism () [Citation70]. The administration via an insulin pump avoided repeated painful injections. The first child presented at 2 weeks with micropenis (7 mm) and small intrascortal testes (< 10 mm diameter) and was also found to have multiple anterior pituitary deficiencies. The second child was referred at 14 weeks for micropenis (12 mm) with small intrascrotal testes (< 10 mm diameter). At baseline, serum LH, FSH, and testosterone were all below the normal range, while inhibin B and AMH were below or marginally above the lower limit of normal. Following initiation of gonadotropin therapy at age 8 and 20 weeks, respectively, serum testosterone, inhibin B, and AMH rose to high normal ranges during the course of treatment, corresponding to a 4-fold increase in penile length and testicular size in both infants. Doses were derived empirically based on authors’ observations: P1 received rLH 56 IU/day and rFSH 67 IU/day and P2 received rLH 50 IU/day and rFSH 125 IU/day. Of note, at the high serum FSH level up to 18-fold, the upper limit of normal was required to attain normal levels of inhibin B and AMH, reflecting a degree of resistance in the Sertoli cells. In the immediate weeks after cessation of gonadotropin therapy, while serum LH, FSH, and testosterone promptly returned to baseline levels, inhibin B fell less sharply to around low-normal range, while AMH persisted at similar concentrations achieved during the treatment phase, mirroring the expanded immature Sertoli cell population.
In a case report by Sarfati et al., a neonate born with micropenis and bilateral microorchidism was given combination rLH 75IU and rFSH 75 IU daily via a subcutaneous infusion pump for 6 months, leading to an increase in the testicular volume (0.33 → 2.3 mL) and penile length (15 → 38 mm) [Citation71]. Given positive family history (mother and maternal uncle), he was tested and confirmed to carry the same KAL1 mutation, consistent with his other phenotypic features including unilateral renal agenesis, synkinesia, and deafness.
5.2. Inducing testicular descent in CHH infants with cryptorchidism
Induction of testicular descent in infants with CHH-associated cryptorchidism is another potential therapeutic advantage of gonadotropin therapy () [Citation72]. First, successful spontaneous descent of testes into the scrotum with hormonal therapy would obviate orchidopexy, thereby avoiding any risk of trauma to the testes during the surgical procedure. Second, gonadotropin therapy seeks to correct the fundamental developmental defects of the testes resulting from deficient minipuberty. This would have important implications on the long-term fertility potential of CHH men, particularly those who are on the more severe end of the spectrum of GnRH deficiency as manifested by cryptorchidism, who typically experience a higher failure rate of spermatogenic response to gonadotropin therapy in adulthood [Citation73].
Table 2. Studies that described testicular descent as an outcome of gonadotropin treatment in CHH neonates/infants
In a French tertiary pediatric center, continuous subcutaneous infusion of rLH and rFSH was employed in 5 infants with CHH/CPHD with micropenis [Citation74]. They were started on treatment at a mean age of around 4 months. Over the course of treatment, the investigators titrated dose and duration according to clinical response and hormonal parameters. On the whole, the doses needed by the 5 infants were similar: rLH 150 IU/24 h and rFSH 75 IU/24 h. Serum testosterone rose to mean 12.1 nmol/L, along with the 3- and 5-fold increase in serum inhibin B and AMH, respectively. Among the 4 CHH patients with cryptorchidism, 3 had unilateral (high-scrotal or inguinal) and 1 had bilateral (high-scrotal) maldescended testes. Gonadotropin therapy led to spontaneous testicular descent in 2/4 infants (1 unilateral and 1 bilateral) and hence reduced the need for orchidopexy by half. The robustness of biochemical response appears to be associated with a better descent rate.
Separately, another case series from France by Lambert and Bougnėres reported favorable experience with the use of subcutaneous combined gonadotropin infusion in inducing testicular descent in bilateral-cryptorchid infants [Citation75]. Eight male infants with CHH, of whom 3 had CPHD, aged 0.25 to 11 months, and had either nonpalpable (5/8) or palpable testes in a high nonscrotal position (3/8). Diagnosis was confirmed by detectably low serum LH and testosterone levels, consistent with that of missing minipuberty. Using an insulin pump system, continuous infusion of rLH and rFSH was delivered at 50 IU and 75–150IU daily, respectively, which served to achieve minipuberty-range gonadotropin and testosterone levels. Similarly, both serum AMH and inhibin B levels reached levels normally expected during minipuberty in most, except in 2 (1 CHH infant with inadequate AMH and inhibin B, and 1 CPHD infant with inadequate inhibin B response). Following therapy, of the 10 nonpalpable (abdominal position) testes in 5 infants, complete descent was observed in 7 testes, whereas incomplete descent occurred in 3 (all high-scrotal position). All the other 3 infants with bilateral high-scrotal testes showed complete descent. Of note, one infant with abdominal testes who had complete response initially experienced reascent of both testes nearly 1 year later, necessitating orchidopexy.
More recently, Papadimitriou et al. reported successful descent of testes into the scrotal position in a cohort of 10 neonates and infants with bilateral cryptorchidism and micropenis following combined gonadotropin therapy (REMAP study) [Citation56]. Prior to treatment, absent minipuberty was all biochemically confirmed by the repeatedly low serum gonadotropins and testosterone levels in the first 3 months after birth. Treatment with a commercially available combined recombinant gonadotropin preparation (LH 75 IU and FSH 150 IU) was initiated at a median age of 0.35 years (0.19–0.78). Parents were trained to administer daily subcutaneous injection at home for 3 months (total of 90 injections). During the course of treatment, the penile length doubled from a median of 2.0 to 3.8 cm, along with a significant rise in serum levels of testosterone, inhibin B, and AMH.
From these published series (), combined LH and FSH therapy has emerged to be a highly promising hormonal regimen in inducing complete testicular descent in infants. Of note, the biochemical response (inhibin B and AMH) to gonadotropin therapy tends to correlate with the success rate of descent.
In contrast, the combination of FSH with exogenous testosterone instead of LH does not appear to exert similar effects [Citation76]. In a cohort of five infants treated with concurrent FSH and testosterone replacement, all required bilateral orchidopexy after cryptorchidism failed to correct in 3/5, and testicular ascent occurred in the other two, despite having elicited a good therapeutic response in both Sertoli cell function and penile lengthening [Citation77]. This provides evidence that the synergistic action of both FSH and LH is essential for generating the hormonal milleu for testicular descent and intrascrotal anchoring. Conceivably, exogenous testosterone therapy is incapable of matching the high concentration of intratesticular Leydig cell-derived androgen and INSL3 under LH drive that is necessary for effective paracrine signaling. The alternative explanation is that the FSH doses used were significantly lower (1/40 or less) compared to other studies, which could be grossly inadequate to drive the growth of Sertoli cells. From the inhibin B levels of the 3 subjects provided graphically, it can be estimated that the peak levels achieved ranged from ~50 to just under 300 pg/mL. In comparison, a mean inhibin B level of 368 pg/mL (normal range 254–513) was achieved in the study by Lambert and Bougnères, which administered a total cumulative FSH dose of 21,000 IU per patient [Citation75]. Sertoli cell resistance has been suggested to be present in CHH infants with cryptorchidism due to the lack of FSH stimulation prenatally, hence necessitating higher therapeutic doses [Citation75].
As alluded to earlier, although surgical orchidopexy by 1 year of age is widely recommended to preserve testicular function and prevent germ cell loss [Citation65,Citation67,Citation78], conservative correction of testicular maldescent by hormonal means would be helpful to avoid surgical risks [Citation42], in addition to the benefit of restoring growth of immature Sertoli cells and spermatogonia. Intriguingly, in a recent retrospective study, history of orchidopexy in cryptorchid CHH men was not associated with a more favorable spermatogenic response to gonadotropin treatment as would normally be expected for cryptorchidism from non-CHH etiologies [Citation79]. A plausible explanation could be the inadequacy of orchidopexy to remediate the absence of gonadotropin-mediated transformation of gonocytes into adult dark (Ad) spermatogonia in undescended testes [Citation80,Citation81]. The supply of stem cells for spermatogenesis relies on Ad spermatogonia. These data may further support the role of targeted hormonal intervention in CHH instead of surgical correction to optimally mitigate the sequelae of deficient minipuberty. Even in less successful cases, the increased testicular size following gonadotropin stimulation would aid in facilitating surgical manipulation, thereby minimizing the risk of trauma to testicular tissue, which could result in long-term damage and further diminishes prospect of fertility.
Our own local experience in adolescent CHH males with cryprorchidism is that, even in late teenage years, combined gonadotropin therapy is invariably successful in inducing descent of testes into the scrotum, provided that there has been no prior attempt at surgical orchidopexy [Citation82], similar to a recent report by Sharma et al. (discussed in section 8.1) [Citation83]. However, for those who underwent unsuccessful surgical exploration earlier in childhood, sometimes repeatedly, our experience with combined gonadotropin treatment is both positive and negative; negativity in that medical descent is not achievable, likely due to postsurgical fibrosis in the line of descent, but positivity in that gonadotropin-induced increase in the testicular volume has invariably permitted definitive localization and allowed successful surgery.
It should be emphasized that the success rate of gonadotropin therapy in the treatment of cryptorchidism relies on careful selection of infants with biochemical evidence of gonadotropin deficiency. Studies performed in the setting of CHH/CHPD have demonstrated favorable outcomes without adverse effects. On the other hand, nonselective use of gonadotropins in cases of idiopathic cryptorchidism demonstrated a negligible effect on testicular descent, with possible deleterious consequences on the viability of testicular germ cells [Citation42,Citation84]. Clinicians unfamiliar with such consideration might have misconceptions that gonadotropin therapy should be avoided entirely.
6. Prognostic factors of fertility induction in CHH men
Adult men with congenital causes of HH typically respond less favorably to gonadotropin therapy with regard to spermatogenic parameters compared to those with acquired causes [Citation85,Citation86]. The fundamental reason is the much earlier onset and greater degree of GnRH/gonadotropin deficiency in CHH, leading to disrupted minipuberty-primed testicular development. The efficacy of combined gonadotropin therapy and pulsatile GnRH therapy is comparable and hence does not significantly affect spermatogenic outcomes [Citation86,Citation87].
In a multicenter study of adolescents and young adults with HH receiving gonadotropin (hCG/rFSH) therapy, factors that are found to be associated with poorer therapeutic response (testicular growth and spermatogenesis) would reflect the above are as follows: history of bilateral cryptorchidism, low baseline testicular volumes, and low serum inhibin B and AMH levels [Citation73]. These clinical features represent long-standing severe gonadotropin deficiency with lack of the minipuberty phase of testicular development. Conversely, among CHH adult men who undergo spermatogenic induction by gonadotropin replacement, predictors of success include testicular volume ≥4 mL (vs. <4 mL), higher inhibin B, and the absence of history of cryptorchidism [Citation88,Citation89].
Overall, nearly one-third of men with severe CHH (testicular volume <4 mL) remain persistently azoospermic even with a prolonged course of combined gonadotropin therapy, while those with partial GnRH deficiency (testicular volume ≥4 mL) tend to respond more favorably, with 80% demonstrating sperm in ejaculate during treatment [Citation90]. Therefore, it is clear that a critical volume of seminiferous tubules and germ cells that attained prepubertal – which would rely on neonatal-infancy minipuberty – at the time of puberty initiation is key to successful testicular maturation and subsequent spermatogenesis.
While testosterone therapy (commonly intramuscular injection of short-acting testosterone formulation) is effective in correcting congenital micropenis, it does not have any significant effect on testicular spermatogenic tissue growth. On the other hand, it was demonstrated in prepubertal boys with CHH in early adolescence that the administration of rFSH alone was successful in inducing at least a 2-fold increase in the testicular volume, along with the rise of serum inhibin B into the healthy range, reflecting the proliferation of Sertoli cells and lengthening of seminiferous tubules that had taken place [Citation91]. Furthermore, the milieu of Leydig cell-derived androgens produced under gonadotropin stimulation may also be beneficial for facilitating spontaneous testicular descent in neonates or infants with CHH-associated cryptorchidism, which conceivably would further improve fertility potential. These changes will not be achieved via exogenous testosterone replacement.
7. Longer-term unmet health needs addressed by early detection and continuous surveillance
The benefit of early diagnosis and intervention extends beyond the provision of targeted hormonal therapy in infancy(36). CHH is a lifelong disorder that would require long-term surveillance for any associated health complications, as well as to ensure appropriate hormonal replacement. Being a rare medical disease, CHH patients would benefit greatly from the watchful eyes of experienced healthcare providers, as nonspecialized centers may lack the expertise to address the gaps in care [Citation92–94]. Evolving genetic landscape in the advent of next-generation sequencing is anticipated to accelerate the understanding of CHH genetics, and patients on follow-up could benefit directly or indirectly from participating in research studies [Citation95].
Psychosocial challenges often confront patients with CHH and have a great impact on quality of life. Surveys conducted revealed high prevalence of psychological comorbidities, moderate to severe depressive symptoms, social isolation, and antidepressant use among CHH men [Citation29,Citation96]. This also negatively impacts the long-term adherence to hormone replacement [Citation28]. Early and continuous engagement with patients and their parents provides opportunities for the multidisciplinary team to intervene and equip them with knowledge and coping strategies. This would allow any issues that could affect their self-esteem and ability to form relationships to be addressed early.
Failure to provide age-appropriate pubertal induction contributes to poor psychological health as well. Issues such as low self-confidence, social withdrawal, poor academic performance, and substance abuse are often associated with delayed puberty [Citation97,Citation98]. A structured follow-up program, therefore, facilitates timely pubertal induction as the children enter adolescence so that they develop secondary sexual characteristics and sexual maturity in tandem with their peers [Citation33], avoiding unnecessary delay due to the diagnostic uncertainty and physician’s hesitancy to treat [Citation97]. Furthermore, it is noteworthy that gonadotropin compared to testosterone therapy appears to be associated with better psychological outcomes including emotional and mental health in one study [Citation99], which informs clinicians the importance to discuss treatment options with patients. Where appropriate and desired, timely discussion on fertility prospects and treatment options, including assisted reproductive techniques, in late adolescence and adulthood would serve well to allay unnecessary anxieties. This illustrates the need to tailor treatment according to needs and priorities at different stages of life [Citation18].
8. Gonadotropin therapy – Novel concepts in adolescents and adults
8.1. Induction of testicular descent beyond infancy
Besides being an effective treatment in inducing puberty and genital development in prepubertal adolescents, gonadotropin therapy appears to be capable of promoting testicular descent even in this older age group akin to that of male infants. Cryptorchidism affects between 30 and 50% of CHH males, of whom up to two-thirds have bilateral disease [Citation20,Citation21,Citation53,Citation100], and thus, the feasibility of gonadotropin-induced testicular descent might be important in those who present late with untreated cryptorchidism.
In a small series of CHH patients aged 1.5 to 26 years who received adequate FSH therapy (given as human menopausal gonadotropin (hMG)) either as pretreatment or in combination with LH (given as hCG) from the outset, 5/6 patients with bilateral cryptorchidism experienced successful correction, with all testes descended from the inguinal canal to scrotal position after a mean treatment duration of close to 5 months () [Citation83]. Of these five patients, long-term follow-up data were available for 3 patients, which showed maintenance of the scrotal position (testicular volume of 5–8 ml) and successful spermatogenesis induction in 2 men. In the only patient with persistent unilateral right undescended testis, it remained undeveloped at 1 ml despite subsequent orchidopexy, whereas the left intrascrotal testis attained a size of 12ml on long-term follow up. It is noteworthy that in two pediatric patients (age 1.5 and 10.4 years), even hMG alone for 2–4 months could demonstrate partial efficacy, which was not observed in another study with patients on FSH monotherapy () [Citation77]. This is presumably due to the LH activity that is provided by hMG in equal potency as its FSH activity.
Table 3. Single-center experience with gonadotropin-induced testicular descent in CHH beyond infancy [Citation84]
8.2. Sequential gonadotropin therapy to maximize fertility potential
Among men with CHH who desire fertility, LH (hCG or rLH) and FSH (hMG or rFSH) therapies are the mainstay of treatment [Citation101]. Spermatogenesis is achieved by the concerted actions of FSH and LH-driven intratesticular testosterone [Citation102]. Its efficacy is at least on par with that of the far more complicated pulsatile GnRH therapy, which is not readily accessible in most tertiary centers. However, it should bear in mind that the order in which gonadotropin therapy is delivered has an impact on the chance of success in achieving spermatogenesis, particularly in those on the more severe end of the CHH disease spectrum.
Early studies of FSH monotherapy in children and adolescents proved its efficacy in promoting testicular growth and Sertoli cell function, as evidenced by increased circulating inhibin B concentrations [Citation91,Citation103]. Additionally, FSH pretreatment before the introduction of hCG demonstrated successful spermatogenesis in 4 out of 5 CHH/CPHD adolescents with a severely low baseline testicular volume of <3 mL [Citation103], which would, otherwise, be typically associated with a substantially poorer response to conventional hCG therapy.
Likewise, FSH priming in adults has been shown in a randomized open-label clinical trial to produce superior spermatogenic outcomes. In this cohort of 13 treatment-naïve CHH men with a prepubertal testicular volume of <4 mL, GnRH treatment was given for 24 months in all subjects, of which seven were randomized to 4-month recombinant FSH pretreatment [Citation104]. During the FSH pretreatment phase, the testicular size doubled along with the rise of inhibin B levels to healthy levels, and all men in this treatment arm developed sperm in ejaculate subsequently on GnRH therapy. In comparison, 2/6 in the GnRH-only treatment arm did not demonstrate spermatogenesis. In addition, the testicular volume, peak sperm counts, and time to appearance of sperm in ejaculate tended to be better in the FSH-primed group. In contrast, hCG monotherapy is typically unsatisfactory in the treatment of CHH men with prepubertal testes (i.e. testis volume <4 mL, comprising 2/3 of CHH males). Chen et al. reported a median treatment duration of 3.5 years before the appearance of sperm ejaculate in those who eventually responded to treatment, and it is not surprising that the testicular volume <4 mL and/or cryptorchidism correlated with poor therapeutic response [Citation105].
Therefore, in adolescent or adult men with prepubertal testes due to severe CHH, current limited evidence suggests that it would be beneficial to first maximize the proliferation of Sertoli and germ cells and elongation of seminiferous tubules, similar to the wave of growth normally present during minipuberty, through a 2–4-month period of unopposed FSH therapy. Importantly, such an approach avoids the risk of causing terminal differentiation of an already depleted pool of Sertoli cells arising from the high concentration of intratesticular testosterone from premature introduction of LH (hCG) therapy. Monotherapy with hCG in complete CHH is essentially pointless as the failure rate is exceedingly high [Citation106,Citation107]. On the other hand, FSH pretreatment before combined gonadotropin therapy may be less crucial in partial CHH men with the testicular volume >4 mL [Citation101,Citation108].
9. Conclusions
Growing literature on the effectiveness and safety of gonadotropin therapy in infancy supports the consideration of such treatment in the care of neonates and infants affected by CHH, particularly in the correction of micropenis and undescended and/or underdeveloped testes. In the longer term, by optimizing the prepubertal development of Sertoli cells and seminiferous tubules through minipuberty replacement, response to spermatogenesis-induction therapy is likely to be enhanced in adulthood, thereby improving the fertility prospect.
As such, it is timely to focus efforts on the early diagnosis in male infants with clinical features suggestive of congenital GnRH/gonadotropin deficiency (”red flags”), so that prompt investigations can be undertaken within the narrow window of opportunity (minipuberty phase).
10. Expert opinion
Male CHH is a challenging condition for two key reasons. First, the diagnosis has not always straightforward, and hence, meaningful treatment is typically delayed until late adolescence and even beyond, with resultant long-term consequences to patients’ physical and mental health. In adult life, these men express levels of distress comparable to patients with end-stage cardiorespiratory diseases [Citation109]. Second, while hypogonadotropic hypogonadism (HH) is generally amenable to gonadotropin therapy for fertility induction, the spermatogenic outcomes in men with CHH have been frustratingly dismal compared to those with adult-onset HH, reflecting the critical role and far-reaching impact that GnRH action during in utero and neonatal-infancy period has on reproductive capacity in adulthood.
Hence, a different approach is clearly necessary to improve clinical outcomes, and this is supported by growing insights into the fundamental role of minipuberty in reproductive tract development. Minipuberty is a key GnRH-driven developmental phase in neonates and infants who stimulate penile and testicular growth, with a priming role for future maturation and acquisition of spermatogenic function during puberty as well. The lack of minipuberty could therefore manifest early in life as micropenis and/or cryptorchidism (”red flags”).
At present, there remains a lack of consensus in undertaking targeted reproductive hormone evaluation in neonates and infants with suspicious phenotypic features. As near-adult levels of gonadotropins and testosterone are normally expected during the phase of minipuberty, biochemical testing within this window of opportunity could reliably identify or exclude underlying HH. Establishing early diagnosis confers the advantage of instituting timely and appropriate hormonal therapy, thereby avoiding surgical orchidopexy in CHH infants with cryptorchidism.
Indeed, gonadotropin therapy in the first year of life has demonstrated to effectively replicate minipuberty by inducing testicular descent as well as correction of microphallus. Importantly, the concomitant testicular growth from the expansion of Sertoli cells and seminiferous tubules following therapy in childhood potentially addresses the gap in spermatogenic response later in life when these men undergo fertility treatment. While long-term outcomes are not yet available, experience in adult men with CHH has corroborated such a minipuberty-like gonadotropin treatment approach in enhancing spermatogenesis. Furthermore, gonadotropin replacement in neonates and infants has a sound physiological basis and is well-tolerated in clinical studies. The key barriers to such treatment presently include the lack of awareness among clinicians, limited published data (in large part due to the rarity of the condition and in part to the costs, regulatory burden and funding-gap for undertaking investigator-led clinical trials in children in areas other than cancer), and the absence of guidance on the most optimal strategy to employ gonadotropin therapy in young children.
As illustrated in , various administration modes and dosing regimens are reported by different investigators, which may inadvertently create uncertainties. For the field to advance, concerted efforts among tertiary centers with the necessary expertise are crucial to generate a framework for research priorities and to provide guiding principles on diagnostic and therapeutic approaches that are feasible for wider clinical adoption. For instance, in major urological guidelines, surgical orchidopexy before the age of 12 months is the recommended treatment of choice for undescended testis, with indiscriminate hormonal therapy being cautioned against. Incorporating a standardized endocrinological evaluation algorithm () would be helpful to guide clinicians in identifying affected children with underlying HH who might better benefit from gonadotropin therapy instead. With greater case detection among boys with suspicious clinical features, larger clinical studies may be carried out to determine the optimal dosing and duration of gonadotropin treatment and to provide longitudinal data on the long-term benefit of minipuberty-hormone replacement. Importantly, early identification allows patients to benefit from structured long-term follow-up of their growth and development in expert centers. With regard to the mode of gonadotropin delivery, the REMAP study demonstrated the feasibility and efficacy of a commercially available fixed rLH:rFSH dosing that could be subcutaneously administered by parents at home with relative ease [Citation56]. Further studies could explore similar principles that will help to reduce the technical barrier to gonadotropin therapy in young children. In the future, with the expansion in the knowledge of CHH genetics in the current era of next-generation sequencing technologies, it is hopeful that these data would improve disease categorization and individualization of treatment.
Article highlights
Congenital hypogonadotropic hypogonadism (CHH) is a rare reproductive disorder characterized by deficiency in gonadotropin-releasing hormone (GnRH) secretion or action that is associated with nonreproductive defects in around 60% of cases; most commonly hyposmia/anosmia defines Kallmann syndrome and more rarely with combined pituitary hormone deficiency.
An early diagnosis of CHH in childhood facilitates not only timely intervention in normalising genital development but also the implementation of a structured follow-up program to optimize long-term reproductive and psychological health by ensuring timely pubertal-induction around 13 years of age, rather than at the typical 18 years of age reported by patients.
GnRH deficiency in late pregnancy and neonatal-infancy is chiefly responsible for the defects of virilization, including micropenis, cryptorchidism, and impaired testicular growth, with micropenis and/or bilateral cryptorchidism occurring in 20-50% of CHH boys.
The presence of a neonatal congenital defect that is strongly associated with CHH should prompt early referral to pediatric endocrinology for timely biochemical evaluation, whether reproductive anomaly (micropenis and/or bilateral cryptorchidism), or nonreproductive (hearing impairment, craniofacial, or digit defect (e.g. syndactyly).
Several clinical studies aiming to replicate minipuberty through combined gonadotropin therapy in neonates and infants with CHH have demonstrated its effectiveness in correcting micropenis, testicular hypoplasia, and cryptorchidism, obviating the need for surgical orchidopexy in almost all cases and potentially improving spermatogenesis-induction outcomes in adulthood.
Declaration of Interest
R Quinton has received speaker honoraria from Bayer UK and Besins UK. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- de Castro F, Seal R, Maggi R. ANOS1: a unified nomenclature for Kallmann syndrome 1 gene (KAL1) and anosmin-1. Brief Funct Genomics. 2017 Jul 1;16(4):205–210.
- Kim JH, Seo GH, Kim GH, et al. Targeted gene panel sequencing for molecular diagnosis of Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Exp Clin Endocrinol Diabetes. 2019 Sep;127(8):538–544.
- Dzemaili S, Tiemensma J, Quinton R, et al. Beyond hormone replacement: quality of life in women with congenital hypogonadotropic hypogonadism. Endocr Connect. 2017 Aug;6(6):404–412.
- Fromantin M, Gineste J, Didier A, et al. [Impuberism and hypogonadism at induction into military service. Statistical study]. Probl Actuels Endocrinol Nutr. 1973 May 3;16: 179–199.
- Laitinen E-M, Vaaralahti K, Tommiska J, et al. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J Rare Dis. 2011 Jun 17;6:41.
- Bosch I Ara L, Katugampola H, Dattani MT. Congenital hypopituitarism during the neonatal period: epidemiology, pathogenesis, therapeutic options, and outcome. Front Pediatr. 2021 Feb 2;8:600962.
- Davis SW, Castinetti F, Carvalho LR, et al. Molecular mechanisms of pituitary organogenesis: in search of novel regulatory genes. Mol Cell Endocrinol. 2010 Jul 8;323(1):4–19.
- Cangiano B, Swee DS, Quinton R, et al. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. 2021 Jan;140(1):77–111.
- Pitteloud N, Acierno JS, Meysing A, et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2006 Apr 18;103(16):6281–6286.
- McCabe MJ, Gaston-Massuet C, Tziaferi V, et al. Novel FGF8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab. 2011 Oct;96(10):E1709–18.
- Men M, Wu J, Zhao Y, et al. Genotypic and phenotypic spectra of FGFR1, FGF8, and FGF17 mutations in a Chinese cohort with idiopathic hypogonadotropic hypogonadism. Fertil Steril. 2020 Jan;113(1):158–166.
- Xu N, Kim HG, Bhagavath B, et al. Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril. 2011 Apr;95(5):1613–20.e1-7.
- Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991 Oct 10;353(6344):529–536.
- Dodé C, Teixeira L, Levilliers J, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006 Oct 20;2(10):e175.
- Monnier C, Dodé C, Fabre L, et al. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum Mol Genet. 2009 Jan 1;18(1):75–81.
- Maione L, Dwyer AA, Francou B, et al. Genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. 2018 Mar;178(3):R55–R80.
- Mitchell AL, Dwyer A, Pitteloud N, et al. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab. 2011 Jul;22(7):249–258.
- Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: european Consensus Statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015 Sep;11(9):547–564.
- Swee DS, Quinton R. Congenital hypogonadotrophic hypogonadism: minipuberty and the case for neonatal diagnosis. Front Endocrinol (Lausanne). 2019 Feb 21;10:97.
- Quinton R, Duke VM, Robertson A, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf). 2001 Aug;55(2):163–174.
- Bonomi M, Vezzoli V, Krausz C, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (IHH). Eur J Endocrinol. 2018 Jan;178(1):23–32.
- Balasubramanian R, Crowley WF. Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am J Med Genet C Semin Med Genet. 2017 Dec;175(4):507–515.
- Xu C, Cassatella D, van der Sloot AM, et al. Evaluating CHARGE syndrome in congenital hypogonadotropic hypogonadism patients harboring CHD7 variants. Genet Med. 2018 Aug;20(8):872–881.
- Jongmans MCJ, Admiraal RJ, Van Der Donk KP, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006 Apr;43(4):306–314.
- Webb EA, Dattani MT. Septo-optic dysplasia. Eur J Hum Genet. 2010 Apr;18(4):393–397.
- Raivio T, Avbelj M, McCabe MJ, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012 Apr;97(4):E694–9.
- Kevelam SHG, Van Harssel JJT, Van Der Zwaag B, et al. A patient with a mild holoprosencephaly spectrum phenotype and heterotaxy and a 1.3Mb deletion encompassing GLI2. Am J Med Genet A. 2012 Jan;158A(1):166–173.
- Dwyer AA, Tiemensma J, Quinton R, et al. Adherence to treatment in men with hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf). 2017 Mar;86(3):377–383.
- Varimo T, Hero M, Laitinen EM, et al. Health-related quality of life in male patients with congenital hypogonadotropic hypogonadism. Clin Endocrinol (Oxf). 2015 Jul;83(1):141–143.
- Quinton R. Phenotypic aspect of Kallmann’s syndrome [MD dissertation]. University of Cambridge; 2000.
- Sedlmeyer IL, Palmert MR. Delayed puberty: analysis of a large case series from an academic center. J Clin Endocrinol Metab. 2002 Apr;87(4):1613–1620.
- Harrington J, Palmert MR. Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab. 2012 Sep;97(9):3056–3067.
- Dunkel L, Quinton R. Transition in endocrinology: induction of puberty. Eur J Endocrinol. 2014 Jun;170(6):R229–39.
- Varimo T, Miettinen PJ, Känsäkoski J, et al. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? An analysis of a large patient series from a single tertiary center. Hum Reprod. 2017 Jan;32(1):147–153.
- Chan YM, Lippincott MF, Sales Barroso P, et al. Using Kisspeptin to predict pubertal outcomes for youth with pubertal delay. J Clin Endocrinol Metab. 2020 Aug 1;105(8):e2717–e2725.
- Dwyer AA, Jayasena CN, Quinton R. Congenital hypogonadotropic hypogonadism: implications of absent mini-puberty. Minerva Endocrinol. 2016 Jun;41(2):188–195.
- Rey RA, Grinspon RP. Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract Res Clin Endocrinol Metab. 2011 Apr;25(2):221–238.
- Bay K, Main KM, Toppari J, et al. Testicular descent: INSL3, testosterone, genes and the intrauterine milieu. Nat Rev Urol. 2011 Apr;8(4):187–196.
- Koskenniemi JJ, Virtanen HE, Wohlfahrt-Veje C, et al. Postnatal changes in testicular position are associated with IGF-I and function of sertoli and leydig cells. J Clin Endocrinol Metab. 2018 Apr 1;103(4):1429–1437.
- Harrison SM, Bush NC, Wang Y, et al. Insulin-like Peptide 3 (INSL3) serum concentration during human male fetal life. Front Endocrinol (Lausanne). 2019 Sep 4;10:596.
- O’Shaughnessy PJ, Baker PJ, Monteiro A, et al. Developmental changes in human fetal testicular cell numbers and messenger ribonucleic acid levels during the second trimester. J Clin Endocrinol Metab. 2007 Dec;92(12):4792–4801.
- Bouvattier C, Maione L, Bouligand J, et al. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2011 Oct 18;8(3):172–182.
- Winter JSD, Hughes IA, Reyes FI, et al. Pituitary-gonadal relations in infancy: 2. Patterns of serum gonadal steroid concentrations in man from birth to two years of age. J Clin Endocrinol Metab. 1976 Apr;42(4):679–686.
- Forest MG, Cathiard AM, Bertrand JA. Evidence of testicular activity in early infancy. J Clin Endocrinol Metab. 1973 Jul;37(1):148–151.
- Johannsen TH, Main KM, Ljubicic ML, et al. Sex-differences in reproductive hormones during mini-puberty in infants with normal and disordered sex development. J Clin Endocrinol Metab. 2018 Aug 1;103(8):3028–3037.
- Chemes HE. Infancy is not a quiescent period of testicular development. Int J Androl. 2001 Feb;24(1):2–7.
- Rey RA, Musse M, Venara M, et al. Ontogeny of the androgen receptor expression in the fetal and postnatal testis: its relevance on sertoli cell maturation and the onset of adult spermatogenesis. Microsc Res Tech. 2009 Nov;72(11):787–795.
- Chemes HE, Rey RA, Nistal M, et al. Physiological androgen insensitivity of the fetal, neonatal, and early infantile testis is explained by the ontogeny of the androgen receptor expression in sertoli cells. J Clin Endocrinol Metab. 2008 Nov;93(11):4408–4412.
- Aksglaede L, Sørensen K, Boas M, et al. Changes in anti-Müllerian hormone (AMH) throughout the life span: a population-based study of 1027 healthy males from birth (cord blood) to the age of 69 years. J Clin Endocrinol Metab. 2010 Dec;95(12):5357–5364.
- Boas M, Boisen KA, Virtanen HE, et al. Postnatal penile length and growth rate correlate to serum testosterone levels: a longitudinal study of 1962 normal boys. Eur J Endocrinol. 2006 Jan;154(1):125–129.
- Cortes D, Müller J, Skakkebaek NE. Proliferation of Sertoli cells during development of the human testis assessed by stereological methods. Int J Androl. 1987 Aug;10(4):589–596.
- Russell LD, Ren HP, Hikim IS, et al. A comparative study in twelve mammalian species of volume densities, volumes, and numerical densities of selected testis components, emphasizing those related to the Sertoli cell. Am J Anat. 1990 May;188(1):21–30.
- Stamou MI, Varnavas P, Kentrou M, et al. Isolated GNRH deficiency: genotypic and phenotypic characteristics of the genetically heterogeneous Greek population. Eur J Endocrinol. 2017 Mar;176(3):L1–L5.
- Mann DR, Gould KG, Collins DC, et al. Blockade of neonatal activation of the pituitary-testicular axis: effect on peripubertal luteinizing hormone and testosterone secretion and on testicular development in male monkeys. J Clin Endocrinol Metab. 1989 Mar;68(3):600–607.
- Kuiri-Hänninen T, Koskenniemi J, Dunkel L, et al. Postnatal testicular activity in healthy boys and boys with cryptorchidism. Front Endocrinol (Lausanne). 2019 Jul 23;10:489.
- Papadimitriou DT, Chrysis D, Nyktari G, et al. Replacement of male mini-puberty. J Endocr Soc. 2019 May 9;3(7):1275–1282.
- Braslavsky D, Grinspon RP, Ballerini MG, et al. Hypogonadotropic hypogonadism in infants with congenital hypopituitarism: a challenge to diagnose at an early stage. Horm Res Paediatr. 2015;84(5):289–297.
- Suomi AM, Main KM, Kaleva M, et al. Hormonal changes in 3-month-old cryptorchid boys. J Clin Endocrinol Metab. 2006 Mar;91(3):953–958.
- Acerini CL, Miles HL, Dunger DB, et al. The descriptive epidemiology of congenital and acquired cryptorchidism in a UK infant cohort. Arch Dis Child. 2009 Nov;94(11):868–872.
- Berkowitz GS, Lapinski RH, Dolgin SE, et al. Prevalence and natural history of cryptorchidism. Pediatrics. 1993 Jul;92(1):44–49.
- John Radcliffe Hospital Cryptorchidism Study Group. Cryptorchidism: a prospective study of 7500 consecutive male births, 1984-8. John Radcliffe Hospital Cryptorchidism Study Group. Arch Dis Child. 1992 Jul;67(7):892–899.
- Ghirri P, Ciulli C, Vuerich M, et al. Incidence at birth and natural history of cryptorchidism: a study of 10,730 consecutive male infants. J Endocrinol Invest. 2002 Sep;25(8):709–715.
- Sijstermans K, Hack WWM, Meijer RW, et al. The frequency of undescended testis from birth to adulthood: a review. Int J Androl. 2008 Feb;31(1):1–11.
- Quinton R, Mamoojee Y, Jayasena CN, et al. Society for Endocrinology UK guidance on the evaluation of suspected disorders of sexual development: emphasizing the opportunity to predict adolescent pubertal failure through a neonatal diagnosis of absent minipuberty. Clin Endocrinol (Oxf). 2017 Feb;86(2):305–306.
- Martin Ritzén E, Bergh A, Bjerknes R, et al. Nordic consensus on treatment of undescended testes. Acta Paediatr. 2007 May;96(5):638–643.
- Ahmed SF, Achermann JC, Arlt W, et al. Society for Endocrinology UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development (Revised 2015). Clin Endocrinol (Oxf). 2016 May;84(5):771–788.
- Kolon TF, Herndon CDA, Baker LA, et al. Evaluation and treatment of Cryptorchidism: AUA guideline. J Urol. 2014 Aug;192(2):337–345.
- Hatipoǧlu N, Kurtoǧlu S. Micropenis: etiology, diagnosis and treatment approaches. J Clin Res Pediatr Endocrinol. 2013;5(4):217–223.
- Main KM, Schmidt IM, Toppari J, et al. Early postnatal treatment of hypogonadotropic hypogonadism with recombinant human FSH and LH. Eur J Endocrinol. 2002 Jan;146(1):75–79.
- Bougnères P, François M, Pantalone L, et al. Effects of an early postnatal treatment of hypogonadotropic hypogonadism with a continuous subcutaneous infusion of recombinant follicle-stimulating hormone and luteinizing hormone. J Clin Endocrinol Metab. 2008 Jun;93(6):2202–2205.
- Sarfati J, Bouvattier C, Bry-Gauillard H, et al. Kallmann syndrome with FGFR1 and KAL1 mutations detected during fetal life. Orphanet J Rare Dis. 2015 Jun 9;10:71.
- Swee DS, Quinton R, Maggi R. Recent advances in understanding and managing Kallmann syndrome. Fac Rev. 2021 Apr 13;10:37.
- Rohayem J, Hauffa BP, Zacharin M, et al. Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? -a multicentre prospective study of hCG/rFSH treatment outcomes during adolescence-. Clin Endocrinol (Oxf). 2017 Jan;86(1):75–87.
- Stoupa A, Samara-Boustani D, Flechtner I, et al. Efficacy and safety of continuous subcutaneous infusion of recombinant human gonadotropins for congenital micropenis during early infancy. Horm Res Paediatr. 2017;87(2):103–110.
- Lambert A-S, Bougneres P. Growth and descent of the testes in infants with hypogonadotropic hypogonadism receiving subcutaneous gonadotropin infusion. Int J Pediatr Endocrinol. 2016;2016:13.
- Schaison G, Young J, Pholsena M, et al. Failure of combined follicle-stimulating hormone-testosterone administration to initiate and/or maintain spermatogenesis in men with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1993 Dec;77(6):1545–1549.
- Kohva E, Huopio H, Hietamäki J, et al. Treatment of gonadotropin deficiency during the first year of life: long-term observation and outcome in five boys. Hum Reprod. 2019 May 1;34(5):863–871.
- Kollin C, Stukenborg JB, Nurmio M, et al. Boys with undescended testes: endocrine, volumetric and morphometric studies on testicular function before and after orchidopexy at nine months or three years of age. J Clin Endocrinol Metab. 2012 Dec;97(12):4588–4595.
- Liu Z, Mao J, Xu H, et al. Gonadotropin-induced spermatogenesis in CHH patients with cryptorchidism. Int J Endocrinol. 2019 Dec 18;2019:6743489.
- Verkauskas G, Malcius D, Eidukaite A, et al. Prospective study of histological and endocrine parameters of gonadal function in boys with cryptorchidism. J Pediatr Urol. 2016 Aug;12(4):238.e1–6.
- Zivkovic D, Bica DTG, Hadziselimovic F. Relationship between adult dark spermatogonia and secretory capacity of Leydig cells in cryptorchidism. BJU Int. 2007 Nov;100(5):1147–1149.
- Santhakumar A, Miller M, Quinton R. Pubertal induction in adult males with isolated hypogonadotropic hypogonadism using long-acting intramuscular testosterone undecanoate 1-g depot (Nebido). Clin Endocrinol (Oxf). 2014 Jan;80(1):155–157.
- Sharma S, Shah R, Patil V, et al. Gonadotropins for testicular descent in cryptorchid congenital hypogonadotropic hypogonadism males beyond infancy. J Pediatr Endocrinol Metab. 2021 Apr 26;34(7):917–924.
- Dunkel L, Taskinen S, Hovatta O, et al. Germ cell apoptosis after treatment of cryptorchidism with human chorionic gonadotropin is associated with impaired reproductive function in the adult. J Clin Invest. 1997 Nov 1;100(9):2341–2346.
- Kirk JM, Savage MO, Grant DB, et al. Gonadal function and response to human chorionic and menopausal gonadotrophin therapy in male patients with idiopathic hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf). 1994 Jul;41(1):57–63.
- Liu L, Banks SM, Barnes KM, et al. Two-year comparison of testicular responses to pulsatile gonadotropin-releasing hormone and exogenous gonadotropins from the inception of therapy in men with isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 1988 Dec;67(6):1140–1145.
- Büchter D, Behre HM, Kliesch S, et al. Pulsatile GnRH or human chorionic gonadotropin/human menopausal gonadotropin as effective treatment for men with hypogonadotropic hypogonadism: a review of 42 cases. Eur J Endocrinol. 1998 Sep;139(3):298–303.
- Pitteloud N, Hayes FJ, Dwyer A, et al. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002 Sep;87(9):4128–4136.
- Liu PY, Baker HWG, Jayadev V, et al. Induction of spermatogenesis and fertility during gonadotropin treatment of Gonadotropin-deficient infertile men: predictors of fertility outcome. J Clin Endocrinol Metab. 2009 Mar;94(3):801–808.
- Burris AS, Rodbard HW, Winters SJ, et al. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J Clin Endocrinol Metab. 1988 Jun;66(6):1144–1151.
- Raivio T, Toppari J, Perheentupa A, et al. Treatment of prepubertal gonadotrophin-deficient boys with recombinant human follicle-stimulating hormone. Lancet. 1997 Jul 26;350(9073):263–264.
- Smith N, Quinton R. Kallmann syndrome. BMJ. 2012 Dec 3;345:e6971.
- Budych K, Helms TM, Schultz C. How do patients with rare diseases experience the medical encounter? Exploring role behavior and its impact on patient-physician interaction. Health Policy. 2012 May;105(2–3):154–164.
- Hannemann-Weber H, Kessel M, Schultz C. Research performance of centers of expertise for rare diseases-The influence of network integration, internal resource access and operational experience. Health Policy. 2012 May;105(2–3):138–145.
- Quaynor SD, Bosley ME, Duckworth CG, et al. Targeted next generation sequencing approach identifies eighteen new candidate genes in normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Cell Endocrinol. 2016 Dec 5;437:86–96.
- Dwyer AA, Quinton R, Morin D, et al. Identifying the unmet health needs of patients with congenital hypogonadotropic hypogonadism using a web-based needs assessment: implications for online interventions and peer-to-peer support. Orphanet J Rare Dis. 2014 Jun 11;9:83.
- Palmert MR, Dunkel L. Delayed Puberty. N Engl J Med. 2012 Feb 2;366(5):443–453.
- Graber JA, Seeley JR, Brooks-Gunn J, et al. Is pubertal timing associated with psychopathology in young adulthood? J Am Acad Child Adolesc Psychiatry. 2004 Jun;43(6):718–726.
- Shiraishi K, Oka S, Matsuyama H. Assessment of quality of life during gonadotrophin treatment for male hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf). 2014 Aug;81(2):259–265.
- Pitteloud N, Hayes FJ, Boepple PA, et al. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002 Jan;87(1):152–160.
- Swee DS, Quinton R. Managing congenital hypogonadotrophic hypogonadism: a contemporary approach directed at optimizing fertility and long-term outcomes in males. Ther Adv Endocrinol Metab. 2019 Feb 10;10:2042018819826889.
- Andersson AM, Müller J, Skakkebæk NE. Different roles of prepubertal and postpubertal germ cells and sertoli cells in the regulation of serum inhibin B levels. J Clin Endocrinol Metab. 1998 Dec;83(12):4451–4458.
- Raivio T, Wikström AM, Dunkel L. Treatment of gonadotropin-deficient boys with recombinant human FSH: long-term observation and outcome. Eur J Endocrinol. 2007 Jan;156(1):105–111.
- Dwyer AA, Sykiotis GP, Hayes FJ, et al. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2013 Nov;98(11):E1790–5.
- Chen Y, Sun T, Niu Y, et al. Correlations among genotype and outcome in chinese male patients with congenital hypogonadotropic hypogonadism under HCG treatment. J Sex Med. 2020 Apr;17(4):645–657.
- Vicari E, Mongioì A, Calogero AE, et al. Therapy with human chorionic gonadotrophin alone induces spermatogenesis in men with isolated hypogonadotrophic hypogonadism‐long‐term follow‐up. Int J Androl. 1992 Aug;15(4):320–329.
- Yang L, Zhang SX, Dong Q, et al. Application of hormonal treatment in hypogonadotropic hypogonadism: more than ten years experience. Int Urol Nephrol. 2012 Apr;44(2):393–399.
- Ortaç M, Hıdır M, Çilesiz NC, et al. Efficacy of follitropin-alpha versus human menopausal gonadotropin for male patients with congenital hypogonadotropic hypogonadism. Turk J Urol. 2020 Nov 29;46(1):13–17.
- Dwyer AA, Quinton R, Pitteloud N, et al. Psychosexual development in men with congenital hypogonadotropic hypogonadism on long-term treatment: a mixed methods study. Sex Med. 2015 Mar;3(1):32–41.