
ABSTRACT
Introduction
During the last 4 decades, registration of patients with primary immunodeficiencies (PID) has played an essential role in different aspects of these diseases worldwide including epidemiological indexes, policymaking, quality controls of care/life, facilitation of genetic studies and clinical trials as well as improving our understanding about the natural history of the disease and the immune system function. However, due to the limitation of sustainable resources supporting these registries, inconsistency in diagnostic criteria and lack of molecular diagnosis as well as difficulties in the documentation and designing any universal platform, the global perspective of these diseases remains unclear.
Areas covered
Published and unpublished studies from January 1981 to June 2020 were systematically reviewed on PubMed, Web of Science and Scopus. Additionally, the reference list of all studies was hand-searched for additional studies. This effort identified a total of 104614 registered patients and suggests identification of at least 10590 additional PID patients, mainly from countries located in Asia and Africa. Molecular defects in genes known to cause PID were identified and reported in 13852 (13.2% of all registered) patients.
Expert opinion
Although these data suggest some progress in the identification and documentation of PID patients worldwide, achieving the basic requirement for the global PID burden estimation and registration of undiagnosed patients will require more reinforcement of the progress, involving both improved diagnostic facilities and neonatal screening.
1. Introduction
Primary immunodeficiencies (PID) are described as heterogeneous diseases with a shortcoming in the development and function of the immune system [Citation1,Citation2]. PID patients usually suffer from more severe and repeated infections [Citation3], however, dysregulation of immunity predisposes the immunodeficient patients to lymphoproliferation, atopy, autoimmunity and malignancy [Citation1,Citation4]. Since the first report of PID in 1952, we have encountered an explosion of knowledge in this field with recognition of more than 400 genetic defects that are connected to even more PID phenotypes [Citation1], that make this group of diseases an independent pursuit within biomedicine.
Presentation of PID varies from potentially benign forms such as IgA deficiency to catastrophic types such as severe combined immunodeficiency (SCID). Complex medical features and the necessity for lifelong and resource-consuming treatments in many patients underscores the importance of qualified epidemiological data in decision making and PID management. Although epidemiological research in this field faces a variety of difficulties due to the novelty of PID and its rarity [Citation5], national and international collaborations help to advance translational research and improve the management and therapeutic strategies [Citation6,Citation7].
Most of the prevalence estimations have been based on selected populations such as tertiary hospitals, or single-center registries. Reports from several PID registries in different countries show a prevalence of 1:8500 to 1:100000 for symptomatic patients [Citation5,Citation8–Citation14]. However, it has been generally accepted that PID is under-diagnosed and under-reported. Furthermore, measurement of the burden of PID is restricted owing to insufficient recording and reporting. In 1981, the registration of patients was first advanced within countries initiated by Japan [Citation15], followed by intracontinental registration efforts such as by the Latin American Society for Immunodeficiencies (LASID) in 1993 [Citation16] and even global initiatives advanced by the Jeffrey Modell Foundation (JMF) in 2011 [Citation17].
Although it has been suggested that obtaining incidence rates from local networks and national registries with proper data documentation could be a way to predict incidence for larger populations, this method cannot create a valid picture of PID in other populations as the prevalence of particular PID varies in different ethnic groups [Citation18]. To consider this issue, several larger networks have been formed to collect the data from independent countries and diverse regions such as the European Society for Immunodeficiencies (ESID), LASID and The United States Immunodeficiency Network (USIDNET) databases [Citation16,Citation19]. However, obvious discrepancies among different online sources and published documents create challenges in measuring accurate global disease burdens. This review is designed to systematically consider all available evidence regarding PID registries and present the frequencies of these diseases in 80 countries and all well-known international PID networks. The overarching objective is to provide deeper insights into the epidemiology of PID by comparing worldwide the prevalence, clinical and molecular diagnoses, and predisposing factors in different populations.
2. Methods
Detailed methods, including search strategy for systematic review, study selection, data extraction and quality assessment are described in the Supplementary Method and Figure S1. The study was approved by the ethics committee of Tehran University of Medical Sciences, Tehran, Iran.
3. Results
3.1. Asia
A total of 15939 PID patients from 18 nationwide registries were surveyed in Asia () representing a diverse distribution of PID in these countries based on PID prevalence (), parental consanguinity (), proportions of PID categories (), gender ratio () and genetic diagnosis (). The first PID registry was reported in Japan, which included 497 patients [Citation15] and the most recent data from Japan has been reported in 2011 by Ishimura et al., in which a total of 1240 patients have been registered with a prevalence of 2.3 per 100000 inhabitants. Predominantly antibody deficiencies (41.2%) were the most frequent type of PID, followed by congenital defects of phagocyte (18.5%) and other well-defined immunodeficiency syndromes (16%). Bruton’s tyrosine kinase (BTK) deficiency was the most prevalent genetic defect amongst Japanese PID patients. Common variable immunodeficiency (CVID) and selective IgA deficiency (SIgAD) comprise only 11% and 4% of the PID patients, respectively [Citation14].
Figure 1. Distribution of primary immunodeficiency prevalence in the world based on the number of reported patients per 100000 individuals (Gray color represents countries without registry or without published report).
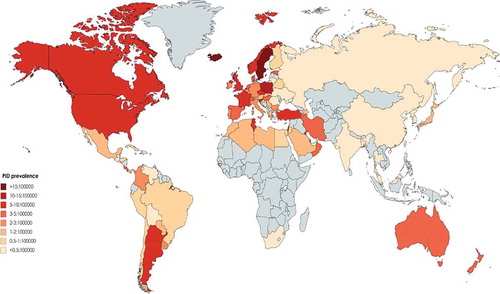
Figure 2. Distribution of parental consanguinity among different registries of patients with primary immunodeficiency in the world. (Gray color represents countries without registry or without appropriate report).
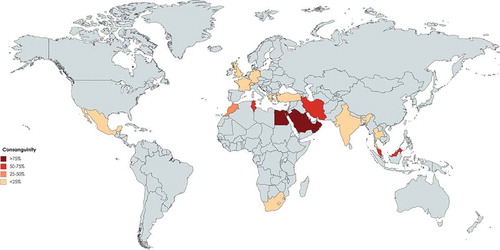
Figure 3. Proportion of predominant antibody deficiencies among different registries of patients with primary immunodeficiency in the world. (Gray color represents countries without registry or without appropriate report).
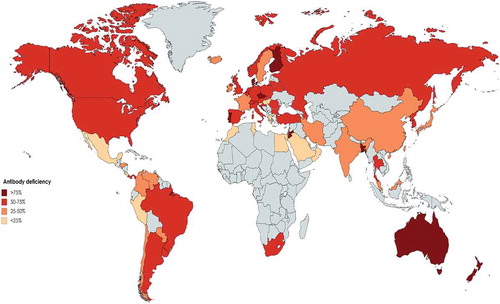
Figure 4. Proportion of male to female ratio among different registries of patients with primary immunodeficiency in the world. (Gray color represents countries without registry or without appropriate report).
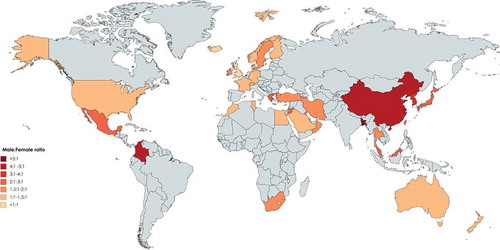
Figure 5. Proportion of molecular diagnosis among different registries of patients with primary immunodeficiency in the world. (Gray color represents countries without registry or without appropriate report).
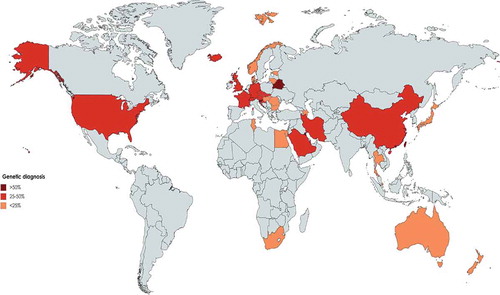
Table 1. The frequency of diagnosed primary immunodeficiency (PID) cases in the world and percentage of Predominantly antibody deficiencies (PAD), immunodeficiencies affecting cellular and humoral immunity (CID), congenital defects of phagocyte number or function, complement deficiencies and other PIDs (including syndromic CID, diseases of immune dysregulation, defects in intrinsic and innate immunity, autoinflammatory disorders and PID phenocopies).
Iran was the second country in Asia to report the national PID registry published in 2002 [Citation25], with updated data reported in 2018 [Citation4,Citation26]. In total, 3056 PID patients were reported by the Iranian PID network centers thus describing the largest cohort of patients in Asia and the fifth-largest national cohort of patients globally. The prevalence of PID has been estimated to be around 3.9 per 100000 in the population. Predominantly antibody deficiencies (29.5%) and combined immunodeficiency (17.3%) were the most common PID subcategories. SCID was reported as the most frequent specific disorder (21.1%) followed by CVID (14.9%). Molecular diagnosis was achieved in 33.1% of registered patients in this highly consanguineous cohort (60.1%).
Wang et al. published the first registry of 201 Chinese PID patients emanating from a single center in 2011. During recent years, efforts of different centers from seven main cities in the mainland China registered 2487 patients (the second-largest PID cohort in Asia, estimated prevalence of <0.2:100000, 45.7% genetically diagnosed). Predominantly antibody deficiency disease was the leading PID type with 27.9% of all patients. Agammaglobulinemia was the most common specific PID disorder and mutations in the BTK gene were the most common genetic defect identified in the Chinese PID population (29%) [Citation27,Citation28].
Kilik et al. in 2013 have reported registry data of 1435 immunodeficient patients from Turkey [Citation29]. However, updated data from the country among 6 documenting centers represented 1837 patients in the 2014 report of the ESID registry [Citation2]. The most recent report, however, provided a more border national view on 6392 patients when the common category of PID was predominantly antibody deficiency which was found in 69.1% of cases followed by autoinflammatory disorders (8.6%). Of note, combined immunodeficiencies occupied only 5.7% of patients in this registry. SIgAD and isolated IgG subclass deficiency were found to be the most common disorders amongst predominantly antibody deficiencies. In this registry, delay in diagnosis was calculated around 26.9 months and the consanguinity rate among reported patients was 14.3% [Citation29,Citation30]. Although several PID genes have been discovered in this cohort there is no national report on molecular diagnosis rate [Citation31].
Al-Saud et al. published a registry report from Saudi Arabia with 502 patients in 2015 and identified a prevalence of all PIDs of 7.2 in 100000 children. The most common PID was combined immunodeficiencies (59.7%), followed by predominantly antibody deficiencies (12.3%) and congenital defects of phagocytosis (9.4%). T-B-SCID was the most common combined immunodeficiencies phenotype (17%). The genetic diagnosis was identified in 152 patients (30.2%) from this cohort in which consanguinity was observed in 75% [Citation32,Citation33].
Madkaikar et al. reported 159 PID cases from a tertiary care center in India in 2013 followed by a more recent report describing 778 registered patients. Frequencies of PIDs were as follows: diseases of immune dysregulation (23.1%), phagocytic defects (21.3%) and predominant antibody deficiency (17.1%), Among patients molecular diagnosis was observed in 40.2% and parental consanguinity was recorded in 19.4% [Citation34–Citation36].
During the last decade, other Asian PID cohorts have been reported containing fewer patients each. Al-Herz et al. in 2019, published a registry report of 314 PID patients in Kuwait with the highest genetically solved rate of 69.1% with 78.0% paternal consanguinity (prevalence of PID 20.2 in 100000, predominantly antibody immunodeficiency of 17.8%) [Citation37–Citation39]. In 2013, Ehlayel et al. have reported a single-center registry of 131 pediatric PID patients from Qatar with the lowest number of classical PID diseases and only 23.7% predominant antibody deficiency (prevalence of PID of 1.1 per 100000 children, genetic diagnosis of 36.6%, parental consanguinity of 61.1%) [Citation40]. Although a cohort from Hong Kong also reported 107 genetically solved PID patients, the current number of patients from this national registry is not updated and spans back to 2014 with 180 reported patients [Citation41–Citation43]. Rhim et al. have reported 152 PID patients from Korea up to 2012 with an estimated prevalence of 1.1 per 100000 children with the highest rate of males to females with PID in Asia (~3.6) [Citation34]. Other registries were reported from Oman (140 patients with high proportion of patients with congenital defects of phagocyte 35%) [Citation44], Taiwan (215 patients with 50.6% molecular diagnosis) [Citation13], Malaysia (150 patients) [Citation43,Citation45], Thailand (67 patients) [Citation46], Jordan (53 patients) [Citation47], Singapore (39 patients) [Citation48], UAE (30 patients) [Citation49] and Bangladesh (13 patients) [Citation50].
3.2. Oceania
PID registries in Oceania have been published only from Australia and New Zealand. The first publication was specific to Australia with 534 reported cases in 1997 [Citation9] and the most recent report included both Australia and New Zealand published by Kirkpatrick et al. including 1209 cases in 2007 (, –). The prevalence of PID was estimated at 5.6 per 100000 for Australia, 12.4 for the state of South Australia, and 4.9 for both Australia and New Zealand. The most frequent subcategory of PID was predominately antibody deficiency syndromes (77.4%), followed by combined immunodeficiency (8.9%), hereditary angioedema (4.5%), and defects of phagocyte function (32%). CVID was the most frequent diagnosis (38.4%) of all patients and the genetic diagnosis was established in 223 patients (18.4%) [Citation12].
3.3. Africa
As the highest estimation, 4509 patients have been recorded in Africa mainly from 7 countries (, –). Initially, South African investigators published a series of PID patients at a single children’s hospital in Cape Town. Patients had been diagnosed for 14 years (between 1983 and 1996). The most frequent disorder of 93 total patients was antibody deficiency (59%) and the delayed diagnosis was noted in majority of patients [Citation51]. Later, in 2011, Naidoo et al. updated this data to span a period of 27 years from 1983 to 2009 with 168 patients. Again, antibody deficiency predominated (50.6%) and the second most common disorder was combined immunodeficiency (25.0%). A family history of PID was observed in 11% of the patients and consanguinity was reported only in 1.1% of the cases. Molecular diagnoses were archived in 7.7% of the cohort [Citation52].
In 2015, Mellouli et al published a Tunisian nationwide registry of 710 PID patients spanning 25-years, thus representing the largest national cohort of the continent. The observed prevalence was 4.3 per 100000 in the population. The frequency of combined immunodeficiency disorders was 28.6%, followed by congenital defects of phagocyte (25.4%) and other well-defined immunodeficiency syndromes (22.7%), and antibody deficiency disorders (17.7%). The consanguinity rate of parents was 58.2% and a genetic diagnosis was identified in 13.8% [Citation53].
Egypt was the second country to establish a PID registry in Africa; Reda et al. published a single-center study of PID patients in 2009 with 64 patients. Antibody deficiency with 35.9% of cases was reported as the most abundant group of PID and combined immunodeficiencies were the second common PID category (29.7%). Consanguinity was reported in 62.5% and the diagnostic delay was 29.9 months [Citation54]. Recently updated records from the Maghreb registry demonstrated that the registered patients in the country have risen to 476 with 106 molecularly diagnosed patients (22.2%) [Citation49,Citation55].
The PID registry next to emerge in Africa was from Morocco; Bousfiha et al. reported 424 PID patients registered by 2014. The reported prevalence of PID was 0.81 in 100000 inhabitants. The most prevalent disorders were reported as follows: well-defined immunodeficiency syndromes (27.4%), antibody deficiencies (22.7%) and combined immunodeficiencies (20.6%). Furthermore, agammaglobulinemia accounted for the majority of antibody deficiencies patients (54.7%) and MHC class II deficiency was reported in 41.2% of combined immunodeficiencies. Parental consanguinity was identified in 43.2% of cases and 19.1% of patients had a positive family history [Citation56]. Three recent additions to the Maghreb registry have released their preliminary reports on the PID patients in Algeria, Libya and Sudan with 600, 106 and 72 patients, respectively [Citation49].
3.4. Europe
In this section, registries of main PID disorders were collected from both published national registries of European countries (highest estimation of 40223 patients) and the ESID registry between 1993 and 2014 (, –). That showed the current total patients from the ESID registry were not queried as part of this evaluation and reliance was placed on that, which could be aggregated by a systematic review of publicly available information. The first report of a European national registry was that of Luzi, et al at in 1983 who described 797 registered patients in the Italian Registry for PIDs. The most prevalent PID entity was humoral defects and SIgAD (66.6%) was found as the most frequent disease, followed by T-cell disorders (14.2%) [Citation57]. More recently the number of Italian patients in the ESID registry has reached 1275 [Citation2,Citation58], but this has not been reflected in other publications from this country with no general information regarding the genetic diagnosis of all PID patients [Citation59,Citation60].
The largest cohort in Europe has been reported from France with 5426 patients within the ESID registry [Citation2,Citation58]. In the latest report from the French PID study group from as 2019, 6602 patients were registered (mainly children) in the National Reference Center of PID. In this study, the total consanguinity was reported as 15% of all patients. The most common PIDs were predominantly B-cell deficiencies (42.8%) and T-cell deficiencies (17.4%), respectively. The most frequent PID diseases were CVID (22.8%) and the molecular diagnosis was established in 43.3% of cases [Citation61,Citation62].
Closely following France in magnitude, Shillitoe et al. reported 4758 registered patients from 2008 to 2018 in the United Kingdom PID Registry, suggesting a doubling of the size of cohort compared to previous ESID registry report in 2014. Antibody disorders formed the largest group (59.6%) of registered patients. The most frequent diseases of PIDs were CVID (36.3%), hereditary angioedema (8%) and agammaglobulinemia (5.4%). Consanguinity was reported at 2.9% and a genetic diagnosis was confirmed in 25.3% of the cohort [Citation63–Citation65]. Abuzakouk, et al at in 2005, registered separately 115 patients from 1996 to 2003 within a national registry for PIDs in the Republic of Ireland (higher than those in the ESID registry 2014 with 107 patients). The most prevalent PID entities were immunoglobulin deficiencies (46%) and complement deficiencies (27.8%). The most common diseases were CVID (24.3%) followed by X-linked agammaglobulinemia (21.1%) and C1 inhibitor deficiencies (13.9%) [Citation66].
In 2005 the Polish Working Group for Immunodeficiencies (PWGID) was established and in their first report in 2016, with 4099 recognized PID patients in Poland, demonstrating a prevalence of 10.6 per 100,000 with a dominance of antibody deficiencies (70.2%) representing the largest cohort among Eastern European countries [Citation67].
The next largest registry in Europe belongs to Germany. El-Helou, et al reported 2453 registered patients spaning 2004 to 2019 in a national registry for PIDs [Citation68], the current data within the ESID database, however, has presented to 1981 cases. In this national registry, consanguinity was reported in 8.0% of the total number of patients and antibody deficiencies formed the largest PID group (56.76%). The most frequent diseases of PIDs were CVID (29.6%), followed by hypogammaglobulinemia (13.5%) and the genetic diagnosis was achieved on 43.3% of patients [Citation69].
Matamoros et al. reported 2030 registered patients in the Spanish national registry for PIDs from 1993 to 2001. According to the ESID registry, the number of reported patients has reached 2211 in 2014 [Citation2,Citation58]. The most prevalent category in this country was antibody disorders (69.1%) mainly due to SIgAD (36.9%) and CVID (19.9%) [Citation18].
Sweden also reported 2727 registered patients in 2019 (most notably with antibody deficiency and phagocytosis disorders, 43.3% and 13.8% respectively) [Citation70,Citation71]; however, due to the inconsistency with the ESID registry, there are only 92 patients included in the continent registry [Citation63,Citation64]. Similarly, other Nordic countries also reported their registries of PID patients separately from ESID registry including Iceland with 66 patients (39.4% antibody deficiency, 33.3% genetic diagnosis, with the highest estimated prevalence of PID in the world ~18:100000) [Citation72], Norway with 372 patients (50.8% antibody deficiency, 8.8% genetic diagnosis) [Citation10,Citation73], Denmark with 179 patients (100% antibody deficiency, 1.6% genetic diagnosis) [Citation74] and Finland with 132 patients (100% antibody deficiency, although in the recent report they did not include previous Finish patients with other PID entities) [Citation75].
Registries from Croatia (226 vs. 24 patients), Czech (518 vs. 259 patients), Portugal (208 vs. 76 patients), Slovenia (272 vs. 16 patients), Slovakia (227 vs. 63 patients), Estonia (99 vs. 42 patients), Belarus (436 vs. 67 patients), Romania (222 vs. 11 patients), Lithuania (172 vs. 8 patients), Hungary (730 vs. 404 patients), Austria (312 vs. 105 patients), Azerbaijan (100 vs. 0 patients), Bulgaria (113 vs. 0 patients), and Israel (294 vs. 4 patients) are not updated in the ESID database and therefore national publications were referred to for the most accurate estimation. In contrast, the ESID registry is the only source for extraction of nationally diagnosed PID patients in many Eastern European countries including Serbia (88 patients), Ukraine (33 patients), as well as the most updated data from Belgium (258 patients), Greece (202 patients), Russia (204 patients), Switzerland (352 patients) and the Netherlands (743 patients) [Citation2,Citation58,Citation76] for which it has been included in these analyses as such ().
3.5. North and South America
The first registry in the United States dates back in 1993 and the first report of this initiative was published in 2000 with only 368 patients. Afterward, following the formation of United States Immunodeficiency Network (USIDNET) for developing a research consortium to advance research in the field of PID, the recent output from their online website presented in 2020 described 5484 diagnosed patients (45.8% genetically diagnosed). The most common diseases of PIDs were CVID (35%), IgA deficiency (26%), IgG subclass deficiencies (9%) and SCID (9%) [Citation77]. Moreover, based on the JMF Physician Education and Public Awareness Campaign (PEPAC) in 2018, 30,227 patients in the United States were identified with specific PIDs. The patients classified within 8 major PID groups where the most prevalent deficiencies were first humoral immunodeficiencies (57.1%) and syndromic cellular immunodeficiencies were second (15.6%) [Citation78]. The same data source from PEPAC shows the data registered from Canada from where 3047 patients have been documented, although detailed data on the PID category and genetic diagnosis has not yet been published from this country [Citation78].
In a parallel effort, the LASID was established in 1993 and their first report was published in 1998 with collected information on a total of 1428 patients from eight countries [Citation79,Citation80]. They later have issued their second report in 2006 with 3321 patients registered from 12 countries and their online evaluations demonstrated that currently 18 countries are included with a total number of 8146 patients. It was reported that 53.0% had predominantly antibody deficiency and 22.6% had other well-defined PID syndromes as the main disease categories of the region. Genetic diagnosis has been made in 18.0% of the registered patients [Citation16,Citation81–Citation85].
The main registered patients of the LASID are from four national registries with more than 1000 patients including Argentina, Brazil, Mexico and Colombia. Nestor et al reported 1319 PID patients from Argentina in 2007 which has now risen to 2730 patients in the LASID database. Reported frequencies were as follows: humoral (69.5%), phagocyte (4.3%), cellular (3.94%) and complement defects (1.4%). Grumach et al. in 1997 reported data on 166 PID patients over 15 years referred to as a national center in Brazil. Now with 1879 patients, this national cohort stands as the second largest of the region where predominantly antibody deficiencies (60.8%), T cell defects (4.9%), and phagocyte disorders (18.7%) are the main PID categories [Citation86]. SIgAD was recorded in 25.1% and CVID in 20.3% of patients within this registry [Citation87]. Mexico had 1744 patients (36.3% antibody deficiencies) and Colombia had 1073 (46.2% antibody deficiencies) patients, each as of prominent countries of the LASID registry that report their outcomes separately [Citation83,Citation84]. represents the PIDs epidemiologic indexes based on reported cases of national and international registers of North and South American countries with an estimated 42067 patients.
4. Discussion
Via systematic review, we considered all evidence of publicly available PID registries world-over based on national reports from 80 countries as well as all well-known international registries. We considered certain clinical parameters and background factors including frequency of specific PIDs, country-specific prevalence, and percentage of consanguinity. Reports from the PID databases of different countries provide an opportunity to understand regional features and epidemiology of PID globally as well as identifying reasons for clinical and epidemiological variations on different continents. Moreover, through the current exercise, we can evaluate previous measures as well as the global report from JMF and highlight potential underestimation and underreporting in different regions [Citation88].
Asia is the most populous continent with 48 countries, but only 18 of them (37.5%) have the reported data of PID registries. It is notable that nearly half of these countries have not established a national registry, but many do have published PID data from single large medical centers including Qatar, Oman, Korea, Thailand and India which make these data relatively unreliable as prevalence estimates. However, this shortcoming to some extent may be overlooked in small and low population countries, but the global estimation of PID is challenged by these types of data originating from large countries such as China and India [Citation89]. Despite limitations in data, the spectrum of the prevalence of PIDs varies from a low of 0.08:100000 in Bangladesh to a high of 20.2:100000 in Kuwait. Collectively, Middle Eastern countries show higher PID prevalence than other countries from Eastern Asia probably due to higher percentages of parental consanguinity. There were also vast variations in the prevalence of individual PIDs throughout the continent. Predominantly antibody deficiencies were the most frequent subcategory of PID in East Asian countries which is parallel to results of Australia [Citation90], LASID [Citation81] and ESID [Citation11] databases, and also the JMF network (55%) [Citation78], but this percentage was lower in the cohorts from the Middle East (). Combined immunodeficiencies were the most common PID entity in Saudi Arabia (59.7%) and the second most prevalent group of PID in other Middle Eastern countries such as Iran [Citation91], and Kuwait [Citation92], which are all much higher than the ESID, LASID [Citation81] and other Western registries [Citation8,Citation11,Citation18,Citation57], Far-East countries [Citation14,Citation27,Citation45], and Australia [Citation12]. This finding is likely explained by the considerable rate of the parental consanguinity which is reported from the Middle East [Citation32,Citation54,Citation91,Citation92] and related to a high rate of autosomal recessive forms of PIDs [Citation93–Citation96]. The latest JMF data describe 8022 patients from Asian countries and thus this global registry has a gap of 7917 patients from currently reported national registries () demonstrating what is likely to be the partial capture of that network [Citation88].
Diagnostic delay, which is defined as the elapsed time between the onset of PID symptoms and the establishment of diagnosis, can reflect the awareness of PID among physicians and the public. The lowest was reported in Iran with a median of 12 months and the highest was in Malaysia at 3.78 years. There was also an increased male to female sex ratio in all registries, but it was greater than 2 in Thailand, Korea, Malaysia, China and Japan suggesting a greater prevalence of X-linked transmission (). Asian countries report a 25.9% molecular diagnostic rate which is the highest globally and is mainly attributed to the Middle East countries (, ).
Oceania is comprised of Australasia, Melanesia, Micronesia and Polynesia and has a population of 43 million living in 15 countries. Reporting of PID patients, however, is available only from 2 countries (13.2%). Although the regional report presented 1209 patients, the recent JMF global report described 1876 patients from this region indicating the need to update registry reported data from Oceania, probably with the inclusion of countries included in the JMF network but otherwise not part of regional registries. Of note, Oceania has an appropriate percentage of genetic diagnosis compared to other continents with 11.8% of reported patients () [Citation88].
Africa accounts for near 15% of the global population [Citation97], but of its 54 countries, only 7 (12.9%) have published registries of PID patients. Furthermore, only Tunis, Morocco and South Africa have established countrywide registries and thus there is not enough experience to establish a continent-wide landscape of PID for Africa [Citation98]. The recent establishment of the Maghreb registry and African Society of Immunodeficiency (ASID), however, could change this condition in the near future [Citation49,Citation98,Citation99]. The reported prevalence of PID in African countries seems to be underestimated due to the general lack of awareness and knowledge of immunodeficiencies, however, a comparison with the JMF global report in which there were only 1836 patients to the 4509 now reported patients in the Maghreb registry suggests progress is accelerating () [Citation88]. Only 5.5% of all reported patients demonstrated a molecular diagnosis, the lowest continental rate globally. However, in North African countries, the appearance of autosomal recessive forms of PID is more frequent and is likely a result of consanguinity (). The pattern of PID in North Africa (mainly with higher percentages of combined immunodeficiency), however, is dissimilar from that identified in South Africa (, ), possibly owing to higher rates of endogamy, geographical alteration, as well as genetic differences. Moreover, differences can be the result of variable data collection methods and awareness of PIDs between African countries.
As of 2014 the ESID registry contained 19355 patients and represented the most robust initial effort in registration. The database, however, does not cover all 51 European countries (58.8%, mainly missing from Nordic countries), although the estimation of the 2019 database reaches 25023 cases (including Turkish patients) [Citation2,Citation58] and 33503 cases on their online website [Citation100]. The current systematic review, however, estimates several missing patients in the registry from individual national reports (). Compared to the JMF global report with 40223 patients from Europe [Citation88], a gap of over 6720 patients is apparent and thus efforts to pursue and integrate data from the missing countries are pressing [Citation69,Citation76,Citation101]. Adaptive immune deficiencies due to predominantly B-cell deficiencies encompass the main category of PIDs in Europe () [Citation61]. Based upon the national registries of European countries, Iceland with its well documented universal medical system reported the highest prevalence of PIDs globally. This has been hypothesized to be due to a sparse population and a small denominator which might skew the comparison to larger nations [Citation72]. However, all Nordic countries cover accessible and universal healthcare, while they also have regional enrichment of rare variants due to previous ‘genetic bottlenecks’. This has led to the enrichment of so-called founder mutations in various monogenic autosomal recessive genes [Citation102,Citation103]. Also, the legislation of most Nordic countries does not presently allow for data exchange with cross-border registries. This does not serve the needs of rare disease patients nor follow the principles behind European General Data Protection (GDPR). According to GDPR, personal data should be designed to serve mankind. The right to the protection of personal data is not absolute; it must be considered in relation to its function in society and be balanced against other fundamental rights (e.g. equity, freedom of science, social rights, the duty of the society to promote public health), following the principle of proportionality [Citation104]. Comparison with other European countries suggests that on average 5:100000 is a reasonable estimation for the PID prevalence in Europe as a whole [Citation5,Citation10,Citation105,Citation106].
Overall, among European countries included in the ESID database, CVID composed approximately 55% and was the most prevalent PIDs registered, whereas, in older studies performed, SIgAD was more commonly reported. The proportion of CVID is lower in France than reported by national registries than in other European countries such as Germany and the UK. There was also a relatively low observed prevalence of PIDs in adults in France. For the five European countries that reported patient consanguinity, France had the highest with 15% along with a concomitant increase of PIDs with an autosomal recessive inheritance pattern (). In general, national European registries reported average consanguinity of 3.4%. Moreover, based on reported cases in the ESID database performed by Gathmann, et al at 2011 [Citation107] focused on eight countries including France, Spain, Turkey, United Kingdom, Germany, Italy, Poland and Netherlands consanguinity was reported in 8.8%, and 18.5% of patients were reported to be familial cases. Genetic diagnosis is only 15.5% of clinically diagnosed patients with Belarus leading (57.3% definite diagnosis) followed by Slovenia (57.0% definite diagnosis) and France (40.0% definite diagnosis, ). The discrepancy amongst European countries suggests substantive opportunity to more broadly advance genetic diagnosis in Europe [Citation76].
Unlike prevalence estimations in many countries based upon their registry reports and diagnosed cases, in the United States, PID epidemiological indices were estimated via a very large geographically and racially diverse population. The USIDNET provides one of the most updated and advanced platforms of evaluation of patients in the country, but a recent report from JMF showed that the participant centers in this database were incomplete (5484 vs. 30227, 24743 missing patients) [Citation88,Citation108]. Moreover, the JMF registry is the only one that covers PID in Canada, a country with 37 million inhabitants and 3047 patients. In contrast, the estimates from Latin American countries via the LASID database are more consistent with data presented in the JMF global report (8146 vs. 8793; only 647 missing patients) [Citation82,Citation88]. Reports from collaborative LASID countries demonstrate that in Latin America the most frequent form of PID is predominantly antibody deficiency, but less frequently than that observed in North America. Other observed differences, however, such as high numbers of patients with CID and MHC class II deficiency in Costa Rica’s and Peru and DiGeorge syndrome in Chile require further evaluation (). The comparison of PID prevalence (5:100000 vs 2:100000) and genetic diagnostic percentages (45.8% vs.18.0%) between North American countries and Latin American countries emphasizes a requirement for in-depth efforts to improve the PID registration quantity and quality.
5. Expert opinion
While many PIDs can be easily diagnosed and have effective treatment options available, awareness of PIDs and their management is low amongst both physicians and the general public in many countries leaving numerous patients undiagnosed. The number of undiagnosed cases is not known, however, because there is no population-based screening process for PID except newborn screening for SCID in certain countries. However, in the present systematic approach as shown in , we estimated possible reported patients of 104614 based on registry reports and diagnosed cases that represent at least 10590 missing patients compared to the JMF global report [Citation88]. The Jeffrey Modell Centers Network (JMCN) consists of 821 Expert Physicians at 379 institutions, in 294 cities, and 86 countries spanning 6 continents. The JMF registry compiles significant data received directly from these reporting Centers from around the world. Of note, the number of individual country registries included in the analysis for each region in this systematic review may not be the same as the number of countries included in the JMF registry for each region. Similarly, the specific countries reporting may vary as well. Still, this number based on registrations remains underestimated as if we simply calculated the incidence of SCID disorders using estimates form newborn screening (1:50000 individuals in a non-consanguineous population [Citation109]) there should be at least 1540000 SCID patients worldwide. Several studies attempted to estimate the total number of PID patients based on hospital and population-based measurements [Citation37]. The upper estimates suggest 6–8 million people may be living with a PID worldwide (based on US population and hospital prevalence [Citation77,Citation110]). It is also estimated that more than 700000 new PID patients might be diagnosed annually (based on US population incidence [Citation111]). In line with the total number of patients we reported based on our systematic review, the majority of experts in the PID filed reported that unfortunately a large portion of PID patients (70–90%) remain undiagnosed and do not have access to appropriate treatment, even in countries with existing PID facilities (the National Institutes of Health [NIH] estimates that as a group, PID may affect 1–2% of the population) [Citation37,Citation112]. The main reasons for high rates of undiagnosed PID patients include a low level of awareness of symptoms of PID among physicians, failure to consider this diagnosis and differences in diagnostic capabilities and treatment of PIDs from country to country on different continents and even within the same region [Citation113]. It should be noted that many PIDs can be diagnosed easily with two simple blood tests (whole blood counts and immunoglobulin levels which diagnose antibody deficiencies and classical immunodeficiencies with a severe lack of lymphocytes or granulocytes) [Citation114]. Prompt PID diagnosis results in better use of health facilities, surveillance of the risk that PID patients can expose for society (e.g. reservoir of Poliovirus challenging the Polio Eradication program [Citation115]) and has been demonstrated to result in lower healthcare costs overall [Citation116].
Both this systematic review and the JMF global report are in agreement that antibody deficiency (51.9% vs. 45.1%, respectively) and combined immunodeficiency (11.8% vs. 6.1%, respectively) are the most often reported PIDs in the world. The number of males affected by PID is slightly higher than females, according to both systematic review and JMF global report (1.5 vs. 1.3, respectively) representing the significant role of X-linked PID disorders in all registries except the UK and Iceland (, ). Parental consanguinity was positive in 6.2% of patients which is less than current estimates of global populations in the world (20%) [Citation117], indicating the low contribution and presence of established PID registries in the countries with consanguinity (). Where present, however, parental consanguinity demonstrates the increased frequency of autosomal recessive PID. In total, 13.2% of patients received a final molecular diagnosis demonstrating the shifting landscape of PID globally (). In an effort to increase the molecular identification of PID patients and significantly increase the genetic diagnostic rate, the JMF is about to embark on a global genetic sequencing initiative. Their current data on global gene defects associated with PID showed that the chromosome 22q11.2 deletion syndrome (5215 cases), MEFV deficiency (2835 cases) and ATM deficiency (2514 cases) are the most common genetic defects worldwide [Citation88]. Collective information and reports on variants are important for long-term therapeutic strategies based on countries or geographical regions. This initiative aims to provide accurate diagnoses, leading to appropriate management and treatment, and ultimately improved patient outcomes and quality of life [Citation118]. Recruitment strategies of PID patients in the national and continent based cohorts should be revised to detect quicker patients with multidisciplinary care for patients presenting different types of infectious or noninfectious complications. Moreover sustainable funding should be provided for long-term maintenance of the currently available databases (societies, registries and websites) and to support highly specialized therapies (e.g. JMF and RITA European research network) [Citation88,Citation119]. Although current data presented cover registries only from 80 countries, the JMF report covers more than 86 countries demonstrating the higher potential for the registry which should be pursued to improve current understanding of PID and increase the chances for improved management. The global burden of PID is clearly underestimated and improved registration efforts could help in more accurately estimating the burden of the disease and allowing for the allocation of appropriate resources for these complex heterogeneous genetic disorders.
Article highlights
Globally, there are more than 6 million children under the age of 15 years die annually despite substantial progress in vaccination, adequate nutrition, exclusive breastfeeding, reduction of household air pollution, providing safe water and food and adequate sanitation and hygiene worldwide. Leading causes of death in children include severe infectious diseases (i.e. pneumonia and diarrhea) which could affect mainly those with underlying immune system defects. Although vaccines are available for some of the most deadly childhood diseases, such as measles, polio, diphtheria, tetanus, and pertussis, pneumonia due to Haemophilius influenzae type B and Streptococcus pneumonia and diarrhea due to rotavirus, they cannot protect immunodeficient children from illness and death.
PIDs are a heterogeneous group of disorders, and no single assay at present will identify all forms of PID, necessitating a challenge of newborn screening paradigms. Therefore, there is no correct estimation of the global PID prevalence in different regions and ethnicities.
This is the first systematic review on PID patients’ registries worldwide summarizing the differences in the distribution and properties of immune system disorders. The authors’ systematic review included 104614 patients registered in 80 countries from five continents.
The authors found that PID patients are at significant risk of misdiagnosis and misclassification in Asia and Africa, the continents with the highest report of childhood death. The findings provide important new insights into the genetic diagnosis of diagnosed patients in different registries and entitlement to molecular approaches worldwide and highlight the need to continue to promote global frameworks for identification of monogenic causes as well as other molecular or environmental modifying factors.
PID burden has been critically underestimated in policymaking and usually, it has been distributed in different categories– mainly infectious diseases, diseases of the blood-forming organs, and neoplasms–because of an inaccurate diagnosis, multiple organ involvement, and different phenotypes. Governments, policymakers, and clinicians must work to develop and enforce measures for early diagnosis of PID, and promote access to appropriate management based on the type of the disease. Existing international frameworks must continue to implement correct clinical, immunological and genetic picture of the PID.
Author contributions
(1) The conception and design of the study
(2) Acquisition of data,
(3) Analysis and interpretation of data,
(4) Drafting the article
(5) Revising it critically for important intellectual content,
(6) Final approval of the version to be submitted
H Abolhassani (2,3,4,5,6), G Azizi, L Sharifi, R Yazdani (2,3,4,6), M Mohsenzadegan, S Delavari, M Sohani, P Shirmast, Z Chavoshzadeh, S Alireza Mahdaviani, A Kalantari, M Tavakol, F Jabbari-Azad, H Ahanchian, T Momen, R Sherkat, M Sadeghi-Shabestari, S Aleyasin, H Esmaeilzadeh (2,3,5,6), W Al-Herz, A Aziz Bousfiha, C Condino-Neto, M Seppänen, K E. Sullivan, L Hammarström, V Modell, F Modell, J Quinn, J S. Orange (1,3,5,6), A Aghamohammadi (1, 3,4,5,6).
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020 Jan;40(1):24–64.
- Seidel MG, Kindle G, Gathmann B, et al. The European Society for Immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J Allergy Clin Immunol Pract. 2019 Jul-Aug;7(6):1763–1770.
- Kobrynski L, Powell RW, Bowen S. Prevalence and morbidity of primary immunodeficiency diseases, United States 2001-2007. J Clin Immunol. 2014 Nov;34(8):954–961.
- Abolhassani H, Kiaee F, Tavakol M, et al. Fourth update on the Iranian National Registry of Primary Immunodeficiencies: integration of molecular diagnosis. J Clin Immunol. 2018 Oct;38(7):816–832.
- Gathmann B, Grimbacher B, Beaute J, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006-2008. Clin Exp Immunol. 2009 Sep;157(Suppl 1):3–11.
- Abolhassani H, Hammarstrom L, Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood. 2020 Feb 27;135(9):656–667.
- Abolhassani H, Aghamohammadi A, Fang M, et al. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med. 2019 Jan;21(1):243–251.
- Fasth A. Primary immunodeficiency disorders in Sweden: cases among children, 1974-1979. J Clin Immunol. 1982 Apr;2(2):86–92.
- Baumgart KW, Britton WJ, Kemp A, et al. The spectrum of primary immunodeficiency disorders in Australia. J Allergy Clin Immunol. 1997 Sep;100(3):415–423.
- Stray-Pedersen A, Abrahamsen TG, Froland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol. 2000 Nov;20(6):477–485.
- Eades-Perner AM, Gathmann B, Knerr V, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004-06. Clin Exp Immunol. 2007 Feb;147(2):306–312.
- Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007 Sep;27(5):517–524.
- Lee WI, Huang JL, Jaing TH, et al. Distribution, clinical features and treatment in Taiwanese patients with symptomatic primary immunodeficiency diseases (PIDs) in a nationwide population-based study during 1985-2010. Immunobiology. 2011 Dec;216(12):1286–1294.
- Ishimura M, Takada H, Doi T, et al. Nationwide survey of patients with primary immunodeficiency diseases in Japan. J Clin Immunol. 2011 Dec;31(6):968–976.
- Hayakawa H, Iwata T, Yata J, et al. Primary immunodeficiency syndrome in Japan. I. Overview of a nationwide survey on primary immunodeficiency syndrome. J Clin Immunol. 1981 Jan;1(1):31–39.
- Condino-Neto A, Sorensen RU, Gomez Raccio AC, et al. Current state and future perspectives of the Latin American Society for Immunodeficiencies (LASID). Allergol Immunopathol (Madr). 2015 Sep-Oct;43(5):493–497.
- Modell V, Gee B, Lewis DB, et al. Global study of primary immunodeficiency diseases (PI)–diagnosis, treatment, and economic impact: an updated report from the Jeffrey Modell Foundation. Immunol Res. 2011 Oct;51(1):61–70.
- Matamoros Flori N, Mila Llambi J, Espanol Boren T, et al. Primary immunodeficiency syndrome in Spain: first report of the National Registry in children and adults. J Clin Immunol. 1997 Jul;17(4):333–339.
- Gathmann B, Mahlaoui N, Ceredih, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014 Jul;134(1):116–126.
- Massaad MJ, Zainal M, Al-Herz W. Frequency and manifestations of autoimmunity among children registered in the Kuwait National Primary Immunodeficiency Registry. Front Immunol. 2020;11:1119.
- Esser MJCA. Immunology C. Primary immunodeficiency-missed opportunities and treatment challenges. Curr Allergy Clin Immunol. 2012;25(4):184–188.
- Cirillo E, Cancrini C, Azzari C, et al. Clinical, immunological, and molecular features of typical and atypical severe combined immunodeficiency: report of the Italian Primary Immunodeficiency Network. Front Immunol. 2019;10:1908.
- Boton Pereira DH, Primo LS, Pelizari G, et al. Primary immunodeficiencies in a Mesoregion of Sao Paulo, Brazil: epidemiologic, clinical, and geospatial approach. Front Immunol. 2020;11:862.
- [cited 2020 Jul 30].https://lasidregistry.org/view/statistics/general/2019-05
- Aghamohammadi A, Moein M, Farhoudi A, et al. Primary immunodeficiency in Iran: first report of the National Registry of PID in children and adults. J Clin Immunol. 2002 Nov;22(6):375–380.
- Aghamohammadi A, Mohammadinejad P, Abolhassani H, et al. Primary immunodeficiency disorders in Iran: update and new insights from the third report of the national registry. J Clin Immunol. 2014 May;34(4):478–490.
- Wang LL, Jin YY, Hao YQ, et al. Distribution and clinical features of primary immunodeficiency diseases in Chinese children (2004-2009). J Clin Immunol. 2011 Jun;31(3):297–308.
- Xia Y, He T, Luo Y, et al. Targeted next-generation sequencing for genetic diagnosis of 160 patients with primary immunodeficiency in south China. Pediatr Allergy Immunol. 2018 Dec;29(8):863–872.
- Kilic SS, Ozel M, Hafizoglu D, et al. The prevalences [correction] and patient characteristics of primary immunodeficiency diseases in Turkey–two centers study. J Clin Immunol. 2013 Jan;33(1):74–83.
- Cekic S, Metin A, Aytekin C, et al. The evaluation of malignancies in Turkish primary immunodeficiency patients; a multicenter study. Pediatr Allergy Immunol. 2020 Feb 14. PMID: 32060950. DOI:10.1111/pai.13231
- Sanal O, Tezcan I. Thirty years of primary immunodeficiencies in Turkey. Ann N Y Acad Sci. 2011 Nov;1238:15–23.
- Al-Saud B, Al-Mousa H, Al Gazlan S, et al. Primary immunodeficiency diseases in Saudi Arabia: a tertiary care hospital experience over a period of three years (2010-2013). J Clin Immunol. 2015 Oct;35(7):651–660.
- Al-Mousa H, Abouelhoda M, Monies DM, et al. Unbiased targeted next-generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol. 2016 Jun;137(6):1780–1787.
- Rhim JW, Kim KH, Kim DS, et al. Prevalence of primary immunodeficiency in Korea. J Korean Med Sci. 2012 Jul;27(7):788–793.
- Jindal AK, Pilania RK, Rawat A, et al. Primary immunodeficiency disorders in India-A situational review. Front Immunol. 2017;8:714.
- [cited 2020 Jul 30].http://ispid.org.in/
- Bousfiha AA, Jeddane L, Ailal F, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. 2013 Jan;33(1):1–7.
- Al-Herz W, Chou J, Delmonte OM, et al. Comprehensive genetic results for primary immunodeficiency disorders in a highly consanguineous population. Front Immunol. 2018;9:3146.
- Al-Herz W, Al-Ahmed M, Al-Khabaz A, et al. The Kuwait National Primary Immunodeficiency Registry 2004-2018. Front Immunol. 2019;10:1754.
- Ehlayel MS, Bener A, Laban MA. Primary immunodeficiency diseases in children: 15 year experience in a tertiary care medical center in Qatar. J Clin Immunol. 2013 Feb;33(2):317–324.
- Lam DS, Lee TL, Chan KW, et al. Primary immunodeficiency in Hong Kong and the use of genetic analysis for diagnosis. Hong Kong Med J. 2005 Apr;11(2):90–96.
- Lau Y. Asian network for molecular diagnosis of primary immunodeficiencies. BMC Proc. 2011;5(Suppl 1):O4.
- Lee P, Lau Y-L. Chapter 55 - Considerations for primary immune deficiency disorders in Asia. In: Sullivan KE, Stiehm ER, editors. Stiehm’s immune deficiencies. Amsterdam: Academic Press; 2014. p. 965–976.
- Al-Tamemi S, Naseem SU, Al-Siyabi N, et al. Primary immunodeficiency diseases in Oman: 10-year experience in a tertiary care hospital. J Clin Immunol. 2016 Nov;36(8):785–792.
- Noh LM, Nasuruddin BA, Abdul Latiff AH, et al. Clinical-epidemiological pattern of primary immunodeficiencies in Malaysia 1987-2006: A 20 year experience in four Malaysian Hospitals. Med J Malaysia. 2013;68(1):13–17.
- Benjasupattananan P, Simasathein T, Vichyanond P, et al. Clinical characteristics and outcomes of primary immunodeficiencies in Thai children: an 18-year experience from a tertiary care center. J Clin Immunol. 2009 May;29(3):357–364.
- Habahbeh ZM, Abu-Shukair ME, Almutereen MA, et al. Primary antibody deficiencies at Queen Rania Children Hospital in Jordan: single center experience. IJI. 2014 Mar;11(1):49–58.
- Lim DL, Thong BY, Ho SY, et al. Primary immunodeficiency diseases in Singapore–the last 11 years. Singapore Med J. 2003 Nov;44(11):579–586.
- Bousfiha AA, Errami A, Jeddane L, et al. Primary Immunodeficiencies: epidemiology in the Maghreb. Tunis Med. 2018 Oct-Nov;96(10–11):672–677.
- Sazzad HM, Rainey JJ, Mach O, et al. The feasibility of identifying children with primary immunodeficiency disorders: preparation for the polio post-eradication era in Bangladesh. Vaccine. 2012 Aug 3;30(36):5396–5400.
- Eley BS, Hughes J, Cooper M, et al. Primary immunodeficiency diseases at Red Cross War Memorial Children’s Hospital. S Afr Med J. 1997 Dec;87(12):1684–1688.
- Naidoo R, Ungerer L, Cooper M, et al. Primary immunodeficiencies: a 27-year review at a tertiary paediatric hospital in Cape Town, South Africa. J Clin Immunol. 2011;31(1):99–105. Febll.
- Mellouli F, Mustapha IB, Khaled MB, et al. Report of the Tunisian Registry of primary immunodeficiencies: 25-years of experience (1988-2012). J Clin Immunol. 2015 Nov;35(8):745–753.
- Reda SM, Afifi HM, Amine MM. Primary immunodeficiency diseases in Egyptian children: a single-center study. J Clin Immunol. 2009 May;29(3):343–351.
- Galal N, Meshaal S, Elhawary R, et al. Patterns of primary immunodeficiency disorders among a highly consanguineous population: Cairo University Pediatric Hospital’s 5-year experience. J Clin Immunol. 2016 Oct;36(7):649–655.
- Bousfiha AA, Jeddane L, El Hafidi N, et al. First report on the Moroccan registry of primary immunodeficiencies: 15 years of experience (1998-2012). J Clin Immunol. 2014 May;34(4):459–468.
- Luzi G, Businco L, Aiuti F. Primary immunodeficiency syndromes in Italy: a report of the national register in children and adults. J Clin Immunol. 1983 Oct;3(4):316–320.
- [cited 2020 Jul 30]. https://esid.org/Working-Parties/Registry-Working-Party/ESID-Database-Statistics
- Pulvirenti F, Sangerardi M, Plebani A, et al. Health-related quality of life and emotional difficulties in chronic granulomatous disease: data on adult and pediatric patients from Italian Network for Primary Immunodeficiency (IPINet). J Clin Immunol. 2020;40(2):289–298.
- Farruggia P, Fioredda F, Puccio G, et al. Idiopathic neutropenia of infancy: data from the Italian Neutropenia Registry. Am J Hematol. 2019;94(2):216–222.
- group CTFPs. The French national registry of primary immunodeficiency diseases. Clin Immunol. 2010 May;135(2):264–272.
- Mahlaoui N, Picard C, Bach P, et al. Genetic diagnosis of primary immunodeficiencies: a survey of the French National Registry. J Allergy Clin Immunol. 2019;143(4):1646–1649. e10.
- Edgar JD, Buckland M, Guzman D, et al. The United Kingdom Primary Immune Deficiency (UKPID) Registry: report of the first 4 years’ activity 2008-2012. Clin Exp Immunol. 2014 Jan;175(1):68–78.
- Shillitoe B, Bangs C, Guzman D, et al. The United Kingdom Primary Immune Deficiency (UKPID) registry 2012 to 2017. Clin Exp Immunol. 2018 Jun;192(3):284–291.
- Thaventhiran JED, Lango Allen H, Burren OS, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. 2020 Jul;583(7814):90–95.
- Abuzakouk M, Feighery C. Primary immunodeficiency disorders in the Republic of Ireland: first report of the national registry in children and adults. J Clin Immunol. 2005 Jan;25(1):73–77.
- Pac M, Bernatowska E. Comprehensive activities to increase recognition of primary immunodeficiency and access to immunoglobulin replacement therapy in Poland. Eur J Pediatr. 2016 Aug;175(8):1099–1105.
- El-Helou SM, Biegner A-K, Bode S, et al. The German National Registry of Primary Immunodeficiencies (2012-2017). Front Immunol. 2019;10:1272.
- Gathmann B, Goldacker S, Klima M, et al. The German national registry for primary immunodeficiencies (PID). Clin Exp Immunol. 2013 Aug;173(2):372–380.
- Bjorkander JF, Brodszki N, Carlsson Å, et al. Best possible treatment for all patients with Primary Immune Deficiency (PID) in Sweden regardless of social factors, sex, age or residence. J Allergy Clin Immunol. 2017 Feb 1;139(2, Supplement):AB249.
- [cited 2020 Jul 30]. http://pidcare.se/
- Ludviksson BR, Sigurdardottir ST, Johannsson JH, et al. Epidemiology of primary immunodeficiency in Iceland. J Clin Immunol. 2015 Jan;35(1):75–79.
- Stray-Pedersen A, Sorte HS, Samarakoon P, et al. Primary immunodeficiency diseases: genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017 Jan;139(1):232–245.
- Westh L, Mogensen TH, Dalgaard LS, et al. Identification and characterization of a Nationwide Danish Adult Common Variable Immunodeficiency Cohort. Scand J Immunol. 2017 Jun;85(6):450–461.
- Selenius JS, Martelius T, Pikkarainen S, et al. Unexpectedly high prevalence of common variable immunodeficiency in Finland. Front Immunol. 2017;8:1190.
- Sediva A, Bataneant M, Belevtsev M, et al. Primary immunodeficiencies in Central and Eastern Europe-the power of networking report on the activity of the Jeffrey Modell Foundation Centers Network in Central and Eastern Europe. Immunol Res. 2019 Oct;67(4–5):358–367.
- Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin Immunol. 2007 Sep;27(5):497–502.
- Cooper MA, Pommering TL, Koranyi K. Primary immunodeficiencies. Am Fam Physician. 2003 Nov 15;68(10):2001–2008.
- Zelazko M, Carneiro-Sampaio M, Cornejo de Luigi M, et al. Primary immunodeficiency diseases in Latin America: first report from eight countries participating in the LAGID. Latin American Group for Primary Immunodeficiency Diseases. J Clin Immunol. 1998 Mar;18(2):161–166.
- Carneiro-Sampaio M. Chapter 53 - Considerations for primary immune deficiency disorders in South America. In: Sullivan KE, Stiehm ER, editors. Stiehm’s immune deficiencies. Amsterdam: Academic Press; 2014. p. 943–955.
- Leiva LE, Zelazco M, Oleastro M, et al. Primary immunodeficiency diseases in Latin America: the second report of the LAGID registry. J Clin Immunol. 2007 Jan;27(1):101–108.
- Wilson S, Ballas Z, Immunology C. Progression of specific antibody deficiency (SAD) to common variable immunodeficiency (CVID). J Allergy Clin Immunol. 2010;125(2):AB73.
- Pedraza A, Vargas-Rumilla MI, Ramirez-Roa JL. [Registry of primary immunodeficiencies in children at a fourth level hospital. Bogota, 2010-2016]. Revista alergia Mexico. 2018 Oct-Dec;65(4):341–348.
- Lugo Reyes SO, Ramirez-Vazquez G, Cruz Hernandez A, et al. Clinical features, non-infectious manifestations and survival analysis of 161 children with primary immunodeficiency in Mexico: a single center experience over two decades. J Clin Immunol. 2016 Jan;36(1):56–65.
- Costa-Carvalho B, Gonzalez-Serrano M, Espinosa-Padilla S, et al. Latin American challenges with the diagnosis and treatment of primary immunodeficiency diseases. Expert Rev Clin Immunol. 2017 May;13(5):483–489.
- Grumach AS, Duarte AJ, Bellinati-Pires R, et al. Brazilian report on primary immunodeficiencies in children: 166 cases studied over a follow-up time of 15 years. J Clin Immunol. 1997 Jul;17(4):340–345.
- Carneiro-Sampaio M, Moraes-Vasconcelos D, Kokron CM, et al. Primary immunodeficiency diseases in different age groups: a report on 1,008 cases from a single Brazilian reference center. J Clin Immunol. 2013 May;33(4):716–724.
- Modell V, Orange JS, Quinn J, et al. Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol Res. 2018 Jun;66(3):367–380.
- Pilania RK, Chaudhary H, Jindal AK, et al. Current status and prospects of primary immunodeficiency diseases in Asia. Genes Dis. 2020 Mar;7(1):3–11.
- Bousfiha A, Jeddane L, Al-Herz W, et al. The 2015 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2015 Nov;35(8):727–738.
- Aghamohammadi A, Abolhassani H, Mohammadinejad P, et al. The approach to children with recurrent infections. Iran J Allergy Asthma Immunol. 2012 Jun;11(2):89–109.
- Al-Herz W, Zainal ME, Salama M, et al. Primary immunodeficiency disorders: survey of pediatricians in Kuwait. J Clin Immunol. 2008 Jul;28(4):379–383.
- Rezaei N, Aghamohammadi A, Moin M, et al. Frequency and clinical manifestations of patients with primary immunodeficiency disorders in Iran: update from the Iranian Primary Immunodeficiency Registry. J Clin Immunol. 2006 Nov;26(6):519–532.
- Moin M, Aghamohammadi A, Kouhi A, et al. Ataxia-telangiectasia in Iran: clinical and laboratory features of 104 patients. Pediatr Neurol. 2007 Jul;37(1):21–28.
- Shabestari MS, Maljaei SH, Baradaran R, et al. Distribution of primary immunodeficiency diseases in the Turk ethnic group, living in the northwestern Iran. J Clin Immunol. 2007 Sep;27(5):510–516.
- al-Attas RA, Rahi AH. Primary antibody deficiency in Arabs: first report from eastern Saudi Arabia. J Clin Immunol. 1998 Sep;18(5):368–371.
- Gudmastad E. 2013 world population data sheet (PDF). Population Reference Bureau; 2013 [cited 2020 Jul 30]. Available from: www.prb.org
- Erjaee A, Bagherpour M, Van Rooyen C, et al. Primary immunodeficiency in Africa - a review. S Afr Med J. 2019 Sep 10;109(8b):3–11.
- Svensson T, Höglund M, Cherif H. Clinical significance of serum immunoglobulin G subclass deficiency in patients with chronic lymphocytic leukemia. Scand J Infect Dis. 2013;45(7):537–542.
- [cited 2020 Jul 30].https://cci-reporting.uniklinik-freiburg.de/#/
- Marschall K, Hoernes M, Bitzenhofer-Gruber M, et al. The Swiss National Registry for primary immunodeficiencies: report on the first 6 years’ activity from 2008 to 2014. Clin Exp Immunol. 2015 Oct;182(1):45–50.
- Norio R. Finnish disease heritage I: characteristics, causes, background. Hum Genet. 2003 May;112(5–6):441–456.
- Norio R. Finnish disease heritage II: population prehistory and genetic roots of Finns. Hum Genet. 2003 May;112(5–6):457–469.
- [cited 2020 Jul 30]. https://gdpr-info.eu
- Grimbacher B, Party ERW. The European Society for Immunodeficiencies (ESID) registry 2014. Clin Exp Immunol. 2014 Dec;178(Suppl 1):18–20.
- Jonkman-Berk BM, van den Berg JM, Ten Berge IJ, et al. Primary immunodeficiencies in the Netherlands: national patient data demonstrate the increased risk of malignancy. Clin Immunol. 2015 Feb;156(2):154–162.
- Gathmann B, Binder N, Ehl S, et al. The European internet-based patient and research database for primary immunodeficiencies: update 2011. Clin Exp Immunol. 2012 Mar;167(3):479–491.
- Abdou NI, Greenwell CA, Mehta R, et al. Efficacy of intravenous gammaglobulin for immunoglobulin G subclass and/or antibody deficiency in adults. Int Arch Allergy Immunol. 2009;149(3):267–274.
- Pyhtila BM, Shaw KA, Neumann SE, et al. Newborn screening for galactosemia in the United States: looking back, looking around, and looking ahead. JIMD Rep. 2015;15:79–93.
- Rubin Z, Pappalardo A, Schwartz A, et al. Prevalence and outcomes of primary immunodeficiency in hospitalized children in the United States. J Allergy Clin Immunol Pract. 2018 Sep - Oct;6(5):1705–1710e1.
- Joshi AY, Iyer VN, Hagan JB, et al. Incidence and temporal trends of primary immunodeficiency: a population-based cohort study. Mayo Clin Proc. 2009;84(1):16–22.
- Chapel H, Prevot J, Gaspar HB, et al. Primary immune deficiencies - principles of care. Front Immunol. 2014;5:627.
- Hernandez-Trujillo HS, Chapel H, Lo Re V 3rd, et al. Comparison of American and European practices in the management of patients with primary immunodeficiencies. Clin Exp Immunol. 2012 Jul;169(1):57–69.
- de Vries E. European Society for Immunodeficiencies m. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol. 2012 Jan;167(1):108–119.
- Aghamohammadi A, Abolhassani H, Kutukculer N, et al. Patients with primary immunodeficiencies are a reservoir of poliovirus and a risk to polio eradication. Front Immunol. 2017;8:685.
- Dias ALA, da Silva RG, Cunha FGP, et al. Managing costs in primary immunodeficiency: minimal immunophenotyping and three national references. APMIS. 2019 Apr;127(4):228–235.
- Bener A, Mohammad RR. Global distribution of consanguinity and their impact on complex diseases: genetic disorders from an endogamous population. Egypt J Med Human Genet. 2017 Oct 1;18(4):315–320.
- Aghamohammadi A, Montazeri A, Abolhassani H, et al. Health-related quality of life in primary antibody deficiency. Iran J Allergy Asthma Immunol. 2011 Mar;10(1):47–51.
- Papa R, Cant A, Klein C, et al. Towards European harmonisation of healthcare for patients with rare immune disorders: outcome from the ERN RITA registries survey. Orphanet J Rare Dis. 2020 Jan 30;15(1):33.