
ABSTRACT
Background
Tocilizumab is a recombinant humanized monoclonal immunoglobulin G1 antibody against the interleukin-6 receptor (IL-6 R). MSB11456 is a proposed tocilizumab biosimilar.
Objectives
To assess the pharmacokinetic and pharmacodynamic similarity of MSB11456 to both US-licensed and EU-approved tocilizumab.
Methods
Healthy adult volunteers (N = 685) received a single 162 mg subcutaneous injection of MSB11456, US-licensed tocilizumab, or EU-approved tocilizumab in this randomized, double-blind, parallel-group study. Blood samples were taken pre-dose and for up to 48 days post-dose. Primary endpoint pharmacokinetic parameters were analyzed using analysis of covariance. Secondary pharmacodynamic measures included serum-soluble IL-6 R and serum C-reactive protein. Safety data were analyzed descriptively.
Results
Pharmacokinetic equivalence (with all corresponding 90% confidence intervals for the geometric least squares mean ratios within the predefined 80.00% to 125.00% equivalence margin) was demonstrated between MSB11456 and both US-licensed and EU-approved tocilizumab, as well as between the reference products. Pharmacodynamic analyses demonstrated similarity of MSB11456 and both US-licensed and EU-approved tocilizumab, as well as between the reference products. Safety, tolerability, and immunogenicity were comparable between treatments.
Conclusion
Pharmacokinetic and pharmacodynamic similarity of MSB11456, US-licensed tocilizumab, and EU-approved tocilizumab were demonstrated, and the three products had comparable immunogenicity and safety, supporting MSB11456 as a biosimilar to tocilizumab.
Plain Language Summary
Tocilizumab is a biologic drug that is used to treat autoimmune diseases, including rheumatoid arthritis. Biologic drugs are very important for the treatment of autoimmune diseases, but their costs limit accessibility. Therefore, the availability of biosimilars, which are biologics that are very similar in structure and function to an existing biologic drug, may provide a significant cost advantage for national healthcare programs and consumers. MSB11456 is a proposed tocilizumab biosimilar. Our study tested the pharmacokinetic and pharmacodynamic similarity of MSB11456 to the approved formulations of tocilizumab in the US and EU (US-licensed and EU-approved tocilizumab) in a large group of healthy adults. Volunteers received a single 162 mg subcutaneous injection of MSB11456, US-licensed tocilizumab, or EU-approved tocilizumab in this randomized, double-blind, parallel-group study. Blood samples were taken before and regularly after the injection, and safety was monitored. We showed that the pharmacokinetics and pharmacodynamics of MSB11456, US-licensed and EU-approved tocilizumab were sufficiently similar to claim equivalence between the three products. Safety and immunogenicity were also comparable between the three treatments. These findings suggest that MSB11456 can be considered as a biosimilar to tocilizumab. Biosimilars have improved price competition and led to a reduction in the net costs of biologics, so tocilizumab biosimilars can be expected to contribute to this and potentially improve access to the best available care.
1. Introduction
Interleukin-6 (IL-6) is a pleiotropic proinflammatory cytokine produced by numerous cell types, including T and B cells, monocytes, neutrophils, and fibroblasts, as well as synovial and endothelial cells [Citation1,Citation2]. It contributes to acute and chronic joint inflammation by activating neutrophils, which further promote inflammatory responses via recruitment of monocytes and leukocytes in the joint; increasing levels of vascular endothelial growth factor, thereby promoting the migration and proliferation of endothelial cells and increasing vascular permeability; stimulating lymphocytes; increasing osteoclast recruitment; and enhancing articular cartilage damage [Citation2]. IL-6 levels have been shown to correlate with disease activity and joint destruction in patients with rheumatoid arthritis [Citation2].
Tocilizumab, a biologic disease-modifying antirheumatic drug (DMARD), was the first recombinant humanized monoclonal immunoglobulin G1 antibody against the IL-6 receptor (IL-6 R) subunit alpha [Citation3]. It is approved for use in a number of autoimmune diseases, including rheumatoid arthritis, systemic sclerosis-associated interstitial lung disease, systemic and polyarticular juvenile idiopathic arthritis, giant cell arthritis, and chimeric antigen receptor T-cell therapy-induced cytokine release syndrome [Citation4,Citation5]. Tocilizumab has proven efficacy in patients with rheumatic disease and the other indications for which it is approved and improves the quality of life of affected patients [Citation1,Citation3]. More recently, the US Food and Drug Administration issued an emergency use authorization for the use of tocilizumab in certain populations of patients with COVID-19 [Citation6] and the European Medicines Agency [Citation7] recommended that tocilizumab be indicated for the treatment of selected adults with COVID-19 after promising clinical trial results [Citation8]. COVID-19 induces the generation of pro-inflammatory cytokines including IL-6 leading to host cell damage [Citation9]. Tocilizumab is available as both an intravenous and subcutaneous formulation, the latter of which might be more convenient for patients because it allows once-weekly self-administration [Citation10].
Biologics are a key cornerstone in the treatment of rheumatoid arthritis and other autoimmune diseases, but access to these drugs is limited due to affordability. Therefore, the availability of biosimilars may provide a significant cost advantage for national healthcare programs and consumers [Citation11]. MSB11456 is a proposed biosimilar to US-licensed tocilizumab and EU-approved tocilizumab. Extensive in vitro testing and functional activity assays showed that MSB11456 was considered highly similar to these reference products prior to initiation of its clinical development program (data on file), in accordance with regulatory requirements for biosimilar compounds [Citation12,Citation13].
This parallel-design study was conducted to determine whether the subcutaneous formulation of tocilizumab MSB11456 (162 mg/0.9 mL) is similar from a pharmacokinetic and pharmacodynamic perspective to both the subcutaneous US-licensed reference product (US-licensed tocilizumab, Actemra®, 162 mg/0.9 mL) and the subcutaneous EU-approved reference medicinal product (EU-approved tocilizumab, RoActemra®, 162 mg/0.9 mL) in healthy adult subjects. The study also investigated the safety, tolerability, and immunogenicity of the three tocilizumab products.
2. Subjects and methods
This randomized, double-blind, parallel-group, single-dose study was conducted at two centers in New Zealand (Auckland Clinical Studies Ltd and Christchurch Clinical Studies Trust, Ltd) between November 2017 and September 2019 (Supplementary Figure 1). Healthy adult subjects were randomized in a 1:1:1 ratio, stratified by body weight (≥60 and ≤80 kg/>80 and ≤100 kg), to receive a single 162 mg dose of MSB11456, US-licensed tocilizumab, or EU-approved tocilizumab as a subcutaneous injection in the lower abdomen on the morning of study day 1. Randomization was performed using an interactive web response system.
All subjects provided written informed consent before study entry. The study was conducted in accordance with ethical principles of the International Council for Harmonization Guideline for Good Clinical Practice and the Declaration of Helsinki as well as with applicable local regulations (ethics committee approval TT50-10,239 (2155)).
2.1. Study population
Eligible subjects were men or non-pregnant, non-breastfeeding women, aged ≥18 to ≤55 years with body mass index (BMI) ≥18.0 to ≤29.9 kg/m2, and in general good health based on a comprehensive medical assessment. All subjects, men and women, were to comply with the contraception requirements specified in the clinical study protocol. Subjects were ineligible if they met any of the exclusion criteria typical of this type of study; had a history of tuberculosis, or active or latent tuberculosis, other infections, or a gastrointestinal condition that might have predisposed them to perforations; had immunodeficiency; or had previous exposure to tocilizumab, IL-6 pathway inhibitors, or other biologics.
2.2. Assessments
Blood samples for pharmacokinetic and pharmacodynamic analysis were taken pre-dose, 2, 8, and 12 hours post-dose, then every 12 hours up to 72 hours post-dose, and at each outpatient visit thereafter to day 48 (end-of-study).Samples for immunogenicity testing were taken pre-dose, and on days 15, 29, and 48. Serum concentrations of tocilizumab were analyzed in a bioanalytical laboratory using a validated method. Pharmacodynamic analyses included changes in soluble IL-6 R (sIL-6 R) and C-reactive protein (CRP), measured using validated analysis methods, as per several other pharmacodynamic assessments of tocilizumab or other anti-IL-6 R antibodies [Citation14–16].
Safety and tolerability (including treatment-emergent adverse effects [TEAEs], adverse events of special interest [AESIs] and serious TEAEs) were assessed throughout the study. AESIs included local tolerability (injection site reactions) that the investigator considered treatment-related, serious infections (those requiring hospitalization, with a fatal outcome or sepsis, or requiring intravenous antibiotics/antimicrobials; or active/latent tuberculosis), and hypersensitivity reactions (Grade ≥3 or serious event).
Immunogenicity was assessed using validated assays and followed a multi-tiered approach: firstly, all samples were assessed for antidrug antibodies (ADAs) using a screening homogeneous electrochemiluminescent (ECL) bridging mesoscale assay, with the biosimilar as both detection and capture reagent. An acid dissociation step was implemented to reach optimal assay sensitivity and drug tolerance. Samples testing putative positive were then subjected to a confirmatory assay, and antibody titers were determined. Finally, all confirmed positive samples were tested in a validated, cell-based neutralizing antibody (NAb) assay, using a cell line expressing the IL-6 receptor, to determine if the ADAs against tocilizumab were neutralizing biological drug activity. For subjects who were ADA positive before receiving any study treatment, a treatment-induced ADA response was defined as a statistically-determined 1.3-fold increase in titers in post-dose assays.
2.3. Endpoints
2.3.1. Primary endpoints
The primary endpoints were the pharmacokinetic parameters area under the concentration–time curve (AUC) from time zero (pre-dose) to the last sampling time at which a quantifiable concentration was obtained (AUC0-t), AUC from time zero extrapolated to infinity (AUC0-∞), and maximum observed concentration (Cmax) from administration of study drug over the entire blood sampling interval.
2.3.2. Secondary endpoints
Secondary endpoints were the additional pharmacokinetic parameters AUC from time zero to 72 hours (AUC0-72), time to Cmax (tmax), apparent terminal half-life (t1/2), apparent total body clearance (CL/F), and time to last observed serum concentration (tlast). The pharmacodynamics of tocilizumab were assessed as area under the effect–time curve (AUE), maximum observed effect (Emax), and time to maximum observed effect (tEmax) for sIL-6 R and CRP, as well as minimum observed effect (Emin) and time to minimum observed effect (tEmin) for CRP. ADA and NAb status, and ADA titers were also recorded, as were TEAEs, including serious TEAEs and AESIs, and abnormal vital signs and laboratory values.
2.4. Sample size
Due to uncertainty in the variability of the primary pharmacokinetic parameters, a blinded sample size re-estimation was pre-specified and performed after 163 subjects completed the study up to day 29. As a result of this blinded sample size re-estimation, a maximum of 696 randomized subjects was planned to ensure 220 evaluable subjects per treatment arm and achievement of 80% overall power, assuming a maximum difference of 5% between any treatments, a coefficient of variation (CV) of up to 65%, and a dropout rate of 5%.
2.5. Analysis set
Pharmacokinetic and pharmacodynamic analyses were performed in all subjects receiving study drug with data for at least one of the primary pharmacokinetic or pharmacodynamic parameters, respectively, and who had no clinically important protocol deviations or events that may have significantly affected the pharmacokinetic or pharmacodynamic assessments. Subgroups were defined based on ADA status (positive/negative), NAb status (positive/negative), baseline body weight (≥60–≤80 kg/>80–≤100 kg), baseline BMI (≤20 kg/m2/>20 and ≤28 kg/m2/>28 kg/m2) and study site. It was expected that BMI would have an impact on PK parameters so a middle range of >20 and ≤28 kg/m2 was selected to ensure lower variability of PK parameters in this particular subgroup, which included the majority of subjects. Immunogenicity and safety were analyzed in all subjects who received study drug.
2.6. Statistical analyses
Pharmacokinetic and pharmacodynamic parameters were calculated using standard noncompartmental methods with the validated software Phoenix® WinNonlin® version 8.0 (Pharsight Corporation, a Certara Company, Princeton, New Jersey, USA). All other statistical analyses were validated and performed using SAS version 9.4.
Pharmacokinetic AUCs were calculated using actual sampling time points and a linear-log trapezoidal method. The primary pharmacokinetic parameters were log-transformed and analyzed using an analysis of covariance (ANCOVA) model that included treatment as a fixed effect, baseline weight category (≥60–≤80 kg/>80–≤100 kg), baseline BMI (continuous variable), and study site as covariates. From this model, the 90% confidence interval (CI) for the difference in mean parameters among the three groups (MSB11456 versus US-licensed tocilizumab, MSB11456 versus EU-approved tocilizumab, and US-licensed tocilizumab versus EU-approved tocilizumab) was calculated, then re-expressed on the original ratio scale to assess equivalence. If the 90% CI for the geometric least squares (LS) mean ratios were entirely within the 80.00% to 125.00% equivalence margin for each primary pharmacokinetic parameter, then pharmacokinetic similarity was concluded for that comparison. All three pharmacokinetic parameter comparisons needed to show statistical significance for bioequivalence to be declared; therefore, no adjustment for multiplicity was required.
Sensitivity analyses were performed for the primary pharmacokinetic endpoints using only the weight stratification factor category as a covariate, and pharmacokinetic parameters were summarized for each subgroup.
Pharmacodynamic parameters were baseline-adjusted, with the exception of CRP Cmin, for which observed values were used. AUE calculations used the linear trapezoidal method. The secondary pharmacodynamic parameters sIL-6 R Emax, sIL-6 R AUE, and CRP Emin were analyzed on the log scale using the same ANCOVA model as the primary pharmacokinetic parameters; CRP Emax and CRP AUE were analyzed in a similar manner but on the original scale without log-transformation, as the maximum effect was a decrease in CRP serum concentration.
Continuous measurements were summarized using descriptive statistics, and categorical data were summarized using frequency tables. Safety and immunogenicity parameters were summarized descriptively, as were subject baseline demographics and characteristics.
3. Results
Of 1055 screened subjects, 685 were randomized and received a single dose of one of the tocilizumab products (Safety population; ). A total of 666 subjects (97.2%) completed the study, and of the 19 subjects who discontinued the study, most were lost to follow-up or withdrew consent (n = 15). Subject demographics and characteristics were similar between treatments ().
Figure 1. Subject disposition.
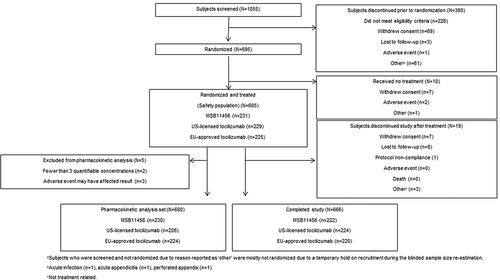
Table 1. Subject demographics and characteristics at baseline, by treatment group and overall (safety analysis seta).
3.1. Pharmacokinetics
Relevant pharmacokinetic data were available from 680 patients and were included in the pharmacokinetic analyses (). Arithmetic mean serum concentration–time profiles of tocilizumab after administration of a single dose of MSB11456, US-licensed tocilizumab, or EU-approved tocilizumab were almost superimposable throughout the entire observation period (). In all treatment groups, mean tocilizumab serum concentrations increased rapidly following a single subcutaneous injection, reached peak concentrations 96 hours post-dose, and then declined in a multi-exponential manner, with the last mean concentration above the lower limit of quantification recorded on day 22 (504 hours). Pharmacokinetic endpoints after a single dose of the three tocilizumab products are summarized in .
Figure 2. Arithmetic mean (SD) tocilizumab serum concentration–time profiles following a single dose of MSB11456, US-licensed tocilizumab or EU-approved tocilizumab in healthy subjects on a linear scale (pharmacokinetic analysis seta).
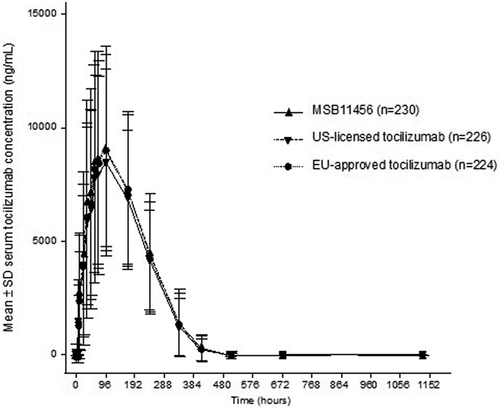
Table 2. Pharmacokinetic results after a single dose of MSB11456, US-licensed tocilizumab or EU-approved tocilizumab in healthy subjects (pharmacokinetic analysis seta).
Pharmacokinetic equivalence was demonstrated between MSB11456 and both US-licensed and EU-approved tocilizumab, as well as between the reference products, since all 90% CIs for the geometric LS mean ratios were within the predefined 80.00% to 125.00% equivalence margin () for the three primary pharmacokinetic parameters in all pairwise treatment comparisons. These results were supported by those of the sensitivity and subgroup analyses. Subgroup analyses showed no apparent difference in primary pharmacokinetic parameters across the ADA-positive, ADA-negative, or NAb-negative subgroups with each treatment (Supplementary Table 1); the NAb-positive subgroup for each treatment was too small to draw any conclusions. Subgroup analyses based on weight and BMI categories showed that tocilizumab exposure was lower in the higher weight and BMI subgroups across all three treatments. ANCOVA models for the primary analysis indicated that BMI had a significant effect on all tocilizumab primary pharmacokinetic parameters (p < 0.0001).
Table 3. Geometric LS mean ratios for primary pharmacokinetic parameters after a single dose of MSB11455, US-licensed tocilizumab, or EU-approved tocilizumab in healthy subjectsa (pharmacokinetic analysis setb).
3.2. Pharmacodynamics
Mean sIL-6 R baseline-adjusted serum concentration–time profiles were similar and mostly overlapping for the three treatments (); mean sIL-6 R pharmacodynamic parameters were also similar across treatments (Supplementary Table 2). All subjects had low baseline CRP levels as expected for healthy subjects (), and no substantial changes in pharmacodynamic profiles were observed in any treatment group (data not shown). Mean CRP pharmacodynamic profiles over time were similar and mostly overlapping for the three treatments and mean CRP pharmacodynamic parameters (Emax, AUE, Emin, tEmax, and tEmin) were generally similar between the three treatments (Supplementary Table 2).
Figure 3. Arithmetic mean (SD) sIL-6 R baseline-adjusted serum concentration–time profiles following a single dose of MSB11456, US-licensed tocilizumab or EU-approved tocilizumab in healthy subjects on a linear scale (pharmacodynamic analysis seta).
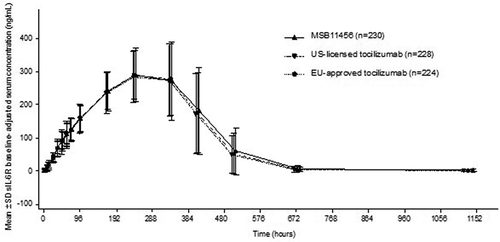
Pharmacodynamic analyses based on sIL-6 R and CRP parameters demonstrated the similarity of MSB11456 and both US-licensed and EU-approved tocilizumab, as well as between the reference products. All corresponding 90% CIs for baseline-adjusted sIL-6 R geometric LS mean ratios were within the predefined 80.00% to 125.00% equivalence margin (). The 90% CIs of CRP LS mean difference for Emax and AUE included zero for all pairwise treatment comparisons (Supplementary Table 3), and the 90% CIs of the baseline-adjusted CRP geometric LS mean Emin ratios were contained within the predefined 80.00% to 125.00% equivalence margin in all pairwise treatment comparisons ().
Table 4. Geometric LS mean ratios for pharmacodynamic parameters after a single dose of MSB11455, US-licensed tocilizumab, or EU-approved tocilizumab in healthy subjectsa (pharmacodynamic analysis setb).
Overall, there was no clear relationship between ADA formation and changes in sIL-6 R and CRP levels (Supplementary Table 4). Subgroup analyses showed no apparent difference in mean sIL-6 R pharmacodynamic parameters across treatments for ADA-positive, ADA-negative, or NAb-negative subgroups; the NAb-positive subgroup for each treatment was too small to draw any meaningful conclusions.
3.3. Safety
Overall, 73.4% of 685 subjects experienced at least one TEAE (), with similar numbers, types and severity of TEAEs reported with each treatment. Few subjects had TEAEs considered related to treatment (22.6%), the most common being headache (in 5.1%), injection site erythema (4.4%), nausea (2.0%), and mouth ulceration (1.9%). The majority of subjects had TEAEs that were at worst mild (70.5%) or moderate (11.8%) in severity; only four TEAEs in four (0.6%) subjects (MSB11456, n = 2; US-licensed tocilizumab, n = 1; EU-approved tocilizumab, n = 1) were considered severe (Grade ≥3), all of which were reported as serious TEAEs and none of which were Grade ≥4. No TEAEs resulted in study discontinuation. A total of five serious TEAEs were reported across treatment groups, all of which resolved before the end of the study; no deaths were reported during the study. No serious TEAEs were reported by more than one subject.
Table 5. Safety and tolerability of tocilizumab, by treatment group and overall (safety analysis seta).
Other than injection site reactions (most commonly erythema and pruritis), AESIs occurred infrequently (in total, 46 events in 35 subjects). A total of 33 subjects (4.8%) had injection site reactions classified as AESIs, all of which were mild, of short duration, and resolved spontaneously, 2 (0.3%) had an infection (both Grade 3, serious perforated appendicitis), and none had a Grade ≥3 or serious hypersensitivity reaction. Changes in vital signs and laboratory values were similar between the three treatments.
Overall, no clinically relevant differences in overall tolerability profiles were noted between the three treatment arms.
3.4. Immunogenicity
The incidence of treatment-induced ADA positivity was similar with each treatment, with 67.1% of subjects who received MSB11456 testing positive at any visit, compared with 53.7% of those who received US-licensed tocilizumab and 65.8% of those who received EU-approved tocilizumab (Supplementary Figure 2). Overall, 2.6%, 1.3%, and 2.7% of subjects, respectively, tested positive for NAb.
4. Discussion
This study was conducted in healthy subjects to reduce variability related to factors other than differences between products, as recommended in regulatory requirements for investigating biosimilarity [Citation17]. In addition, the final population size was adequately powered to determine biosimilarity, based on blinded size re-estimation after study initiation that was preplanned because of uncertainty in actual variability of the primary pharmacokinetic parameters. In this study, subject disposition and demographic and baseline characteristics were comparable between the three treatment arms.
Importantly, the primary objective of demonstrating pharmacokinetic equivalence of MSB11456 and both US-licensed tocilizumab and EU-approved tocilizumab was achieved, as well as pharmacokinetic similarity between the reference products, as 90% CIs of the geometric LS mean ratios were entirely within the 80.00% to 125.00% equivalence margin for each primary pharmacokinetic parameter and comparison. These findings were based on a single subcutaneous injection of each study drug at the approved adult dose for tocilizumab of 162 mg [Citation4,Citation5]. Across all treatments, BMI was shown to have a similar and significant effect on all tocilizumab primary pharmacokinetic parameters.
In support of pharmacokinetic equivalence of the three tocilizumab formulations, pharmacodynamic similarity was also demonstrated for sIL-6 R parameters between MSB11456 and both US-licensed tocilizumab and EU-approved tocilizumab, as well as between US-licensed tocilizumab and EU-approved tocilizumab. Based on Emax and AUE, sIL-6 R exposure following a single subcutaneous injection of MSB11456 was similar to that of both reference formulations, as was exposure for US-licensed tocilizumab versus EU-approved tocilizumab; 90% CIs of the geometric LS mean ratio were contained within the predefined 80.00% to 125.00% equivalence margin in all pairwise treatment comparisons. CRP profiles and related parameters were also similar across the three treatments. As such, pharmacokinetic equivalence and pharmacodynamic similarity could be concluded for all primary pharmacokinetic and pharmacodynamic parameters for all three pairwise treatment comparisons, and the objectives of the study were met.
Results of this study also confirmed the comparable safety and tolerability of the three tocilizumab treatments, as assessed by the incidence and type of TEAEs, serious TEAEs, AESIs including hypersensitivity reactions, vital signs, laboratory parameters, and other relevant measures of clinical safety in a large number of subjects. The most frequently reported TEAEs were comparable between treatment arms and were consistent with what would be anticipated in a study of healthy subjects. Only a small number of subjects reported serious TEAEs during the study, all of which were expected events, except one event of spontaneous abortion. The majority of AESIs reported were injection site reactions; these were generally mild and resolved without intervention. Injection site erythema and injection site bruising were the most commonly reported local events, occurring in fewer than 5.0% of subjects overall. US-licensed tocilizumab was associated with a lower frequency of injection site erythema and injection site bruising; however, this finding was considered to be incidental and not clinically relevant, since all injection site reactions were mild, of short duration and resolved spontaneously. In addition, injection site bruising was most likely attributable to injection technique rather than treatment. No subject experienced systemic hypersensitivity.
Immunogenicity, as measured by the incidence of treatment-induced ADA and NAb against tocilizumab, was also comparable between treatment arms; however, the proportions of subjects who were NAb-positive in each group were too low to allow any conclusions to be drawn. More than 50% of subjects in each treatment group were ADA positive at any visit, but no relationship was observed between ADA status and pharmacokinetic or pharmacodynamic outcomes. Moreover, ADA status has been reported to have no effect on the efficacy or safety of tocilizumab in patients with rheumatoid arthritis [Citation18]. The ADA positivity rate was considerably higher than the reported 0.8–1.6% of patients with rheumatoid arthritis who became ADA positive in clinical trials with subcutaneous tocilizumab formulations [Citation4,Citation5,Citation18] but more in line with the proportions of healthy subjects who were ADA positive in other biosimilarity studies of intravenous tocilizumab formulations (14–42%) [Citation19,Citation20]. It is possible that this disparity in reported incidence of ADA positivity is a result of the use of more sensitive and drug tolerant ADA assay technologies in biosimilarity studies. It should be noted that ADA findings are highly dependent on the sample collection methods, and type, design and conduct of the assay used, with recent assays being more sensitive and specific than historically used assays; in addition, most immunoassays cannot be universally calibrated or validated, all of which limit the value of between-study comparisons [Citation21,Citation22]. The incidence of tocilizumab-specific NAbs was low for all three treatments (<3% for each).
Tocilizumab is an established treatment option for adults with moderate-to-severe rheumatoid arthritis who respond inadequately to synthetic or other biologic DMARDs [Citation1]. The observed benefit with tocilizumab in patients in the early phases of rheumatoid arthritis [Citation10], and knowledge that IL-6 contributes to the first phase of this disease, suggest that initiating tocilizumab in patients with newly diagnosed rheumatoid arthritis may be of particular benefit [Citation1]. Updated European League Against Rheumatism (EULAR) recommendations indicate that IL-6-pathway inhibitors may have some advantages over other biologics in patients with contraindications to/intolerance of conventional synthetic DMARDs and that less costly drugs, such as biosimilars, should be preferred over more costly ones provided they are similarly efficacious and safe, and in line with treatment recommendations [Citation23]. Currently, several effective treatment options for rheumatoid arthritis are available on the market, but these drugs are costly, which limits widespread use and contributes to inequities in access to best care both across regions and countries [Citation24]. The development of biosimilars has introduced price competition and led to a reduction in the net costs of biologics [Citation24]; tocilizumab biosimilars can be expected to contribute to this improved cost-effectiveness of therapy.
5. Conclusions
In this study, conducted in a large group of healthy subjects, pharmacokinetic equivalence was demonstrated for all three pairwise treatment comparisons of subcutaneously administered MSB11456, US-licensed tocilizumab, and EU-approved tocilizumab for all primary pharmacokinetic parameters. Pharmacodynamic results further support the similarity of MSB11456, US-licensed tocilizumab, and EU-approved tocilizumab. The safety profiles, local tolerability and immunogenicity of the three tocilizumab formulations have likewise been proven to be comparable. This study therefore supports the biosimilarity of MSB11455 to both US-licensed tocilizumab and EU-approved tocilizumab.
Abbreviations
ADA, antidrug antibody
AESI, adverse event of special interest
ANCOVA, analysis of covariance
AUC, area under the plasma concentration–time curve
AUC0-72, AUC from time zero to 72 hours
AUC0-∞, AUC from time zero extrapolated to infinity
AUC0-t, AUC from time zero (pre-dose) to the last sampling time at which a quantifiable concentration was obtained
AUE, area under the effect–time curve
BMI, body mass index
CI, confidence interval
CL/F, apparent total body clearance
Cmax, maximum plasma drug concentration
CRP, C-reactive protein
CV, coefficient of variation
DMARD, disease-modifying antirheumatic drug
Emax, maximum observed effect
Emin, minimum observed effect
EULAR, European League Against Rheumatism
GCV%, geometric coefficient of variation
IL-6, interleukin-6
IL-6 R, IL-6 receptor
LLOQ, lower limit of quantification
LS, least squares
MedDRA, Medical Dictionary for Regulatory Activities
Nab, neutralizing antibody
NR, not reported
SC, subcutaneous
SD, standard deviation
sIL-6R, soluble IL-6 R
t1/2, apparent terminal half-life
TEAE, treatment-emergent adverse effect
tEmax, time to Emax
tEmin, time to Emin
Author contributions
All authors were involved in conception and design, or analysis and interpretation of the data; the drafting of the paper or revising it critically for intellectual content; the final approval of the version to be published; and all authors agree to be accountable for all aspects of the work. All authors agree for the final version of the manuscript to be published.
Data availability and deposition
All data relevant to this study have been presented in this publication.
Declaration of interest
C Schwabe is an employee and shareholder of ACS. C Wynne is a shareholder of Christchurch Clinical Studies Trust Ltd. A Illes, M Ullmann, E Vincent, C Petit-Frere, and J Monnet are employees of Fresenius Kabi SwissBioSim GmbH. V Ghori and A Racault were employed by Fresenius Kabi SwissBioSim GmbH at the time of the study. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Research Involving Human Participants: Ethics approval
Prior to commencement of the study, the Clinical Study Protocol and all amendments, together with associated documents, were approved by the responsible Independent Ethics Committee.
All procedures performed in studies involving human participants were in accordance with ethical standards of the ICH guideline for GCP, applicable local regulations, and the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all individuals participating in the study before any study procedures were performed.
Prior presentation
Details of this study were presented as a poster at the American College of Rheumatology Convergence 2021 Online Conference (November 1–10, 2021) (Session: RA - Treatments Poster I: Comparative Effectiveness, Biosimilars, Withdrawal, & the Real World, Abstract ID: 1,060,194).
Supplemental Material
Download MS Word (104 KB)Acknowledgments
The sponsors acknowledge the participation of all healthy volunteers and the study site personnel involved in the clinical trial.
Medical writing support was provided by Caroline Spencer and Dr Sue Chambers (Rx Communications, Mold, UK), funded by Fresenius Kabi SwissBioSim.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
References
- Biggioggero M, Crotti C, Becciolini A, et al. Tocilizumab in the treatment of rheumatoid arthritis: an evidence-based review and patient selection. Drug Des Devel Ther. 2019;13:57–70
- Srirangan S, Choy EH. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2010;2:247–256.
- Rubbert-Roth A, Furst DE, Nebesky JM, et al. A review of recent advances using tocilizumab in the treatment of rheumatic diseases. Rheumatol Ther. 2018;5:21–42*A
- Genentech, Inc. Actemra® (tocilizumab) injection, for intravenous or subcutaneous use. Highlights of prescribing information. South San Francisco (CA): Genentech, Inc.; 2021 cited 2021 Apr 7]. Available from 2021 Apr 7: https://www.gene.com/download/pdf/actemra_prescribing.pdf
- Roche Registration Gmb H. RoActemra 20 mg/mL concentrate for solution for infusion. Summary of product characteristics. Grenzach-Wyhlen: Roche Pharma AG; 2020 cited 2021 Apr 7]. Available from 2021 Apr 7: https://www.ema.europa.eu/en/documents/product-information/roactemra-epar-product-information_en.pdf
- US Food & Drug Administration. Coronavirus (COVID-19) update: FDA authorizes drug for treatment of COVID-19. June 24, 2021. Available at: cited 2021 Apr 07https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-drug-treatment-covid-19.
- European Medicines Agency. EMA recommends approval for use of RoActemra in adults with severe COVID-19. 06 December 2021. Available at: cited 2022 Feb 23https://www.ema.europa.eu/en/news/ema-recommends-approval-use-roactemra-adults-severe-covid-19.
- Gupta S, Padappayil RP, Bansal A, et al. Tocilizumab in patients hospitalized with COVID-19 pneumonia: systematic review and meta-analysis of randomized controlled trials. J Investig Med. 2022;70(1):55–60.
- Conti P, Pregliasco FE, Calvisi V, et al. Monoclonal antibody therapy in COVID-19. J Biol Regul Homeost Agents. 2021;35:423–427.
- Scott LJ. Tocilizumab: a review in rheumatoid arthritis. Drugs. 2017;77:1865–1879.
- Ahmed I, Kaspar B, Sharma U. Biosimilars: impact of biologic product life cycle and European experience on the regulatory trajectory in the United States. Clin Ther. 2012;34:400–419.
- US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. guideline for industry. Rockville (MD): US FDA; April 2015 [cited 2021 Apr 7]. Available from 2021 Apr 7: https://www.fda.gov/media/82647/download*Guidance for evaluating biosimilars
- European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. London: EMA; 2014 cited 2021 Apr 7]. Available from 2021 Apr 7: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf
- Abdallah H, Hsu JC, Lu P, et al. Pharmacokinetic and pharmacodynamic analysis of subcutaneous tocilizumab in patients with rheumatoid arthritis from 2 randomized, controlled trials: SUMMACTA and BREVACTA. J Clin Pharmacol. 2017;57:459–468.
- Paccaly AJ, Kovalenko P, Parrino J, et al. Pharmacokinetics and pharmacodynamics of subcutaneous sarilumab and intravenous tocilizumab following single-dose administration in patients with active rheumatoid arthritis on stable methotrexate. J Clin Pharmacol. 2021;61:90–104.
- Zhang X, Georgy A, Rowell L. Pharmacokinetics and pharmacodynamics of tocilizumab, a humanized anti-interleukin-6 receptor monoclonal antibody, following single-dose administration by subcutaneous and intravenous routes to healthy subjects. Int J Clin Pharmacol Ther. 2013;51:443–455.
- European Medicines Agency. Guideline on the investigation of bioequivalence. London: EMA; 2010 cited 2021 Apr 12]. Available from 2021 Apr 12: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf
- Burmester GR, Choy E, Kivitz A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:1078–1085.
- Zhang H, Li X, Liu J, et al. A randomized phase-I pharmacokinetic trial comparing the potential biosimilar tocilizumab (QX003S) with the reference product (Actemra®) in Chinese healthy subjects. Ann Med. 2021;53:375–383.
- Zhang H, Wang H, Wei H, et al. A phase I clinical study comparing the tolerance, immunogenicity, and pharmacokinetics of proposed biosimilar BAT1806 and reference tocilizumab in healthy Chinese men. Front Pharmacol. 2021;11:609522.
- Schreitmüller T, Barton B, Zharkov A, et al. Comparative immunogenicity assessment of biosimilars. Future Oncol. 2019;15:319–329.
- Pineda C, Castañeda Hernández G, Jacobs IA, et al. Assessing the immunogenicity of biopharmaceuticals. BioDrugs. 2016;30:195–206.
- Smolen JS, Landewé RBM, Bijlsma JWJ, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79:685–699.
- Kay J, Schoels MM, Dörner T, et al. Consensus-based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis. 2018;77:165–174. •An evidence and consensus. •An evidence and consensusbased statement regarding the evaluation and use of biosimilars to treat rheumatological diseases from an international multidisciplinary task force on biosimilars.