
ABSTRACT
Introduction
The treatment of cutaneous melanoma has been revolutionized by the development of small-molecule inhibitors targeting the MAPK pathway, including inhibitors of BRAF (BRAFi) and MEK (MEKi), and immune checkpoint blockade antibodies, occurring in tandem. Despite these advances, the 5-year survival rate for patients with advanced melanoma remains only around 50%. Although not designed to alter immune responses within the tumor microenvironment (TME), MAPK pathway inhibitors (MAPKi) exert a range of effects on the host immune compartment that may offer opportunities for therapeutic interventions.
Areas covered
We review the effects of MAPKi, especially BRAFi, on the TME, focusing on alterations in inflammatory cytokine secretion, recruitment of immune cells and their functions, both during response to BRAFi treatment and as resistance develops. We outline potential combinations of MAPKi with established and experimental treatments.
Expert opinion
MAPKi in combination or in sequence with established treatments such as checkpoint inhibitors, anti-angiogenic agents, or new therapies such as adoptive cell therapies, may augment their immunological effects, reverse tumor-associated immune suppression, and offer the prospect of longer-lived clinical responses. Refining therapeutic tools at our disposal and embracing ‘old friends’ in the melanoma treatment arsenal, alongside new target identification, may improve the chances of therapeutic success.
1. Introduction
The incidence of cutaneous melanoma is increasing, with an estimated 300,000 cases reported globally per annum [Citation1]. While melanoma constitutes less than 5% of all skin cancers diagnosed, it remains the most lethal. Within the last decade, there has been significant expansion of the therapeutic arsenal for melanoma with the introduction of molecularly targeted therapies, in the form of mitogen-activated protein kinase (MAPK) pathway inhibitors, and immune modulatory therapies, in the form of checkpoint inhibitors, occurring in parallel [Citation2]. The MAPK pathway has been identified as a critical signaling cascade underlying disease pathogenesis, with 50% of patients presenting with mutations in the signaling molecule BRAF [Citation3]. Mutations in BRAF lead to constitutive activation of the MAPK pathway, promoting cell proliferation and inhibiting apoptosis, thus driving tumorigenesis [Citation4]. Tumors are tested at diagnosis for BRAF mutations, and BRAF inhibitors (BRAFi) including Vemurafenib, introduced in 2011, and Dabrafenib, approved in 2013, initially demonstrated high efficacy as treatments, with a significant proportion of patients showing profound tumor regression. However, these responses are typically short-lived and development of acquired resistance follows [Citation5,Citation6]. This led to the development of MEK inhibitors (MEKi), which target the MAPK pathway downstream of BRAF. The combination of BRAFi and MEKi is now used as first-line treatment for unresectable or metastatic BRAFV600E/K mutant melanoma, leading to greater initial tumor response and delaying MAPK-driven acquired resistance compared with BRAFi treatment alone [Citation7–9]. Although combinations of BRAFi and MEKi improve progression-free survival (PFS) when compared to monotherapy [Citation10], therapeutic resistance remains a challenge.
Melanoma is well recognized as the archetypal immunogenic tumor, and the interactions between melanoma cells and immune cells within the TME contribute to tumor pathogenesis, invasion, and immune evasion [Citation2]. The importance of these interactions has been demonstrated with the success of immune checkpoint inhibition. Immune checkpoint inhibitors (ICIs), developed at the same time as BRAFi, are monoclonal antibodies that target and block the cell-surface marker cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or the programmed death-1 pathway (PD-1/PD-L1). CTLA-4 is expressed on T cells, including regulatory T cells (T-regs), and inhibits T cell activation through binding of CD80 and CD86 expressed on antigen-presenting cells (APCs). Blocking this interaction with the antibody Ipilimumab results in T cell activation and enhanced effector function [Citation11]. PD-L1 is highly expressed by tumor and stromal cells within melanoma lesions [Citation12], while PD-1 is expressed on the T cell surface [Citation11]. PD-L1/PD-1 interaction leads to negative regulation of T cells. Anti-PD-1 antibodies, such as Nivolumab or Pembrolizumab, and antibodies against PD-L1, such as Atezolizumab, inhibit PD-L1/PD-1 signaling, allowing T cell activation and reducing the number of exhausted T cells within the TME, augmenting anti-tumor immune responses [Citation11,Citation13]. The introduction of ICIs has resulted in significant improvements in PFS in advanced melanoma with up to 50% of patients experiencing unprecedented durable responses at 5 years following treatment with Ipilimumab and Nivolumab [Citation14,Citation15]. The success of ICIs, including in BRAF mutant melanoma [Citation16], demonstrates that the manipulation of the immune system is a valuable therapeutic tool.
Although MAPK pathway inhibitors (MAPKi) were not designed to manipulate the immune system, they too appear to exert a range of immunological effects, which forms the focus of this review. Understanding the interactions between BRAF mutant melanoma cells and immune cells, both during response to MAPKi treatment and as resistance develops, may uncover new therapeutic avenues, to ultimately lead to more sustained responses and improved clinical outcomes. This may include combining new treatments with MAPKi with the aim of identifying new synergistic effects or optimizing the combination and sequencing of already successful therapies, including ICIs.
2. The MAPK pathway in melanoma and targeting with inhibitor drugs
The MAPK pathway is a family of intracellular signaling pathways activated through the binding of growth factors to receptor tyrosine kinases (RTK) [Citation17]. Downstream events linked to these pathways include cellular differentiation, proliferation, apoptosis, and regulation of both innate and adaptive immune responses [Citation18,Citation19].
The pathway is arranged in three tiers containing sequentially acting serine threonine kinases: MAP kinase kinase kinase (MAPKKK), MAP kinase kinase (MAPKK), and a downstream effector MAP kinase (MAPK) [Citation20]. Sequential phosphorylation of these kinases (outlined in ) leads to the phosphorylation of downstream targets in the cytosol and nucleus, which mediate biological responses [Citation18]. To prevent persistent activation of MAPK, upstream negative feedback occurs. In the absence of RTK-ligand engagement, members of the MAPK pathway remain in their non-phosphorylated, inactive states, preventing downstream signaling.
Figure 1. The mitogen-activated signaling pathway, from left to right: a) normal function of MAPK pathway signaling; b) BRAFV600E mutation leading to constituently activated MAPK signaling and enhanced pathogenesis of melanoma; c) targeting of MAPK pathway, with BRAFi and MEKi, reducing melanoma pathogenicity; and d) mechanisms by which resistance can develop. Created with BioRender.com.
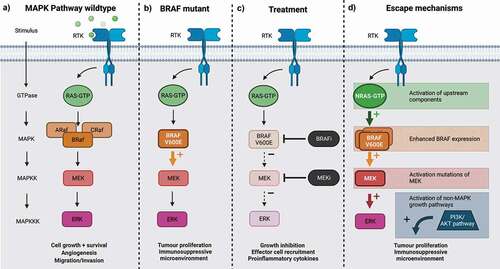
Aberrant activation of the MAPK pathway is a central step in melanoma pathogenesis [Citation21], often driven by activating mutations of RAF (MAPKKK). The RAF family consists of ARAF, BRAF, and CRAF. While mutants of ARAF and CRAF are rare, activating mutations of BRAF are found in different malignancies including colorectal cancer, ovarian cancer, lung cancer, and melanoma [Citation22]. The most common BRAF mutation, accounting for 90% of melanomas containing MAPK mutations, BRAFV600E, remains active in the absence of RAS, resulting in a constitutively active MAPK pathway that can drive aberrant cell growth and proliferation [Citation4,Citation18], and influence the TME [Citation23]. This makes the MAPK pathway a valuable therapeutic target for melanoma.
Selective BRAF inhibitors with a high affinity for the BRAFV600E mutant include Vemurafenib, Dabrafenib, and Encorafenib [Citation23]. Preclinical data for Vemurafenib (PLX4032) and Dabrafenib (GSK2118436) demonstrated significant antitumor activity against BRAFV600E mutant melanoma cell lines, including G1 cell cycle arrest, apoptosis, and blockage of ERK phosphorylation, reducing proliferation [Citation23,Citation24]. Both were also shown to inhibit other BRAFV600 mutant cell lines without inhibiting either wild-type BRAF or non-V600 BRAF mutants [Citation24,Citation25].
Encorafenib (LGX818), a second-generation BRAFi, produced prolonged target suppression and efficacy compared to first-generation BRAFi such as Vemurafenib and Dabrafenib [Citation26]. Studies in xenograft mouse models of BRAFV600E expressing melanoma revealed that exposure to Vemurafenib, Dabrafenib, or Encorafenib resulted in a dose-dependent inhibition of tumor growth and in some cases tumor regression [Citation24,Citation25,Citation27]. Clinical trials of BRAFi monotherapy demonstrated high rates of objective responses, improved PFS, and overall survival (OS) when compared to conventional cytotoxic chemotherapy [Citation28]. Despite these results, a median PFS of only 6–7 months was seen on BRAFi monotherapy due to both intrinsic and acquired resistance and subsequent relapse [Citation5,Citation6,Citation29].
Resistance to BRAFi monotherapy is mediated by the reactivation of the MAPK pathway, which can occur through MAPK-dependent or MAPK-independent mechanisms. Common patterns of resistance include activating mutations of upstream components (NRAS mutants) and downstream components (MEK mutants) [Citation30,Citation31], activation of non-MAPK growth pathways (phosphatidylinositol-3-kinase/AKT pathway) [Citation32], splice variants of BRAFV600E [Citation33], and enhanced BRAF expression [Citation34]. Vemurafenib, Dabrafenib, and, to a lesser extent, Encorafenib can all cause RAS-dependent paradoxical activation of the MAPK pathway, particularly in those tumors with preexisting RAS mutations [Citation35,Citation36]. Paradoxical activation is associated with hyperactive MAPK signaling, resulting in the emergence of hyperproliferative cutaneous events [Citation37]. Resistance may also be precipitated by BRAFi treatment, creating an environment that favors the survival of non-BRAFV600 cells [Citation35].
Preclinical studies demonstrate that acquired resistance to BRAFi therapy was associated with rapid recovery of MAPK signaling; this suggests that complete pathway inhibition may be required for therapeutic effect and thus MEK inhibitors (MEKi) are now used as first-line treatment in combination with BRAFi. This combination allows further inhibition of the MAPK pathway downstream of BRAF, and thus long-term inhibition of ERK (MAPK). Three phase III clinical trials demonstrated the clinical efficacy of BRAFi/MEKi combination therapy: the COLUMBUS trial compared the combination of Encorafenib + Binimetinib to Vemurafenib or Encorafenib monotherapy; and the COMBI-d and COMBI-v trials compared Trametinib and Dabrafenib combination therapy to Dabrafenib monotherapy. All demonstrated significant and clinically meaningful differences in OS when combinations were used, leading to these combinations to be approved as first-line therapy [Citation38,Citation39]. Furthermore, MEKi monotherapy was shown to block BRAFi-induced hyperproliferative cutaneous events in squamous cell carcinoma mouse models, indicating that addition of MEKi may have a role in preventing both BRAFi resistance and paradoxical MAPK activation [Citation40].
As well as directly contributing to melanoma cell growth and survival, a constitutively active MAPK pathway contributes to the immunosuppressive TME seen in melanoma tumors. Inhibiting this pathway with BRAFi and MEKi can therefore have immunomodulatory effects, not just by targeting MAPK signaling in melanoma cells but by causing paradoxical activation in immune and stromal cells within the TME. The immunomodulatory activity of BRAFi may help to overcome the immunosuppressive microenvironment found in BRAF mutant melanomas [Citation23], an effect reversed with inhibitor resistance.
3. Immunosuppressive environment created by BRAF mutant melanoma
BRAF mutant melanomas can create a TME that promotes both tumor growth and immune evasion by a range of mechanisms, including the secretion of immunosuppressive factors and the recruitment of immunoregulatory cells, outlined in .
Figure 2. Potential immunological effects of BRAFi, BRAFi resistance, and mechanisms to therapeutically enhance BRAFi: a) The immunosuppressive TME created in BRAF mutant melanoma includes the recruitment and expansion of immunosuppressive cells, effector cells with reduced cancer killing potency and the secretion of factors by immune cells that can increase cancer growth and angiogenesis. b) The proinflammatory TME created by BRAFi includes increased effector immune cell recruitment and expansion, reduced immunosuppressive immune cell recruitment, and increased ability of immune effector cells to trigger cancer killing. c) As BRAFi resistance develops, the reversal of BRAFi immune effects is observed. d) There are multiple mechanisms by which BRAFi resistance can be overcome using current and future therapies. These include depleting immunosuppressive cells such as tumor-associated macrophages (TAMs), targeting angiogenesis and growth promoting vascular endothelial growth factor (VEGF), targeting immune exhaustion molecules with checkpoint inhibitors (PD-1/PD-L1), and enhancing cancer killing with adoptive cell therapy (ACT), including the use of chimeric antigen receptor T cell therapies (CAR-T), as well as stimulating effector cells, for example, by using toll-like receptor agonists (TLR). Created with BioRender.com.
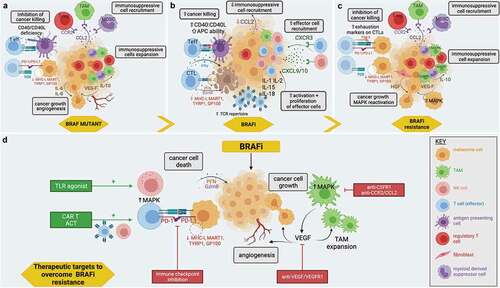
BRAF mutant melanomas may recruit immunosuppressive immune cells via the secretion of chemokines, such as CCL2 [Citation41]. The receptor for CCL2, CCR2, is expressed by myeloid-derived suppressor cells (MDSCs), regulatory T cells (T-regs), and monocytes, the latter of which are able to differentiate into macrophages upon entry into tumor tissue [Citation42]. Together, these cells aid the establishment of a protumor TME. For example, tumor-associated macrophages (TAMs) are skewed towards a more immunoregulatory phenotype in BRAF mutant melanoma, as demonstrated by the expression of marker genes such as Arg1, Chi3l3, Mr1, and Mmp9 [Citation43]. TAMs secrete factors such as IL-10, which can promote the expansion of T-regs and establish a positive feedback loop with further polarization of TAMs toward an immunosuppressive phenotype [Citation44]; vascular endothelial growth factor (VEGF), which stimulates tumor growth and angiogenesis and promotes immunosuppressive TAMs [Citation45]; and metalloproteases, which aids tumor growth and invasion [Citation44]. As well as the recruitment of immunosuppressive cells, which secrete regulatory factors such as IL-6, IL-8, IL-10, and TGF-β [Citation46,Citation47], cells within the TME demonstrate altered cell–cell signaling, for instance, through impaired CD40:CD40L interactions. This prevents the maturation of APCs and CD8+ T cell activation [Citation43]. The resultant effect of the combination of all the above mechanisms is increased immune evasion and therefore enhanced pathogenicity of melanoma lesions.
4. The immune effects of BRAF inhibition
Although not specifically designed to trigger anti-tumor immune responses, several preclinical and clinical studies (summarized in ) suggest that MAPK pathway inhibition can alter the TME to improve anti-melanoma immune responses. It has been suggested that these effects may contribute to the anti-tumor efficacy of BRAFi. Indeed, immune-related adverse events (irAEs), such as vitiligo and panniculitis, have been postulated as a predictive marker of response to BRAFi treatment [Citation48]. The effects of BRAF inhibition on cancer cell death may lead to the release of danger signals and of cancer-associated antigens. These could enhance anti-tumor immune responses by: (1) increasing inflammation and effector immune cell recruitment, e.g., cytotoxic T lymphocytes (CTLs); (2) altering the balance of cytokines to promote a more proinflammatory TME; and (3) enhancing the effector function of such immune cells, for example, by promoting antigen presentation. These effects are outlined in .
Table 1. BRAFi effect on immune function: preclinical data of the effect of BRAFi on the tumor microenvironment, specifically changes in immune mechanisms and pathways. TILS – tumor infiltrating lymphocytes; NK cells – natural killer cells; T-regs – regulatory T cells; MDSCs – myeloid-derived suppressor cells; APCs- antigen-presenting cells
4.1. BRAF inhibition and immune cell recruitment
Multiple studies, in samples from both preclinical models of cancer and from patients undergoing BRAFi therapy, have shown that BRAFi promote tumor infiltration by lymphocytes [Citation41,Citation43,Citation46,Citation49,Citation50]. Increased lymphocyte infiltration of melanoma, in particular CD8+ T cells, is correlated with more favorable outcomes [Citation51]. BRAFi treatment has been associated with an increased ratio of cytotoxic CD8+ to CD4+ T cells, the latter of which may include immunosuppressive T-regs [Citation41]. As well as promoting CTL infiltration, one small study looking at sequential melanoma biopsies demonstrated a direct effect of BRAFi on the expansion of T cell populations within the TME [Citation52]. Together, these mechanisms may create a T cell population with a richer and more diverse T cell receptor repertoire [Citation52]. Furthermore, immunosuppressive MDSC populations decrease with BRAFi [Citation43,Citation53,Citation54]. BRAFi cause a reduction in CCL2 secreted by tumor cells [Citation41]. Although CCR2 is expressed on T-regs and MDSCs, it is not expressed on natural killer (NK) cells and CD8+ T cells [Citation41]. The reduction in CCL2 caused by BRAFi, therefore, may support the preferential recruitment of effector cells over immunoregulatory cells, aiding the anti-tumor response. Aside from changes in the CCL2/CCR2 chemokine pathway, BRAFi can also increase the expression of the chemokines CXCL9 and CXCL10, which promote the recruitment of NK cells and T cells [Citation50,Citation55]. Together these effects compound a change in the TME toward one which promotes anti-tumor responses.
Interestingly, the immune cell compartment can also be interrogated to predict response to MAPKi. One study of pre-treatment biopsies of melanomas from patients who subsequently were treated with BRAFi, with or without additions MEKi, demonstrated a correlation between high intratumoural CD8+ T cells with concomitantly low CD163+ myeloid cells, likely representing TAMs, and higher probability of response as well as longer PFS and OS, when compared to those with fewer infiltrating CD8+ T cells and high frequency of TAMs [Citation56].
4.2. Changing the balance to create a more proinflammatory and less immunoregulatory TME
As well as altering the immune cell compartment, BRAFi can influence the inflammatory milieu of the TME, upregulating proinflammatory cytokines, such as IL-1 and IL-2, and IL-15 and IL-18, which promote the activation of T cells and NK cells [Citation50,Citation55], while decreasing the secretion of cytokines such as IL-6, which contributes to cancer cell invasion, and IL-8, an important chemokine in melanoma growth, cell motility, invasion, and angiogenesis [Citation46,Citation57,Citation58]. BRAFi can reduce VEGF [Citation55,Citation59,Citation60], a strong angiogenic mediator normally secreted by melanoma and tumor-associated stromal cells. Tumor-associated neovasculature is largely abnormal and tortuous. It can restrict the penetration of drugs, impair the infiltration of immune effector cells [Citation61], and increase hypoxia within the TME which promotes immunosuppressive TAMs, T-regs, and MDSCs [Citation55,Citation62]. Aside from this, VEGF has a direct effect on melanoma cell growth [Citation63]. The reduced expression of VEGF seen with BRAF inhibition, along with the decreased secretion of chemokines that attract ‘protumor’ immune cells, can therefore create a more hospitable environment for effector cell trafficking and effector function against cancer cells [Citation45].
4.3. BRAFi and immune cell function
Not only can BRAFi recruit effector immune cells and create a more favorable environment for their activation and proliferation, but they can also improve the cancer killing mechanisms of such cells. The increased infiltration of CD8+ T cells seen with BRAFi is associated with tumor shrinkage and necrosis and improved survival in preclinical models, and BRAFi have been shown to actively improve CTL function, as measured by an increase of granzyme B and perforin, co-localized with CD8+ T cells [Citation46,Citation49]. A key mechanism driving enhanced CTL function is improved antigen presentation: BRAFi promote the maturation of APCs, such as TAMs and dendritic cells (DCs), through enhanced CD40:CD40L signaling [Citation43]; BRAFi decreases major histocompatibility complex (MHC-I) internalization from the melanoma cell surface, thereby increasing the expression of MHC-I on tumor cells [Citation64]; and BRAF inhibition has been reported to lead to increased expression of melanoma specific antigens: glycoprotein 100 (gp100), melanoma antigen recognized by T cells (MART-1), dopachrome tautomerase (DCT), and tyrosinase-related protein 1 (TYRP1), enhancing immune recognition of melanoma cells [Citation46]. Aside from improving CTL function, it has been demonstrated that CD4+ T effector function (through the secretion of IFNγ) and CD4+ cell helper function (via signaling through CD40L) are increased with BRAFi [Citation43].
5. Development of BRAFi resistance – the contribution of immune cells
Although a favorable TME following BRAFi treatment might suggest the creation of an effective immune response to the tumor, as mentioned, BRAFi resistance is common and typically observed clinically after around 6 months of treatment [Citation10]. An important resistance mechanism is the paradoxical activation of MAPK pathways in BRAF wild-type cells in the TME, including immune and stromal cells, as outlined in . For example, there is evidence to suggest that tumor-associated fibroblasts can be paradoxically activated by BRAFi and MEKi, leading to matrix remodeling [Citation65] and the secretion of growth factors, such as hepatocyte growth factor (HGF), enabling the survival and growth of melanoma cells [Citation66]. Importantly, due to their abundance in the TME and their protumor functions, TAMs can also be paradoxically activated and are thought to be important contributors to BRAFi resistance, secreting growth factors such as VEGF, which can in turn cause reactivation of MAPK pathway and enhance tumor growth [Citation67].
As tumors develop resistance to MAPKi, the initial upregulation of melanoma antigens seen on initiation of BRAFi treatment is reversed, and melanoma antigen expression is reduced [Citation68]. This results in reduced CD8+ cell activation and CTL function. Aside from this, the reactivation of MAPK leads to increased expression of PD-L1 on melanoma cells [Citation69], which can impair T cell activation and aid immune evasion. Despite an initial increase in CD8+ T cell infiltration, at the point of progression on BRAFi, patient samples demonstrate a low immune infiltrate [Citation70], and CD8+ T cells with an exhausted phenotype, with increased TIM-3 (T cell immunoglobulin and mucin domain-containing protein 3) and PD-1 expression [Citation46].
6. Overcoming BRAF resistance
Combinations of BRAFi and MEKi have been used to overcome BRAFi resistance, yet, since both inhibitor types act on the same pathway, resistance still develops by compensatory pathway activation, eventually leading to disease progression. The immunomodulatory effects of MAPKi create the potential for combining BRAFi with treatments that target cancer immunity, to promote a more sustained response to MAPK pathway inhibition, thus prolonging the time to resistance. We outline below potential combinations of both new and established treatments, which could offer the chance of longer-lived responses in patients (summarized in ) and have summarized ongoing clinical trials in .
Table 2. Clinical trials of combination therapies in metastatic melanoma with BRAFi/MEKi with another therapeutic intervention
6.1. Combination with ICIs
The rationale for combining BRAFi/MEKi with ICI is attractive given the frequent but short-lived responses to BRAFi/MEKi, and the less predictable but more durable responses seen with ICIs. Preclinical studies have demonstrated that markers of T cell exhaustion, such as PD-1, are upregulated by BRAFi and MEKi. Combined with the fact that these targeted drugs do not impact negatively on the viability, proliferation, or signaling of human lymphocytes suggests that BRAFi/MEKi could be utilized to prime the tumor and TME for therapy with ICIs [Citation46,Citation71]. Furthermore, there is evidence to suggest that BRAFi may in fact increase the effector function of human lymphocytes, potentially making the TME more amenable to immune activation with ICIs [Citation72]. As discussed, BRAFi can be shown to increase CD8+ T cell tumor infiltration; however, this infiltration diminishes with time. Timing of administration of ICIs may therefore be optimized to coincide with this proinflammatory response to maximize potential anti-tumor efficacy [Citation70]. Studies published to date have focused on concurrent administration of ICI and BRAFi/MEKi, the efficacy of which has been suggested in preclinical models but in practice has been limited by unacceptable toxicity [Citation73,Citation74].
Overlapping side effect profiles have caused several initial trials examining the combination of ICI and BRAFi/MEKi to be discontinued early. A phase I study combining Vemurafenib and Ipilimumab closed to recruitment after six of the first ten patients developed grade 3 hepatic adverse events [Citation75]. A further trial examining the role of doublet therapy of Dabrafenib and Ipilimumab and triplet therapy of Dabrafenib, Trametinib, and Ipilimumab demonstrated that two of the seven patients receiving triplet therapy developed colitis with intestinal perforation [Citation76]. The lower toxicity of BRAFi when given in combination with MEKi, along with the development of anti-PD-L1/PD-1 agents (themselves associated with lower rates of irAEs than CTLA-4 inhibition), has renewed interest in combination therapy, with a number of clinical trials in progress or recently completed.
The KEYNOTE-022 (NCT02130466) trial compared the combination of Dabrafenib, Trametinib, and Pembrolizumab to Dabrafenib, Trametinib, and placebo [Citation77]. While statistical significance was not reached for the primary end point of PFS, there was a numerical trend toward improved PFS. The median duration of responses was also numerically longer in triplet therapy than in the placebo arm. However, this was associated with a 58% rate of grade 3–5 treatment-related adverse events (trAEs), which would not typically be accepted in clinical practice [Citation78]. Further to this, the findings of the TRIDeNT (NCT02910700) study, adding Nivolumab to Dabrafenib and Trametinib, showed encouraging efficacy signals at the cost of a 78% rate of grade 3–4 trAEs [Citation79]. The COMBI-I trial (NCT02967692) did not demonstrate any benefit from the addition of the anti-PD-1 monoclonal antibody Spartalizumab to Dabrafenib and Trametinib, with the authors commenting that adverse event management was challenging and frequent dosing adjustments were required [Citation80].
For BRAFi/MEKi/ICI combinations to be safely used in clinical practice, there is a need to identify predictive biomarkers for toxicity as well as response. Preplanned subgroup analyses within the TRILOGY/IMSpire150 trial (NCT0290867) evaluating combinations of Vemurafenib plus Cobimetinib with or without Atezolizumab () suggested benefit in those patients with high serum LDH, more than three sites of metastasis and M1c disease, the latter referring to metastatic spread to locations other than the central nervous system [Citation81]. It may be that the benefits of anti-tumor efficacy outweigh those of trAEs in patients with a higher disease burden [Citation77,Citation80]. In addition to improved patient selection, modifications to treatment dose and schedule with ‘lead in’ or priming therapy, whereby BRAFi/MEKi are given prior to ICIs, or ‘on–off’ dosing regimens where BRAFi/MEKi are given for short periods while patients are maintained on ICIs or vice versa, may lead to better tolerated regimens and more durable responses.
As discussed, preclinical studies support the principle of lead in/priming therapy with BRAFi/MEKi prior to ICI therapy. In a phase 1b study (NCT01656642) including a BRAFi/MEKi lead in/priming phase with Vemurafenib and Cobimetinib, followed by the addition of the antibody Atezolizumab (anti-PD-L1), CD4+ T cell proliferation was increased without an increase in T-regs, priming the TME for immunotherapy as demonstrated by increased helper T cells and CTLs in the TME following the administration of Atezolizumab [Citation82]. This has been recapitulated in the IMSpire150 trial, which demonstrated an improved median PFS, when Atezolizumab was added after a BRAF/MEKi run in, with little difference in adverse events reported. The US Food and Drug Administration (FDA) have now approved the combination, but it is yet to be approved by the European Medicines Agency (EMA) or the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK [Citation81] ().
The IMPemBra trial (NCT02625337) examined the impact of short-term/intermittent dual MAPK pathway inhibition (Dabrafenib and Trametinib) with Pembrolizumab in the hope of reducing the high frequency of grade 3–4 trAEs in triplet therapy and high rates of treatment discontinuation. This trial demonstrated improved median PFS of patients from 10.6 months in the Pembrolizumab monotherapy arm to 27.0 months in patients with Pembrolizumab combined with short-term intermittent Dabrafenib and Trametinib therapy with improved tolerability compared to traditional triplet therapy [Citation83]. There are ongoing studies exploring the appropriate sequencing of these agents within the DREAMseq (NCT02224781), SECOMBIT (NCT02631447), and NEOTRIO (NCT02858921) studies, and preliminary results have been recently published.
In contrast to preclinical studies, early data from the DREAMseq trial seem to demonstrate that patients, in fact, benefit from ICI treatment prior to BRAF/MEKi. Their unique trial design enrolled patients to either receive Nivolumab/Ipilimumab or Dabrafenib/Trametinib until disease progression, whereby they switched to the other treatment. Superior OS was seen in the patients receiving ICI first, which became evident at 10 months, with more ongoing responses than patients who started with MAPKi [Citation84]. In addition, preliminary results from the phase 2 SECOMBIT trial, which compared a similar treatment sequencing, appears to support the use of ICI prior to MAPK inhibition [Citation85]. In addition to the treatment arms used in DREAMseq, SECOMBIT has a third arm, in which patients receive priming with BRAFi/MEKi for eight weeks, before switching to ICI therapy until disease progression, and switching back to treatment with BRAFi/MEKi thereafter. This third arm also demonstrated better outcomes compared with patients receiving BRAFi/MEKi until disease progression as their first treatment.
It is important to note that the initial advantageous early infiltration of T cells observed with BRAFi is no longer present at the time of disease progression, with a reduced CD8+ T cell population and a more pronounced protumor, anti-inflammatory TAM gene expression profile [Citation49,Citation70,Citation86]. In order to use BRAFi/MEKi to prime the TME for ICI treatment, a shorter priming phase, such as in the third arm of SECOMBIT, might be included in trials. In this way, it may be possible to capture the effects of MAPKi at an optimum timepoint for activating the patient’s immune response and thus augmenting ICI treatment, but before the onset of resistance.
6.2. Targeting immune cells that contribute to BRAFi resistance: myeloid cells
The importance of the infiltrating myeloid cell compartment in the development of BRAFi resistance has been demonstrated in studies which show how CCL2/CCR2 signaling is restored as BRAFi resistance develops, increasing the numbers of MDSCs and the frequency of TAMs in the TME [Citation87]. Aside from CCL2/CCR2, signaling through colony-stimulating factor 1 receptor (CSF1R), expressed on myeloid cells, is essential for their recruitment and maintenance in the TME. Small-molecule inhibitors against CSF1R in combination with BRAFi have demonstrated a reduction in tumor size in preclinical studies [Citation88]. However, anti-CSF1R is less efficacious than BRAFi when used alone and is also expressed on other myeloid cells that may play beneficial roles in anti-tumor immunity [Citation67]. Despite this, there are a two ongoing early-phase clinical trials exploring the role of CSF1R inhibitors as monotherapy (NCT02071940, NCT02975700) and two in combination with BRAFi and MEKi (NCT03455764, NCT03101254) ().
6.3. Angiogenesis and BRAF resistance in melanoma
Angiogenesis is a recognized hallmark of cancer, and within melanoma it is associated with increased aggressiveness and worse prognosis [Citation89]. The importance of VEGF signaling within tumorigenesis has been demonstrated across multiple cancer types including colorectal, ovarian, and uterine cancers [Citation89]. Antiangiogenic drugs bind either angiogenic factors (e.g., VEGF-A) or their receptors (e.g., VEGFR1/2). Antiangiogenic agents not only remodel irregular and leaky vessels in cancer lesions, resulting in improved tumor penetration of chemotherapy agents, but may also impact on the TME through relieving tissue hypoxia, which can alter immune cell composition [Citation89]. The first trials involving antiangiogenic drugs within melanoma date back to almost two decades ago, when chemotherapy was the only available treatment for advanced stage disease; these trials often involved small population sizes [Citation89]. Prior to the introduction of targeted and ICIs, the FDA, but not the EMA or the MHRA, had approved Bevacizumab, a monoclonal antibody recognizing VEGF, plus cytotoxic chemotherapy as a first-line treatment for unresectable melanoma [Citation90,Citation91]. There is significant interest in reexamining the role of VEGF in optimizing current regimens with BRAFi/MEKi and ICIs. In the setting of BRAFi resistance, it is recognized that BRAFi can lead to the paradoxical activation of the MAPK pathway in non-mutant cells in the TME, including TAMs [Citation67], driving these cells toward immunosuppressive phenotypes and, in the case of TAMs, increasing the secretion of VEGF, which in turn, can reactivate MAPK in melanoma cells [Citation67,Citation92]. Blocking VEGF and its interaction with VEGF-R-expressing cells, in conjunction with BRAFi, delayed the time to treatment resistance in a mouse model of melanoma [Citation92].
A number of clinical trials are studying this further (NCT01495988, NCT03175432) [Citation89]. There have also been initial promising results within clinical trials combining immunotherapy and antiangiogenic agents which are now progressing to phase III studies (NCT04356729, NCT02681549, and NCT03239145) [Citation93,Citation94] (). Exploring combinations of inhibitors such as anti-VEGF may still reveal novel mechanisms in the future that may help understand BRAFi resistance.
6.4. Enhancing Immune Effector Function: Adoptive Cell Therapies
Personalized cell therapy targeting tumor-associated antigens with expanded tumor-infiltrating lymphocytes (TILs) has shown some promise in metastatic melanoma since the early 1990s [Citation95]. Adoptive cell therapy (ACT) of T cells, involving the allogenic transplant of TILs, and CAR-T therapy, whereby genetically modified T cells expressing novel T cell receptors, chimeric antigen receptors (CARs) are expanded and reinfused into the patient, have both shown promise in treating hematological malignancies but efficacy is yet to be realized in solid tumors, including melanoma [Citation96].
In the latest Phase 2 study examining ACT using TILs in patients with treatment refractory melanoma, 36% patients had a partial response (22 of 66), with two patients having a complete response [Citation97]. The safety profile was comparable to lymphodepleting chemotherapy, and this has led to studies evaluating combinations of ACT with other treatments. MAPKi have been suggested to enhance the clinical efficacy of ACT. In a preclinical study, BRAFi was shown to induce the upregulation of the mannose 6 phosphate receptor (M6PR) in a dose-dependent fashion in both BRAFi-sensitive and -resistant melanoma cells. M6PR increases cancer cell uptake of granzyme B, a main component of the cytotoxic activity of CTLs. Thus, it has been suggested that it may be beneficial to treat with BRAFi prior to TIL therapy, as a means of enhancing ACT-mediated elimination of tumor cells [Citation98]. A clinical study of 13 patients with checkpoint inhibitor-resistant melanoma evaluated priming treatment with Vemurafenib prior to ACT with TILs. This study demonstrated significant clinical responses (one complete, eight partial responses, three patients with stable disease) and no unexpected toxicity [Citation99]. While this may serve as proof of principle, larger trials with longer follow-up times are required in order to identify patients most likely to benefit, identify the most appropriate point of intervention, and to establish durability of responses.
There has been significant interest in examining the role of CTL populations in anti-cancer therapy, however other cell types within the TME, including NK cells, can also engender tumor cell destruction. NK cells are of particular interest since their effector functions are not impaired as BRAFi resistance develops, and indeed their ability to lyse melanoma cells can increase in models of BRAFi-resistant melanoma [Citation100,Citation101]. Boosting the effector function of NK cells may delay BRAFi resistance and increase an effective anti-tumor response. The TLR7 agonist, imiquimod, already used for the topical treatment of non-melanoma skin cancers, has been shown to increase the activity of both NK cells and T cells in a murine model of melanoma [Citation50], and this is currently being tested in clinical trials as an adjuvant to melanoma vaccine therapies and checkpoint inhibitors (NCT0436423, NCT04401995).
Aside from ACT, several large-scale clinical trials using CAR-T cells for melanoma are ongoing as a second-line therapy after treatment failure with ICI or BRAFi/MEKi; however, the use of CAR-T in BRAFi-resistant melanoma remains underexplored [Citation102].
7. Conclusion
Although designed to impact the growth, proliferation, and survival of melanoma cells, MAPK pathway small-molecule inhibitors such as BRAFi in clinical use for the treatment of BRAF mutant melanoma can influence the immune composition of the TME. With immunotherapy dominating the landscape of melanoma therapeutics development, it is worth understanding how BRAFi can manipulate the immune response and how new and established treatments may be used sequentially or in combination to take advantage of cancer vulnerabilities and improve outcomes for patients.
8. Expert opinion
The past two decades have witnessed an unprecedented expansion in our understanding of the biology of melanoma, underpinning the development of new therapeutic classes: targeted oncogene inhibitors and ICIs. These developments have significantly improved the historically low five-year survival rates in advanced melanoma. Within cancer therapeutics, however, the development of resistance is a frustrating, yet unsurprising, phenomenon, given the selection pressure that therapies apply on cancer cells.
The success of ICIs has highlighted the importance of understanding the dynamic interactions between tumor cells and the immune component of the TME. Research has largely focused on the role of BRAF as an oncogenic target; however, there is evidence that MAPK inhibition significantly impacts on the TME by its effects in both immune and nonimmune cells. In the next decade, research will reveal previously unappreciated immune mechanisms which accompany response and resistance to BRAFi. These will enable us to better select treatment combinations or refine the sequence and timing of administering therapeutic agents to unlock more durable responses for our patients.
BRAFi initially leads to profound tumor regression; however, responses are short lived. This contrasts with ICI, where responses are less predictable but more durable. The approach within current clinical practice suggests that these treatments are mutually exclusive as first-line therapy, with debate over which treatments should be used first. New evidence suggests that ICIs may prolong survival in patients with BRAF mutant melanoma over MAPK inhibition [Citation16]; however, it does not follow that BRAFi and MEKi should be relegated to second-line therapies, especially given that there are still significant limitations to ICI therapy. There remains a degree of unpredictability about which patients will respond to ICIs and whether responses to treatment differ between patient cohorts. For example, patients of female sex and increasing age appear to derive less benefit from ICI than their younger and male counterparts [Citation103,Citation104], while at the same time BRAFi appear to have comparable efficacy across age groups [Citation105]. An arsenal of multiple first-line therapies and patient stratification may help ensure more patients, from multiple cohorts, respond to treatment. In addition to this, it is important to understand how MAPKi can modulate the immune system, and with the correct timing, could be successfully used as an adjuvant not only to ICIs, but to other therapies too.
It is possible that different regimens of existing drugs, or better stratification of patients receiving them, may lead to better outcomes quicker than the development of novel therapies. For example, since BRAFi resistance in melanoma can lead to the upregulation of VEGF pathways, and VEGF inhibitors have shown efficacy in a range of solid tumors including renal, urological and ovarian cancers, it may be timely to reconsider the use of VEGF inhibitors alongside BRAFi. Early trials in melanoma were disappointing, a better understanding of why these therapies failed and how new treatments interact with them, may reveal how best to use them in the future. To take the first point, since the early initial trials of VEGF inhibitors, it has been demonstrated that the expression of VEGF decreases with aging [Citation106]. This has been shown to reduce responses to anti-VEGF treatments in older patients, while the age-related proangiogenic factor secreted frizzled-related protein 2 (sFRP2) may be responsible for driving angiogenesis in older aged groups. Thus, better stratification of patients may lead to more successful implementation of tailored anti-angiogenic therapies and combinations. Alongside, since BRAFi resistance is characterized by the paradoxical increased secretion of VEGF, there is an opportunity for VEGF or other targeted anti-angiogenic inhibitors to work synergistically with a treatment which had not been discovered when VEGF inhibitors were initially trialed.
The success of ICIs has invariably led a focus on how to improve and augment the clinical efficacy of this class of drug, including how MAPKi can be used alongside ICIs. Increased understanding in how both MAPKi and ICIs alter the TME may provide answers to the current clinical problems arriving from combination treatment, namely: how can toxicity be overcome and how can sequencing be optimized to prolong patient survival? New trial design, as seen in SECOMBIT and DREAMseq, whereby different sequences of therapy are compared, will be invaluable in answering some of these questions. As well as clinical outcomes, the examination of biomarkers and biopsies throughout these studies will enable a better insight into the synergistic immune mechanisms that could underpin successful combinations of the two most revolutionary melanoma treatments of the decade.
The success of ICIs may represent the end of the beginning for drug discovery in melanoma rather than the beginning of the end. In cancers where ICI have failed, clinical trials reevaluating old drugs, as well as those testing new therapies abound. In contrast, the focus within melanoma treatment appears to be optimizing ICI regimens when we know that responses are unpredictable and not universal. However, refining and augmenting established treatment strategies, such as MAPK pathway inhibition hold significant merit.
Importantly, there must be continued investment and research into new druggable targets for melanoma. The discovery of new drugs will contribute to the transformation of therapeutic strategies for melanoma patients we have witnessed over the past decade. Revisiting old therapies in the context of recent conceptual and translational advances, may unearth combinations or sequential therapeutic protocols that can improve patient care and outcomes immeasurably.
Article highlights
Mutations in the MAPK pathway, the most common of which is the BRAFV600E mutation, contribute to melanoma pathogenesis and progression.
The development of BRAF inhibitors (BRAFi), and later MEK inhibitors (MEKi), has led to improved survival for patients with melanoma; however, therapeutic resistance typically develops after months of treatment.
BRAFi can alter the immune response to create a proinflammatory tumour microenvironment. BRAFi increase the ratio of effector to immunosuppressive immune cells, increase proinflammatory cytokines, and reduce cytokines which contribute to the pathogenesis of melanoma and promote effector cell function by improving antigen presentation and subsequent T cell activation. As BRAFi resistance occurs, these immune changes begin to reverse.
The effects of BRAFi on host immunity may be harnessed therapeutically to achieve more durable treatment responses.
Using MAPK pathway inhibitors in combination or sequentially with established or experimental therapies, including checkpoint inhibitor antibodies, may augment the immunological effects of both BRAFi and immunotherapies, improving the effector function of immune cells within the tumour microenvironment and creating more successful and longer-lasting treatments for patients with melanoma.
Nomenclature
Declaration of interest
S Karagiannis is founder and shareholder of Epsilogen Ltd. and declares patents on antibody technologies. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosure
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- CRUK. Melanoma skin cancer statistics, cancer research UK February 2021. [cited 2022 Jan]. Available from: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/melanoma-skin-cancerhttps://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/melanoma-skin-cancer. https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/melanoma-skin-cancerhttps://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/melanoma-skin-cancer.
- Schadendorf D, Fisher DE, Garbe C, et al. Melanoma. Nat Rev Dis Primers. 2015 04;1:15003.
- Menzies AM, Haydu LE, Visintin L, et al. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin Cancer Res. 2012 Jun;18(12):3242–3249.
- Ascierto PA, Kirkwood JM, Grob -J-J, et al. The role of BRAF V600 mutation in melanoma. J Transl Med. 2012;10(1):85.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011 Jun;364(26):2507–2516.
- Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012 Feb 23;366(8):707–714.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014 Nov;371(20):1877–1888.
- Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014 Nov;371(20):1867–1876.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015 Jan;372(1):30–39.
- Moreira A, Heinzerling L, Bhardwaj N, et al. Current melanoma treatments: where do we stand? Cancers (Basel). 2021 Jan 9;13(2):221.
- Willsmore ZN, Coumbe BGT, Crescioli S, et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: treatment of melanoma and immune mechanisms of action. Eur J Immunol. 2021 Mar;51(3):544–556.
- Hartley GP, Chow L, Ammons DT, et al. Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. 2018 10; 6(10): 1260–1273.
- Sahni S, Valecha G, Sahni A. Role of Anti-PD-1 antibodies in advanced melanoma: the era of immunotherapy. Cureus. 2018 Dec 07;10(12):e3700.
- Hodi FS, Chiarion-Sileni V, and Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018 Nov;1911:1480–1492.
- Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Five-Year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019 10; 381(16): 1535–1546.
- Moser JC, Chen D, Hu-Lieskovan S, et al. Real-world survival of patients with advanced BRAF V600 mutated melanoma treated with front-line BRAF/MEK inhibitors, anti-PD-1 antibodies, or nivolumab/ipilimumab. Cancer Med. 2019 12; 8(18): 7637–7643.
- Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001 Apr;22(2):153–183.
- Amaral T, Sinnberg T, Meier F, et al. The mitogen-activated protein kinase pathway in melanoma part I - Activation and primary resistance mechanisms to BRAF inhibition. Eur J Cancer. 2017 Mar;73:85–92.
- Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases–regulating the immune response. Nat Rev Immunol. 2007 Mar;7(3):202–212.
- Coulombe P, Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta. 2007 Aug;1773(8):1376–1387.
- Shain AH, Yeh I, Kovalyshyn I, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373(20):1926–1936.
- Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy.Pharmacol Ther. 2014 2014 May 01;142(2):176–182.
- Proietti I, Skroza N, Michelini S, et al. BRAF inhibitors: molecular targeting and immunomodulatory actions. Cancers (Basel). 2020;12(7):1823.
- King AJ, Arnone MR, Bleam MR, et al. Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS ONE. 2013;8(7):e67583.
- Yang H, Higgins B, Kolinsky K, et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical Melanoma models. Cancer Res. 2010;70(13):5518–5527.
- Koelblinger P, Thuerigen O, Dummer R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr Opin Oncol. 2018;30(2):125–133.
- Tsai J, Lee JT, and Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105( 8):3041–3046.
- Broman KK, Dossett LA, Sun J, et al. Update on BRAF and MEK inhibition for treatment of melanoma in metastatic, unresectable, and adjuvant settings. Expert Opin Drug Saf. 2019 2019 May 04;18(5):381–392.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012 Jul 28;380(9839):358–365.
- Trunzer K, Pavlick AC, Schuchter L, et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013 May 10;31(14):1767–1774.
- Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014 Jan;4(1):80–93.
- Penna I, Molla A, Grazia G, et al. Primary cross-resistance to BRAFV600E-, MEK1/2- and PI3K/mTOR-specific inhibitors in BRAF-mutant melanoma cells counteracted by dual pathway blockade. Oncotarget. 2016 Jan 26;7(4):3947–3965.
- Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011 Nov 23;480(7377):387–390.
- Welsh SJ, Rizos H, Scolyer RA, et al. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: where to next? Eur J Cancer. 2016 Jul;62:76–85.
- Kim A, Cohen MS. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin Drug Discov. 2016;11(9):907–916.
- Karoulia Z, Gavathiotis E, Poulikakos PI. New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer. 2017;17(11):676–691.
- Subbiah V, Baik C, Kirkwood JM. Clinical development of BRAF plus MEK inhibitor combinations. Trends Cancer. 2020;6(9):797–810.
- Robert C, Grob JJ, Stroyakovskiy D, et al. Five-Year outcomes with dabrafenib plus trametinib in metastatic Melanoma. N Engl J Med. 2019 Aug 15;381(7):626–636.
- Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (Columbus): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018 05; 19(5): 603–615.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012 Jan 19;366(3):207–215.
- Knight DA, Ngiow SF, Li M, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest. 2013 Mar;123(3):1371–1381.
- Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32(1):659–702.
- Ho PC, Meeth KM, Tsui YC, et al. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNγ. Cancer Res. 2014 Jun 15;74(12):3205–3217.
- Michielon E, López González M, Burm JLA, et al. Micro-environmental cross-talk in an organotypic human melanoma-in-skin model directs M2-like monocyte differentiation via IL-10. Cancer Immunol Immunother. 2020 Nov;69(11):2319–2331.
- Comunanza V, Corà D, Orso F, et al. VEGF blockade enhances the antitumor effect of BRAFV600E inhibition. EMBO Mol Med. 2017 02; 9(2): 219–237.
- Frederick DT, Piris A, and Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013 Mar 1;19(5):1225–31.
- Zhou J, Tang Z, Gao S, et al. Tumor-Associated macrophages: Recent insights and therapies. Front Oncol. 2020;10:188.
- Consoli F, Manganoni AM, Grisanti S, et al. Panniculitis and vitiligo occurring during BRAF and MEK inhibitors combination in advanced melanoma patients: Potential predictive role of treatment efficacy. PLoS One. 2019;14(4):e0214884.
- Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012 Mar;18(5):1386–1394.
- Bellmann L, Cappellano G, Schachtl-Riess JF, et al. A TLR7 agonist strengthens T and NK cell function during BRAF-targeted therapy in a preclinical melanoma model. Int J Cancer. 2020 03; 146(5): 1409–1420.
- Weiss SA, Han SW, Lui K, et al. Immunologic heterogeneity of tumor-infiltrating lymphocyte composition in primary melanoma. Hum Pathol. 2016 11;57:116–125.
- Peiffer L, Farahpour F, Sriram A, et al. BRAF and MEK inhibition in melanoma patients enables reprogramming of tumor infiltrating lymphocytes. Cancer Immunol Immunother. 2021 Jun;70(6):1635–1647.
- Schilling B, Sucker A, Griewank K, et al. Vemurafenib reverses immunosuppression by myeloid derived suppressor cells. Int J Cancer. 2013 Oct;133(7):1653–1663.
- Schilling B, Paschen A. Immunological consequences of selective BRAF inhibitors in malignant melanoma: neutralization of myeloid-derived suppressor cells. Oncoimmunology. 2013 Aug;2(8):e25218.
- Whipple CA, Boni A, and Fisher JL, et al. The mitogen-activated protein kinase pathway plays a critical role in regulating immunological properties of BRAF mutant cutaneous melanoma cells. Melanoma Res. 2016 06;26(3):223–35
- Massi D, Rulli E, Cossa M, et al. The density and spatial tissue distribution of CD8+ and CD163+ immune cells predict response and outcome in melanoma patients receiving MAPK inhibitors. J Immunother Cancer. 2019 Nov 15;7(1):308.
- Weber R, Riester Z, Hüser L, et al. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J Immunother Cancer. 2020 08; 8(2): e000949.
- Jobe NP, Rösel D, Dvořánková B, et al. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem Cell Biol. 2016 Aug;146(2):205–217.
- Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013 Jan;19(2):393–403.
- Liu L, Mayes PA, Eastman S, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015 Apr;21(7):1639–1651.
- Huang H, Langenkamp E, Georganaki M, et al. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-κB-induced endothelial activation. FASEB J. 2015 Jan;29(1):227–238.
- Laoui D, Van Overmeire E, Di Conza G, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014 Jan;74(1):24–30.
- Graells J, Vinyals A, Figueras A, et al. Overproduction of VEGF concomitantly expressed with its receptors promotes growth and survival of melanoma cells through MAPK and PI3K signaling. J Invest Dermatol. 2004 Dec;123(6):1151–1161.
- Bradley SD, Chen Z, Melendez B, et al. BRAFV600E co-opts a Conserved MHC class i internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of Melanoma. Cancer Immunol Res. 2015 Jun;3(6):602–609.
- Hirata E, Girotti MR, Viros A, et al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell. 2015 Apr 13;27(4):574–588.
- Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012 Jul;487(7408):500–504.
- Wang T, Xiao M, Ge Y, et al. BRAF inhibition stimulates Melanoma-Associated macrophages to drive tumor growth. Clin Cancer Res. 2015 Apr;21(7):1652–1664.
- Pieper N, Zaremba A, Leonardelli S, et al. Evolution of melanoma cross-resistance to CD8 + T cells and MAPK inhibition in the course of BRAFi treatment. Oncoimmunology. 2018;7(8):e1450127.
- Jiang X, Zhou J, Giobbie-Hurder A, et al. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013 Feb;19(3):598–609.
- Cooper ZA, Reuben A, Spencer CN, et al. Distinct clinical patterns and immune infiltrates are observed at time of progression on targeted therapy versus immune checkpoint blockade for melanoma. Oncoimmunology. 2016 Mar;5(3):e1136044.
- Comin-Anduix B, Chodon T, Sazegar H, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010 Dec;16(24):6040–6048.
- Rossi A, Roberto M, Panebianco M, et al. Drug resistance of BRAF-mutant melanoma: review of up-to-date mechanisms of action and promising targeted agents. Eur J Pharmacol. 2019 Nov 05;862:172621.
- Hu-Lieskovan S, Mok S, and Homet Moreno B, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015 Mar;7279:279ra41.
- Cooper ZA, Juneja VR, Sage PT, et al. Response to BRAF inhibition in melanoma is enhanced when combined with immune checkpoint blockade. Cancer Immunol Res. 2014 Jul;2(7):643–654.
- Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–1366.
- Minor DR, Puzanov I, Callahan MK, et al. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015;28(5):611–612.
- Ribas A, Lawrence D, and Atkinson V, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med. 2019;25(6):936–940.
- Ascierto PA, Ferrucci PF, Fisher R, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med. 2019 2019 Jun 01;25(6):941–946.
- Burton EM, Amaria RN, Glitza IC, et al. Safety and efficacy of TRIplet combination of nivolumab (N) with dabrafenib (D) and trametinib (T) [TRIDeNT] in patients (pts) with BRAF-mutated metastatic melanoma (MM): a single center phase II study. Ann Oncol. 2019;30:v534–v535.
- Nathan P, Dummer R, Long GV, et al. LBA43 spartalizumab plus dabrafenib and trametinib (Sparta-DabTram) in patients (pts) with previously untreated BRAF V600–mutant unresectable or metastatic melanoma: results from the randomized part 3 of the phase III COMBI-i trial. Ann Oncol. 2020;31:S1172.
- Gutzmer R, Stroyakovskiy D, Gogas H, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF V600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020 Jun 13;;395(10240):1835–1844.
- Sullivan RJ, Hamid O, Gonzalez R, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med. 2019 2019 Jun 01;25(6):929–935.
- Rozeman EA, Versluis JM, Sikorska K, et al. The IMPemBra trial, a phase II study comparing pembrolizumab with intermittent/short‐term dual MAPK pathway inhibition plus pembrolizumab in melanoma patients harboring the BRAFV600 mutation. J clin oncol. 2020;38(15_suppl):10021.
- Atkins MB, Lee SJ, Chmielowski B, et al. DREAMseq (doublet, randomized evaluation in advanced melanoma sequencing): a phase III trial—ECOG-ACRIN EA6134. J clin oncol. 2021;39(36_suppl):356154.
- LBA40 SECOMBIT: The best sequential approach with combo immunotherapy ipilimumab (I) nivolumab (N) and combo target therapy encorafenib (E)/binimetinib (B) in patients with BRAF mutated metastatic melanoma: a phase II randomized study. Ann Oncol. 2021;32(suppl_5):S1316–S1317.
- Hugo W, Shi H, Sun L, et al. Non-genomic and immune evolution of melanoma acquiring MAPKI resistance. Cell. 2015 Sep 10;162(6):1271–1285.
- Steinberg SM, Shabaneh TB, Zhang P, et al. Myeloid cells that impair immunotherapy are restored in melanomas with acquired resistance to BRAF Inhibitors. Cancer Res. 2017 Apr 1;77(7):1599–1610.
- Mok S, Tsoi J, Koya RC, et al. Inhibition of colony stimulating factor-1 receptor improves antitumor efficacy of BRAF inhibition. BMC Cancer. 2015 May 05;15(1):356.
- Quaresmini D, Guida M. Neoangiogenesis in Melanoma: An issue in biology and systemic treatment [mini review]. Front Immunol. 2020 2020 Oct 29; 11(2836). https://doi.org/10.3389/fimmu.2020.584903
- Del Vecchio M, Mortarini R, Canova S, et al. Bevacizumab plus fotemustine as first-line treatment in metastatic melanoma patients: clinical activity and modulation of angiogenesis and lymphangiogenesis factors. Clin Cancer Res. 2010 Dec 1;16(23):5862–5872.
- Kottschade LA, Suman VJ, Perez DG, et al. A randomized phase 2 study of temozolomide and bevacizumab or nab-paclitaxel, carboplatin, and bevacizumab in patients with unresectable stage IV melanoma: a North central cancer treatment group study, N0775. Cancer. 2013 Feb 1;119(3):586–592.
- Atzori MG, Ceci C, Ruffini F, et al. Role of VEGFR-1 in melanoma acquired resistance to the BRAF inhibitor vemurafenib. J Cell Mol Med. 2020 01; 24(1): 465–475.
- Taylor MH, Lee CH, Makker V, et al. Phase IB/II Trial of lenvatinib plus pembrolizumab in patients with advanced renal cell Carcinoma, endometrial cancer, and other selected advanced solid tumors. J Clin Oncol. 2020 Apr 10;38(11):1154–1163.
- Arance AM, Ldl C-M, Petrella TM, et al. Lenvatinib (len) plus pembrolizumab (pembro) for patients (pts) with advanced melanoma and confirmed progression on a PD-1 or PD-L1 inhibitor: updated findings of LEAP-004. J clin oncol. 2021;39(15_suppl):9504.
- Borch TH, Andersen R, Ellebaek E, et al. Future role for adoptive T-cell therapy in checkpoint inhibitor-resistant metastatic melanoma. J Immunother Cancer. 2020;8(2):e000668.
- Simon B, Uslu U. CAR-T cell therapy in melanoma: a future success story? Exp Dermatol. 2018 12;27(12):1315–1321.
- Sarnaik AA, Hamid O, Khushalani NI, et al. Lifileucel, a tumor-Infiltrating lymphocyte therapy, in metastatic Melanoma. J Clin Oncol. 2021 Aug 20;39(24):2656–2666.
- Atay C, Kwak T, Lavilla-Alonso S, et al. BRAF targeting sensitizes resistant melanoma to cytotoxic T cells. Clin Cancer Res. 2019;25(9):2783–2794.
- Borch TH, Harbst K, Rana AH, et al. Clinical efficacy of T-cell therapy after short-term BRAF-inhibitor priming in patients with checkpoint inhibitor-resistant metastatic melanoma. J Immunother Cancer. 2021 Jul;9(7):e002703.
- Sottile R, Pangigadde PN, Tan T, et al. HLA class I downregulation is associated with enhanced NK-cell killing of melanoma cells with acquired drug resistance to BRAF inhibitors. Eur J Immunol. 2016 Feb;46(2):409–419.
- Frazao A, Rethacker L, Jeudy G, et al. BRAF inhibitor resistance of melanoma cells triggers increased susceptibility to natural killer cell-mediated lysis. J Immunother Cancer. 2020 09; 8(2): e000275.
- Dafni U, Michielin O, Lluesma SM, et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Ann Oncol. 2019;30(12):1902–1913.
- Jang SR, Nikita N, Banks J, et al. Association between sex and immune checkpoint inhibitor outcomes for patients with Melanoma. JAMA Network Open. 2021 Dec 01;;4(12):e2136823.
- Safi M, Al-Azab M, Jin C, et al. Age-Based disparities in metastatic melanoma patients treated in the immune checkpoint inhibitors (ICI) versus Non-ICI Era: a population-based study. Front Immunol. 2021;12:609728.
- Kohtamäki LM, Hernberg M, Jaakkola M, et al. BRAF inhibitor treatment is feasible in the oldest-old advanced melanoma patients. Melanoma Res. 2021 Jun 01;;31(3):218–223.
- Fane ME, Ecker BL, Kaur A, et al. sFRP2 supersedes VEGF as an Age-related driver of angiogenesis in Melanoma, affecting response to Anti-VEGF therapy in older patients. Clin Cancer Res. 2020 Nov 01;26(21):5709–5719.