
ABSTRACT
Introduction: Systemic sclerosis (SSc) is a rare and complex connective tissue disease characterized by fibrosis of the skin and internal organs. Interstitial lung disease (ILD) is a common complication of SSc and the leading cause of SSc-related death. No drugs are licensed for the treatment of SSc-ILD.
Areas covered: This review provides an overview of the current treatment of SSc-ILD and a perspective on investigational therapies, focusing on those studied in randomized controlled trials.
Expert opinion: There is substantial room for improvement in the treatment of SSc-ILD. Current treatment focuses on immunosuppressant therapies, particularly cyclophosphamide and mycophenolate. Hematopoietic stem cell transplantation has been shown to improve long-term outcomes, but the risk of treatment-related mortality restricts its use to select patients at specialized centers. Modifying the course of disease to improve outcomes remains the goal for new therapies. Several drugs are under investigation as potential therapies for SSc-ILD, providing hope that the limited treatment armamentarium for SSc-ILD will be expanded and improved in the near future. Expert consensus is needed on how to screen for and monitor SSc-ILD and on when to initiate and escalate therapy.
1. Introduction
Systemic sclerosis (SSc) is a rare and complex connective tissue disease (CTD) characterized by fibrosis of the skin and internal organs [Citation1]. While the exact cause of SSc is unknown, it is believed to involve an abnormal response to microvascular injury in individuals with genetic susceptibility and/or epigenetic modifications, which leads to immune dysregulation, inflammation, microvasculopathy and fibrosis [Citation2,Citation3]. SSc may be classified according to the degree of skin involvement into limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc) [Citation4]. The clinical course of SSc is highly variable, necessitating regular monitoring for the development and progression of specific organ manifestations [Citation1,Citation5].
Interstitial lung disease (ILD) occurs in the majority of patients with SSc and is a leading cause of SSc-related death [Citation6]. Risk factors for the development of SSc-ILD include dcSSc [Citation7], African-American ethnicity [Citation8], shorter disease duration [Citation9], older age at disease onset [Citation7], and the presence of anti-topoisomerase I antibody and/or absence of anticentromere antibody [Citation7]. Cough and dyspnea are common in patients with SSc-ILD [Citation10] and may compromise patients’ quality of life [Citation11,Citation12]. Pulmonary hypertension, which may or may not be associated with the underlying SSc or ILD, is common in patients with SSc-ILD and is associated with high mortality [Citation13,Citation14].
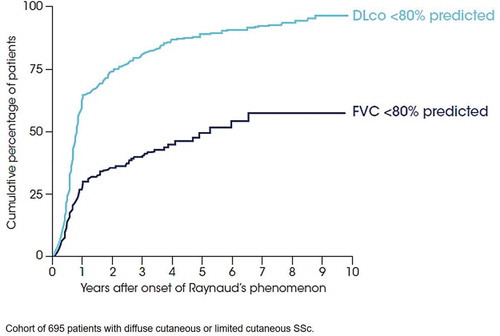
ILD typically occurs early in the course of SSc, particularly in patients with dcSSc () [Citation7,Citation9]. The clinical course of SSc-ILD is variable: some patients show stability in FVC while others show a progressive decline in lung function [Citation15]. Pulmonary function tests (PFTs) alone may not be sensitive enough to detect SSc-ILD [Citation16] and visual analysis of high-resolution computed tomography (HRCT) images is the gold standard tool for screening. The histological pattern most commonly observed in SSc-ILD is nonspecific interstitial pneumonia (NSIP), which is observed in approximately two-thirds of patients [Citation17]. Usual interstitial pneumonia (UIP) is present in a minority of individuals with SSc-ILD [Citation17–Citation19] and may be associated with poorer outcomes [Citation20]. There is growing interest in the use of quantitative analysis of HRCT to assess disease extent and monitor progression in patients with SSc-ILD [Citation21,Citation22] but this is not yet feasible for use in everyday clinical practice.
Figure 1. ILD occurs early in the course of SSc
The extent of fibrosis on HRCT predicts the progression of ILD [Citation23]. In a seminal study, the presence of extensive disease (disease extent on HRCT >30%, or 10-30% plus FVC <70% predicted) was a strong predictor of early mortality [Citation24]. In a study following 172 patients with SSc-ILD over 3.5 years, a disease extent on HRCT >20% was associated with a three-fold increased risk of progression of SSc-ILD (defined as the use of home oxygen or lung transplantation) or death compared to patients with a disease extent <20% on HRCT [Citation25]. In an analysis of 519 patients with SSc who had HRCT scans at baseline, the standardized mortality rate (compared to a control group matched by gender, age, and year of birth) increased with the extent of lung fibrosis on HRCT, from 2.2 in patients with no fibrosis to 8.0 in patients with an extent of lung fibrosis >25% [Citation26]. Decline in lung function is also a predictor of mortality in patients with SSc-ILD [Citation27,Citation28]. A study of 49 patients with extensive lung fibrosis showed that a decline in FVC of ≥10%, or a decline in FVC of 5–9% with a decline in DLco of >15%, was the most important predictor of mortality over a 15-year follow-up [Citation27]. In an analysis of data from patients treated in Scleroderma Lung Study (SLS) I (n = 158) and SLS II (n = 142), FVC % predicted and DLco % predicted measured as time-varying covariates over 2 years were independently associated with long-term mortality [Citation28].
In this article, we provide an overview of the current treatment of SSc-ILD and a perspective on investigational treatments, focusing on the therapies studied in randomized controlled trials.
2. Current treatment of SSc-ILD
There is no established algorithm for the management of SSc-ILD, but patients with advanced ILD on HRCT, or ILD plus a decline in lung function or symptoms, generally undergo treatment with immunosuppression [Citation29–Citation33]. The treatment recommendations for SSc issued by the European League Against Rheumatism (EULAR) use a stringent selection of randomized controlled trials as supportive evidence. The latest recommendations, issued in 2016, support the use of tailored therapy with cyclophosphamide (CYC), particularly for patients with progressive ILD, while acknowledging the need to consider the risk-to-benefit ratio for individual patients [Citation34,Citation35]. A recommendation for mycophenolate mofetil (MMF) was not included in the latest EULAR treatment guidelines, because the results of the SLS II [Citation36] were not published when the recommendations were developed.
The selection of therapy for SSc-ILD is highly variable. An online survey of 445 pulmonologists and rheumatologists from 7 countries conducted in 2017 found that while almost half of respondents used corticosteroids as first-line therapy, there was great heterogeneity in second- and third-line therapies, with CYC, azathioprine, MMF, methotrexate, and rituximab all being chosen as second-line therapies by at least 9% of respondents [Citation37]. In algorithms for the treatment of SSc-ILD developed in 2016–2017, the consensus of experts was that MMF, intravenous CYC and rituximab should be used as first-, second- and third-line induction therapy, and MMF, azathioprine and CYC (intravenous or oral) should be used as first-, second- and third-line maintenance therapy, respectively [Citation38]. Recent Delphi consensus studies conducted among rheumatologists and pulmonologists with expertise in managing patients with SSc-ILD also supported the use of MMF as first-line therapy [Citation39,Citation40]. In clinical practice, selection of therapy is based on a variety of factors, including access to therapies, a belief in the superiority of a specific agent, as well as a desire to provide patients with a treatment of some kind even in the absence of approved therapies.
2.1. Cyclophosphamide
The EULAR recommendation for use of CYC in the treatment of SSc-ILD was based on the results of two randomized placebo-controlled trials: the Fibrosing Alveolitis in Scleroderma Trial (FAST) [Citation41] and SLS I [Citation42]. In FAST, in which 45 subjects received oral prednisolone on alternate days plus monthly intravenous infusions of CYC for 6 months followed by oral azathioprine for 6 months, or placebo for 12 months, there was an improvement of 2.4% in FVC % predicted with active treatment versus a decline of 3.0% with placebo (p = 0.08) [Citation41]. In SLS I, there was a significant benefit of CYC versus placebo in the primary endpoint of change from baseline in FVC % predicted at month 12 (−1.0 versus −2.6% predicted), as well as in a number of secondary endpoints, including total lung capacity (TLC) % predicted, radiographic extent of fibrosis, dyspnea and health-related quality of life [Citation42]. Differences in FVC % predicted with CYC versus placebo at month 12 were greater in subjects with FVC <70% predicted at baseline [Citation43]. Further, a retrospective regression analysis showed that patients with a greater extent of disease on HRCT and/or greater extent of skin disease (higher score on the modified Rodnan skin score) were more likely to respond to CYC [Citation44]. Adverse events that were more common in the CYC group included leukopenia (26.0% versus 0%), hematuria (12.3% versus 4.2%), neutropenia (9.6% versus 0%), pneumonia (6.8% versus 1.4%), and anemia (2.7% versus 0%) [Citation42]. Except for a sustained improvement in dyspnea, the effects of CYC had dissipated 12 months after discontinuing treatment [Citation43]. Long-term use of cyclophosphamide is limited due to its cumulative toxicity.
2.2. Mycophenolate mofetil
In SLS II, in which 142 subjects at 14 US centers received oral MMF for 24 months or oral CYC for 12 months followed by placebo for 12 months, there was no significant difference between groups in the change in FVC % predicted over the course of 24 months (the primary endpoint). Subjects in both treatment arms experienced an improvement in FVC % predicted at month 24 (MMF: 2.2% versus CYC: 2.9%), as well as improvements in dyspnea [Citation36] and a reduction in the extent of ILD on HRCT [Citation22]. MMF had better tolerability than CYC, with fewer premature withdrawals due to adverse events (35% versus 42%), fewer treatment-related serious adverse events (4% versus 10%), and lower rates of leukopenia (41% versus 6%) and thrombocytopenia (6% versus 0%). Over twice as many deaths occurred in the CYC group (11 versus 5) with most attributed to progression of ILD [Citation36].
2.3. Autologous hematopoietic stem cell transplantation
The latest treatment guidelines issued by EULAR included a recommendation for use of autologous hematopoietic stem cell transplantation (HSCT) in patients with rapidly progressive SSc at risk of organ failure, while emphasizing the need for careful assessment of the risk-benefit profile for the individual patient [Citation35]. In high-risk patients with progressive dcSSc, HSCT has been associated with a substantial improvement in long-term survival, but with significant mortality in the year following transplant. In the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) trial in 156 patients with early dcSSc (including 125 patients with ILD), HSCT was associated with high treatment-related mortality in the first 12 months (10.1% versus 0% with CYC) [Citation45]. Causes of treatment-related death were variable and included Epstein-Barr virus, lymphoma, acute respiratory distress syndrome, heart failure, and myocardial infarction. However, at month 12, event-free survival was significantly greater with HSCT than CYC (HR 0.52 [95% CI 0.28, 0.96]). At month 24, there were significant improvements from baseline with HSCT versus CYC in FVC % predicted (6.3% versus −2.8%) and total lung capacity % predicted (5.1% versus −1.3%) and non-significant changes in DLco % predicted (−4.7% versus −4.1%) and residual volume (−4.8% versus −2.1%) [Citation45]. In the Scleroderma: Cyclophosphamide or Transplantation (SCOT) trial in 75 subjects with dcSSc and renal or pulmonary involvement (including 73 subjects with ILD), HSCT was associated with improved event-free (74% versus 47%) and overall (86% versus 51%) survival at month 72 versus CYC [Citation46]. Of note, treatment-related mortality in the first year in patients who received HSCT was lower in the SCOT trial than in the ASTIS trial (0% versus 10%). The reasons for this difference in treatment-related mortality in the first year are unknown, but may be due to differences in how patients were selected for inclusion, differences in the pre-transplant conditioning protocols, the higher proportion of smokers in the ASTIS trial, and/or that the SCOT trial was conducted at more highly specialized SSc centers.
2.4. Lung transplantation
The use of lung transplantation in patients with SSc requires discussion due to concerns over whether esophageal dysfunction and gastro-esophageal reflux may cause damage to the transplanted lung, and whether the multimorbidity of patients with SSc will impact outcomes. However, recent data suggest that post-transplant outcomes, including graft dysfunction and survival, are similar in selected patients with SSc-ILD as in those with other types of ILD [Citation47–Citation49].
3. Therapies in clinical development
There is a high unmet need for effective and well-tolerated treatments for SSc-ILD. Modifying the course of disease to improve outcomes for patients remains the aspiration for the development of new treatments. A large number of therapies, with a variety of mechanisms of action, are under investigation (). Here we summarize the clinical trials that have/had change in lung function as a primary endpoint or demonstrated potentially meaningful changes in lung function in a Phase 2 study. Ongoing and recently completed Phase 2/3 trials of potential therapies for SSc-ILD are shown in and .
Figure 2. Nintedanib reduces the rate of decline in forced vital capacity in patients with SSc-ILD: results of the randomized placebo-controlled SENSCIS trial [Citation55]. From Distler et al. Copyright © (2019) Massachusetts Medical Society
![Figure 2. Nintedanib reduces the rate of decline in forced vital capacity in patients with SSc-ILD: results of the randomized placebo-controlled SENSCIS trial [Citation55]. From Distler et al. Copyright © (2019) Massachusetts Medical Society](/cms/asset/44d61501-f615-44e3-97ac-189bb2f85223/ierm_a_1668269_f0002_oc.jpg)
Table 1. Ongoing and recently (2019) completed randomized controlled Phase II/III trials of investigational therapies for SSc-ILD in which change in lung function is the primary endpoint
Table 2. Ongoing and recently (2019) completed randomized controlled Phase II/III trials of investigational therapies for SSc-ILD in which change in lung function is a secondary endpoint
3.1. Nintedanib
Nintedanib, an inhibitor of tyrosine kinases involved in the pathophysiology of fibrosis, slows lung function decline in patients with idiopathic pulmonary fibrosis (IPF) [Citation50] and is an approved treatment for IPF. Nintedanib was investigated as a potential treatment for SSc-ILD based on the clinical and mechanistic similarities between IPF and SSc-ILD [Citation51]. Across a variety of animal models of SSc, including the bleomycin model, Tsk-1 model, GvHD model and Fra-2 transgenic mice, nintedanib has shown consistent antifibrotic effects, in addition to effects on inflammation and vascular remodeling [Citation51–Citation54].
The efficacy and safety of nintedanib in patients with SSc-ILD were assessed in the Phase III SENSCIS® trial [Citation55]. Subjects with onset of first non-Raynaud symptom ≤7 years before screening, ≥10% extent of fibrosis of the lungs on HRCT, FVC ≥40% predicted and DLco 30–89% predicted were randomized to receive oral nintedanib (n = 288) or placebo (n = 288) stratified by the presence of anti-topoisomerase I antibody. Patients receiving low-dose prednisone and/or stable background therapy with MMF or methotrexate were allowed to remain on these therapies. Nintedanib significantly reduced the annual rate of decline in FVC assessed over 52 weeks (the primary endpoint) versus placebo (−52.4 versus −93.3 mL/year; difference 41.0 mL/year [95% CI 2.9, 79.0] p = 0.04) () [Citation55]. Nintedanib reduced the rate of decline in FVC both in patients who were and were not receiving mycophenolate at baseline, with a numerically greater treatment effect in patients who were not taking mycophenolate. The adverse event profile of nintedanib was similar to that previously observed in patients with IPF, being characterized mainly by gastrointestinal events, particularly diarrhea of mild or moderate severity [Citation56].
The efficacy and safety of nintedanib in patients with progressive fibrosing ILDs have also been assessed in the INBUILD® trial. This trial enrolled subjects with ILD diagnoses other than IPF, including SSc-ILD, who had >10% extent of fibrosis on HRCT and met criteria for progression of ILD in the prior 24 months [Citation57,Citation58]. As in the SENSCIS® trial, the primary endpoint was the annual rate of decline in FVC (mL/year) assessed over 52 weeks. Results will be reported in September 2019.
3.2. Pirfenidone
Pirfenidone, a pyridone derivative, is an approved treatment for IPF that has been shown to slow lung function decline in this patient population [Citation59]. Pirfenidone has shown anti-fibrotic and anti-inflammatory effects in vitro and in animal models of lung fibrosis [Citation60–Citation62] but has not been tested specifically in animal models of SSc. An analysis of lung tissue from patients with SSc-ILD suggests that pirfenidone may exert anti-fibrotic effects by interfering with the hedgehog signaling pathway [Citation63]. In the LOTUSS trial, 16 weeks’ open-label pirfenidone was generally well tolerated in patients with SSc-ILD, but no conclusions could be drawn about its efficacy given the lack of a comparator group [Citation64]. The efficacy and safety of pirfenidone plus MMF versus MMF alone in subjects with SSc-ILD are being investigated in SLS III (NCT03221257). Target enrollment is 150 subjects with a disease duration of ≤7 years from first non-Raynaud’s symptom, dyspnea, ground-glass opacification on HRCT, and FVC ≤80% predicted. The primary endpoint is change from baseline in FVC % predicted at month 18 and the study is due to be completed in 2021.
3.3. Rituximab
Rituximab is a monoclonal cytolytic antibody against CD20 that acts to deplete B cells [Citation65]. Small open-label trials of rituximab compared to conventional therapy suggest that rituximab therapy leads to improvements in FVC and DLco in patients with SSc-ILD [Citation66,Citation67]. In a post-hoc analysis of 9 subjects with SSc-ILD in the European Scleroderma Trial and Research (EUSTAR) cohort, there were improvements from baseline in FVC % predicted with rituximab versus matched controls (0.4% versus −7.7%) after a median follow-up of 6 months [Citation68]. A follow-up study of 254 patients in the EUSTAR database treated with rituximab, of whom 58% were treated for lung involvement, did not show effects on FVC or DLco. After a median follow-up of 2 years, compared to 9575 control patients matched for propensity score, rituximab-treated patients were more likely to have an improvement in skin fibrosis, but did not have significantly different rates of decline in FVC or DLco [Citation69]. An ongoing multicenter randomized trial in the UK, RECITAL, is assessing the efficacy and safety of rituximab given intravenously at baseline and week 2 versus monthly intravenous CYC in 116 subjects with ILD associated with SSc, idiopathic inflammatory myositis, or mixed connective tissue disease [Citation70]. Changes from baseline in FVC (mL) at week 24 (primary endpoint) and week 48 will be assessed. The EvER-ILD trial (NCT02990286) is assessing the efficacy and safety of rituximab, given at baseline and week 2, added to MMF, versus MMF alone, in 122 subjects with CTD-ILD, interstitial pneumonia with autoimmune features (IPAF), or idiopathic ILD, plus NSIP on HRCT or histology, who have not responded to immunosuppressant therapy; the primary endpoint is the change from baseline in FVC % predicted at 6 months.
3.4. Tocilizumab
Tocilizumab is an anti-IL-6R monoclonal antibody. IL-6 levels have been associated with declines in FVC and DLco, as well as mortality, in patients with SSc-ILD [Citation71]. In the randomized Phase II FaSScinate trial, in which 87 subjects with progressive SSc received subcutaneous tocilizumab or placebo for 48 weeks, a clinically meaningful but not statistically significant reduction in change in mRSS at week 24 (primary endpoint) was observed with tocilizumab (−3.92 versus −1.22) [Citation72]. Although ILD was not an inclusion criterion, in an exploratory analysis, fewer patients treated with tocilizumab than placebo had an FVC decline of >10% predicted at week 24 (3% versus 19%). In a Phase III trial, FocuSSced, in which 210 subjects with SSc received subcutaneous tocilizumab for 48 weeks, there was no significant difference between tocilizumab and placebo in the primary endpoint of change in mRSS at week 48 (−6.14 versus −4.41, respectively). In exploratory analyses of change in FVC % predicted at week 48, the mean change from baseline at week 48 was −0.4% predicted in the tocilizumab group versus −4.6% predicted in the placebo group [Citation73].
4. Supportive care
Although limited evidence is available on the effectiveness of non-pharmacologic interventions in patients with SSc [Citation74], patients with SSc-ILD should be offered supportive care individualized to the needs of the patient [Citation75–Citation77]. This should include patient education and may also include oxygen supplementation, pulmonary rehabilitation, and vaccinations. In addition, management of extra-pulmonary manifestations of SSc and of comorbidities is an important part of the overall care of patients with SSc-ILD [Citation78].
5. Future perspectives
The therapies commonly used in the treatment of SSc-ILD have, at best, modest efficacy and/or are not supported by robust evidence from clinical trials. MMF is the most widely used therapy but has not been tested in a placebo-controlled trial. The duration of treatment with CYC is limited due to its toxicity. The use of HSCT, while it has shown efficacy, is restricted to a small subset of patients at high risk of severe organ damage and mortality and should be performed at specialized centers due to concerns over treatment-related mortality. Targeted therapies that inhibit key pathways in the pathogenesis of SSc-ILD are needed to improve patient outcomes. Although a number of drugs with different molecular targets are under investigation, a better understanding of pathophysiological targets and how these might interact with each other is needed. The potential influence of immunosuppressant therapy on the activity of drugs targeting other pathogenic pathways also needs to be investigated. It is likely that different therapeutic regimens, probably based on combination therapy, will be most effective for sub-groups of patients with SSc. In addition, more research is needed to discover biomarkers, including those based on molecular expression patterns, which can identify the patients most likely to benefit from specific therapies and bring a precision medicine approach to the treatment of SSc [Citation79–Citation81].
6. Conclusions
ILD is a common manifestation of SSc associated with high mortality, but at present, there is a limited evidence base to inform best practice in its monitoring and management. Patients with advanced and/or progressive ILD usually receive immunosuppressants, but there is a lack of robust evidence to support their efficacy or guide their use. The results of clinical trials will provide insights into the potential role of new therapies in the management of SSc-ILD.
7. Expert opinion
There is substantial room for improvement in the screening, diagnosis, and management of SSc-ILD. Delayed diagnosis of SSc often leads to delayed referral to specialty care, preventing patients from receiving the monitoring and management needed to optimize outcomes [Citation82]. A lack of consensus guidelines for screening or early diagnosis of ILD in patients with SSc results in discrepancies in diagnosis and to delayed and missed diagnosis of SSc-ILD in clinical practice. Although there is some promising evidence on the use of biomarkers, these are not yet validated to an extent that would allow them to be incorporated into clinical practice. More evidence is needed on clinical, radiological, serological and molecular markers of SSc-ILD and its progression to inform identification of patients; decision-making related to initiation, choice and escalation of treatment; and individualized counseling of patients. There is a clear need to establish an expert consensus on the drivers and timing of screening for SSc-ILD, the detection and prediction of disease progression, and the initiation, change and escalation of treatment.
Patients with SSc who have a limited extent of ILD on HRCT, or have stable lung function and minimal symptoms, may not undergo treatment with immunosuppression. However, patients whose lung function initially appears to be stable might show progression during follow-up. Further research is needed to define the optimal frequency of monitoring of such patients and when and how to re-evaluate treatment initiation. Further analysis of trajectories of FVC decline in individual patients, and how such trajectories can be predicted, is needed to improve patient care. The current practice of treating patients once worsening has occurred might be sub-optimal, as the opportunity to treat patients whose disease is progressing might already have been missed and these patients might be stable during further follow-up.
As a rare and heterogeneous disease, SSc presents challenges for clinical trial design, particularly regarding the selection of patients, the generalizability of results, and the detection of differences between treatment groups within a relatively short time frame. Recently, the tyrosine kinase inhibitor nintedanib was shown to reduce the rate of progression of ILD in a broad population of patients with SSc-ILD. Regulatory authorities are now considering applications for the approval of nintedanib as a treatment for SSc-ILD. In addition to trials of new therapies, further research is needed to assess the efficacy and safety of therapies that are currently used. In the next few years, it is likely that treatment options will increase as new therapies tested in clinical trials show efficacy on lung fibrosis. If so, further research questions will arise. For example, precision medicine approaches to predict response to a specific treatment will be required. Research will also be needed into whether different treatment strategies have additive effects, and whether immediate combination therapy, rather than sequential use of individual medications, provides better outcomes with an acceptable toxicity profile.
Article highlights
SSc is a rare and complex connective tissue disease with an unpredictable clinical course. ILD is a common manifestation of SSc and is the leading cause of death related to SSc.
There is no established treatment algorithm for SSc-ILD, but patients with advanced or progressive disease generally receive immunosuppression. CYC and MMF are the most commonly used therapies, based on the results of randomized controlled trials.
Hematopoietic stem cell transplantation (HSCT) has been shown to improve outcomes in patients with rapidly progressive SSc at risk of organ failure, but its use is restricted to highly selected patients treated at specialized centers due to the risk of treatment-related mortality.
There is a high unmet need for disease-modifying treatments for SSc-ILD with an acceptable side-effect profile. Several therapies are under investigation as treatments for SSc-ILD. Nintedanib has recently been shown to slow the progression of ILD in patients with SSc-ILD, with a side-effect profile that was manageable for most patients.
The discovery of biomarkers would facilitate the identification of patients most likely to benefit from specific therapies and so enable a precision medicine approach to treatment.
Declaration of interest
O Distler has/had consultancy relationship and/or has received research funding from Acceleron, Actelion, AnaMar, Bayer, Biogen Idec, Blade Therapeutics, Boehringer Ingelheim, ChemomAb, espeRare foundation, Genentech/Roche, GlaxoSmithKline, Inventiva, Italfarmaco, Lilly, medac, MedImmune, Mitsubishi Tanabe Pharma, Novartis, Pharmacyclics, Pfizer, Sanofi, Sinoxa and UCB in the area of potential treatments for SSc and its complications, and has a patent mir-29 for the treatment of SSc filed. ER Volkmann has consultancy relationships with Boehringer Ingelheim and has received research grants from the Rheumatology Research Foundation, Scleroderma Foundation, and EMD/Serono. AM Hoffmann-Vold has obtained research support from Boehringer Ingelheim; acted as a scientific consultant for Actelion and Boehringer Ingelheim; and received travel expenses from GlaxoSmithKline, Actelion and Boehringer Ingelheim for activities related to SSc and related diseases. TM Maher has served as a board member, consultant or has worked on clinical trials for: Apellis, Bayer, Biogen ldec, Blade, Boehringer Ingelheim, Bristol-Myers Squibb, Galecto, Galapagos, GlaxoSmithKline, Indalo, Novartis, Respivent, Roche, Trevi and UCB. TM Maher has also, via his institution, received research funding from GlaxoSmithKline and UCB. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Acknowledgments
Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Melanie Stephens and Wendy Morris of FleishmanHillard Fishburn, London, UK during the preparation of this article. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and have approved the final version.
Additional information
Funding
References
- Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390:1685–1699.
- Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: recent insights of molecular and cellular mechanisms and therapeutic opportunities. J Scleroderma Relat Disord. 2017;2:137–152.
- Henderson J, Distler J, O’Reilly S. The role of epigenetic modifications in systemic sclerosis: a druggable target. Trends Mol Med. 2019;25:395–411.
- Wolheim FA. Classification of systemic sclerosis. Visions and reality. Rheumatology (Oxford). 2005;44:1212–1216.
- van Den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum Dis. 2013;72:1747–1755.
- Elhai M, Meune C, Boubaya M, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis. 2017;76:1897–1905.
- Nihtyanova SI, Schreiber BE, Ong VH, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol. 2014;66:1625–1635.
- Steen V, Domsic RT, Lucas M, et al. A clinical and serologic comparison of African American and Caucasian patients with systemic sclerosis. Arthritis Rheum. 2012;64:2986–2994.
- Jaeger VK, Wirz EG, Allanore Y, et al. Incidences and risk factors of organ manifestations in the early course of systemic sclerosis: a longitudinal EUSTAR study. PLoS One. 2016;11:e0163894.
- Tashkin DP, Volkmann ER, Tseng CH, et al. Improved cough and cough-specific quality of life in patients treated for scleroderma-related interstitial lung disease: results of Scleroderma Lung Study II. Chest. 2017;151:813–820.
- Saketkoo LA, Mittoo S, Frankel S, et al. Reconciling healthcare professional and patient perspectives in the development of disease activity and response criteria in connective tissue disease-related interstitial lung diseases. J Rheumatol. 2014;41:792–798.
- Mittoo S, Frankel S, LeSage D, et al. Patient perspectives in OMERACT provide an anchor for future metric development and improved approaches to healthcare delivery in connective tissue disease related interstitial lung disease (CTD-ILD). Curr Respir Med Rev. 2015;11:175–183.
- Lefèvre G, Dauchet L, Hachulla E, et al. Survival and prognostic factors in systemic sclerosis-associated pulmonary hypertension: a systematic review and meta-analysis. Arthritis Rheum. 2013;65:2412–2423.
- Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015;46:903–975.
- Man A, Davidyock T, Ferguson LT, et al. Changes in forced vital capacity over time in systemic sclerosis: application of group-based trajectory modelling. Rheumatology (Oxford). 2015;54:1464–1471.
- Suliman YA, Dobrota R, Huscher D, et al. Pulmonary function tests: high rate of false-negative results in the early detection and screening of scleroderma-related interstitial lung disease. Arthritis Rheumatol. 2015;67:3256–3261.
- Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165:1581–1586.
- Kim DS, Yoo B, Lee JS, et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19:121–127.
- Fischer A, Swigris JJ, Groshong SD, et al. Clinically significant interstitial lung disease in limited scleroderma: histopathology, clinical features, and survival. Chest. 2008;134:601–605.
- Winstone TA, Assayag D, Wilcox PG, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest. 2014;146:422–436.
- Jacob J, Bartholmai BJ, Rajagopalan S, et al. Evaluation of computer-based computer tomography stratification against outcome models in connective tissue disease-related interstitial lung disease: a patient outcome study. BMC Med. 2016;14:190.
- Goldin JG, Kim GHJ, Tseng CH, et al. Longitudinal changes in quantitative interstitial lung disease on computed tomography after immunosuppression in the Scleroderma Lung Study II. Ann Am Thorac Soc. 2018;15:1286–1295.
- Hoffmann-Vold AM, Aaløkken TM, Lund MB, et al. Predictive value of serial high-resolution computed tomography analyses and concurrent lung function tests in systemic sclerosis. Arthritis Rheumatol. 2015;67:2205–2212.
- Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177:1248–1254.
- Moore OA, Goh N, Corte T, et al. Extent of disease on high-resolution computed tomography lung is a predictor of decline and mortality in systemic sclerosis-related interstitial lung disease. Rheumatology (Oxford). 2013;52:155–160.
- Hoffmann-Vold AM, Fretheim H, Halse AK, et al. Tracking impact of interstitial lung disease in systemic sclerosis in a complete nationwide cohort. Am J Respir Crit Care Med. (ePub ahead of print). DOI:10.1164/rccm.201903-0486OC
- Goh NS, Hoyles RK, Denton CP, et al. Short-term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol. 2017;69:1670–1678.
- Volkmann ER, Tashkin DP, Sim M, et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann Rheum Dis. 2019;78:122–130.
- Wells AU, Denton CP. Interstitial lung disease in connective tissue disease–mechanisms and management. Nat Rev Rheumatol. 2014;10:728–739.
- Cappelli S, Bellando Randone SB, Camiciottoli G, et al. Interstitial lung disease in systemic sclerosis: where do we stand? Eur Respir Rev. 2015;24:411–419.
- Volkmann ER, Tashkin DP. Treatment of systemic sclerosis-related interstitial lung disease: a review of existing and emerging therapies. Ann Am Thorac Soc. 2016;13:2045–2056.
- Adler S, Huscher D, Siegert E, et al. Systemic sclerosis associated interstitial lung disease – individualized immunosuppressive therapy and course of lung function: results of the EUSTAR group. Arthritis Res Ther. 2018;20:17.
- Wijsenbeek M, Kreuter M, Olson A, et al. Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin. (ePub ahead of print). DOI:10.1080/03007995.2019.1647040
- Kowal-Bielecka O, Landewé R, Avouac J, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR). Ann Rheum Dis. 2009;68:620–628.
- Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76:1327–1339.
- Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4:708–719.
- Kreuter M, Olson A, Fischer A, et al. Current treatment of patients with non-IPF progressive fibrosing interstitial lung disease. Am J Respir Crit Care Med. 2018;197:A4273. [ [abstract]].
- Fernández-Codina A, Walker KM, Pope JE, Scleroderma Algorithm Group. Treatment algorithms for systemic sclerosis according to experts. Arthritis Rheumatol. 2018;70:1820–1828.
- Rahaghi FF, Strek ME, Southern BD, et al. Expert consensus on the screening, treatment, and management of patients with SSc-ILD, and the potential future role of anti-fibrotics in a treatment paradigm for SSc-ILD: a Delphi consensus study. Poster presented at the American Thoracic Society (ATS) International Conference; 2019 May; Dallas, USA. Available from: http://www.usscicomms.com/respiratory/ats2019/rahaghi
- Hoffmann-Vold AM, Maher TM, Philpot EE, et al. Evidence-based consensus recommendations for the identification and management of interstitial lung disease in systemic sclerosis. Oral presentation at the European League Against Rheumatism (EULAR) Congress 2019; Madrid, Spain.
- Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54:3962–3970.
- Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655–2666.
- Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176:1026–1034.
- Roth MD, Tseng CH, Clements PJ, et al. Predicting treatment outcomes and responder subsets in scleroderma-related interstitial lung disease. Arthritis Rheum. 2011;63:2797–2808.
- van Laar JM, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311:2490–2498.
- Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. 2018;378:35–47.
- Crespo MM, Bermudez CA, Dew MA, et al. Lung transplant in patients with scleroderma compared with pulmonary fibrosis. Short- and long-term outcomes. Ann Am Thorac Soc. 2016;13:784–792.
- Miele CH, Schwab K, Saggar R, et al. Lung transplant outcomes in systemic sclerosis with significant esophageal dysfunction. a comprehensive single-center experience. Ann Am Thorac Soc. 2016;13:793–802.
- Pradère P, Tudorache I, Magnusson J, et al. Lung transplantation for scleroderma lung disease: an international, multicenter, observational cohort study. J Heart Lung Transplant. 2018;37:903–911.
- Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–2082.
- Wollin L, Distler JHW, Denton CP, et al. Rationale for the evaluation of nintedanib as a treatment for systemic sclerosis-associated interstitial lung disease. J Scleroderma Relat Disord. 2019. Epub ahead of print. DOI:10.1177/2397198319841842
- Wollin L, Maillet I, Quesniaux V, et al. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349:209–220.
- Huang J, Beyer C, Palumbo-Zerr K, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. 2016;75:883–890.
- Huang J, Maier C, Zhang Y, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. 2017;76:1941–1948.
- Distler O, Highland KB, Gahlemann M, et al. Nintedanib in systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019;380:2518–2528.
- Lancaster L, Crestani B, Hernandez P, et al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials. BMJ Open Respir Res. 2019;6:e000397.
- Flaherty KR, Brown KK, Well AU, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res. 2017;4:e000212.
- Flaherty KR, Wells AU, Clerisme-Beaty E, et al. Characteristics of patients with progressive fibrosing interstitial lung diseases (ILDs) in the INBUILD trial of nintedanib. Poster presented at the American Thoracic Society (ATS) International Conference; 2019 May; Dallas, USA. Available from: http://ildposters2019.com/PDF/ATS_INBUILD_Flaherty.pdf
- King TE Jr, WZ B, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–2092.
- Iyer SN, Gurujeyalakshmi G, Giri SN. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J Pharmacol Exp Ther. 1999;291:367–373.
- Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590:400–408.
- Visner GA, Liu F, Bizargity P, et al. Pirfenidone inhibits T-cell activation, proliferation, cytokine and chemokine production, and host alloresponses. Transplantation. 2009;88:330–338.
- Xiao H, Zhang GF, Liao XP, et al. Anti-fibrotic effects of pirfenidone by interference with the hedgehog signalling pathway in patients with systemic sclerosis-associated interstitial lung disease. Int J Rheum Dis. 2018;21:477–486.
- Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol. 2016;43:1672–1679.
- Daoussis D, Liossis SN, Yiannopoulos G, et al. B-cell depletion therapy in systemic sclerosis: experimental rationale and update on clinical evidence. Int J Rheumatol. 2011;214013:2011.
- Daoussis D, Liossis SN, Tsamandas AC, et al. Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatology (Oxford). 2010;49:271–280.
- Daoussis D, Melissaropoulos K, Sakellaropoulos G, et al. A multicenter, open-label, comparative study of B-cell depletion therapy with rituximab for systemic sclerosis-associated interstitial lung disease. Semin Arthritis Rheum. 2017;46:625–631.
- Jordan S, Distler JH, Maurer B, et al. EUSTAR Rituximab study group. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2015;74:1188–1194.
- Elhai M, Boubaya M, Distler O, et al. Outcomes of patients with systemic sclerosis treated with rituximab in contemporary practice: a prospective cohort study. Ann Rheum Dis. 2019;78:979–987.
- Saunders P, Tsipouri V, Keir GJ, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials. 2017;18:275.
- De Lauretis A, Sestini P, Pantelidis P, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol. 2013;40:435–446.
- Khanna D, Denton CP, Jahreis A, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387:2630–2640.
- Khanna D, Lin CJF, Kuwana M, et al. Efficacy and safety of tocilizumab for the treatment of systemic sclerosis: results from a phase 3 randomized controlled trial. Oral presentation at the American College of Rheumatology (ACR) Annual Meeting 2018; Chicago, USA.
- Willems LM, Vriezekolk JE, Schouffoer AA, et al. Effectiveness of nonpharmacologic interventions in systemic sclerosis: a systematic review. Arthritis Care Res. 2015;67:1426–1439.
- Lanken PN, Terry PB, Delisser HM, et al. An official American Thoracic Society clinical policy statement: palliative care for patients with respiratory diseases and critical illnesses. Am J Respir Crit Care Med. 2008;177:912–927.
- Kreuter M, Bendstrup E, Russell AM, et al. Palliative care in interstitial lung disease: living well. Lancet Respir Med. 2017;5:968–980.
- Milette K, Thombs BD, Maiorino K, et al. Challenges and strategies for coping with scleroderma: implications for a scleroderma-specific self-management program. Disabil Rehabil. 2018;9:1–10.
- Denton CP, Wells AU, Coghlan JG. Major lung complications of systemic sclerosis. Nat Rev Rheumatol. 2018;14:511–527.
- Dobrota R, Mihai C, Distler O. Personalized medicine in systemic sclerosis: facts and promises. Curr Rheumatol Rep. 2014;16:425.
- Martyanov V, Whitfield ML. Molecular stratification and precision medicine in systemic sclerosis from genomic and proteomic data. Curr Opin Rheumatol. 2016;28:83–88.
- Taroni JN, Martyanov V, Mahoney JM, et al. A functional genomic meta-analysis of clinical trials in systemic sclerosis: toward precision medicine and combination therapy. J Invest Dermatol. 2017;137:1033–1041.
- Distler O, Allanore Y, Denton CP, et al. Factors influencing early referral, early diagnosis and management in patients with diffuse cutaneous systemic sclerosis. Rheumatology (Oxford). 2018;57:813–817.