
ABSTRACT
Introduction
Despite advances in the medical management of ulcerative colitis (UC), a subgroup of patients does not respond to currently available therapies. A number of novel drugs are in late stages of clinical development or have recently received regulatory approval for UC.
Areas covered
This review focuses on three drug classes that have recently been approved or are awaiting approval for UC: antibodies against interleukin (IL)-23, sphingosine-1-phosphate receptor (S1PR) modulators, and selective inhibitors of Janus kinases (JAK). We provide an overview of their mechanism of action and summarize available evidence for their efficacy and safety. Finally, we discuss expected future challenges in UC management.
Expert opinion
The evaluated drugs have demonstrated efficacy with an acceptable safety profile. IL-23 antagonists appear to be safest with very few (serious) adverse events, while the use of S1PR modulators or JAK inhibitors has been associated with infectious and cardiovascular/thromboembolic events, albeit in low numbers. Although advances in drug development are promising, there is an urgent need for (validated) biomarkers to guide rational treatment selection. The scarcity of head-to-head trials also complicates comparisons between available drugs. Breaking the therapeutic ceiling of efficacy in UC will require marked advances in management extending well beyond drug development.
1. Introduction
Ulcerative colitis (UC) is an inflammatory bowel disease (IBD) characterized by contiguous chronic mucosal inflammation of the colon with a relapsing-remitting course, resulting in high morbidity and impaired quality of life [Citation1]. Treatment goals in UC are stepwise: symptomatic response is the immediate treatment goal, symptomatic remission the intermediate goal, while endoscopic and even histologic remission in conjunction with symptomatic remission is the long-term target [Citation2]. Histologic remission is considered a potential adjunct to endoscopic remission representing a deeper level of healing. Molecular remission is currently being investigated.
Tumor necrosis factor (TNF) antagonists were the key therapeutic agent for moderate-to-severe UC for more than a decade [Citation3], with vedolizumab, a gut-selective anti-integrin antibody [Citation4], tofacitinib, a pan-Janus kinase (JAK) inhibitor [Citation5], and ustekinumab, an antibody against the p40 subunit of interleukin (IL)-12/23 [Citation6], entering clinical practice in recent years. Despite their proven efficacy, some patients either do not respond to treatment or lose response after initial improvement [Citation7]. Furthermore, particularly anti-TNF agents and tofacitinib are associated with potentially serious adverse effects [Citation8,Citation9], underscoring the necessity for further drug development in UC.
Management of moderate-to-severe UC in recent years has shifted from hospital- or practice-based infusion units to patients’ homes by reformulating existing biologics for subcutaneous use (infliximab, vedolizumab) [Citation10,Citation11], development of subcutaneously administered monoclonal antibodies (ustekinumab, adalimumab, golimumab) [Citation6], and the use of orally administered small molecules (tofacitinib, ozanimod) [Citation5]. Beyond convenience, small molecules also have the advantage of avoiding immunogenicity, a somewhat limiting factor in the efficacy of monoclonal antibodies, particularly TNF-antagonists [Citation12]. It should be noted, however, that novel biologics have lower immunogenicity. A number of novel agents have entered late stages of clinical development or have recently been approved for UC. In this review, we provide an overview of the efficacy and safety of emerging therapeutic agents for UC that have entered or completed Phase 3 trials: anti-IL-23 antibodies, sphingosine-1-phosphate receptor (S1PR) modulators, and selective JAK inhibitors (). We also outline clinical dilemmas surrounding their optimal use in practice and identify research priorities.
Table 1. Characteristics and primary endpoints from phase 3 trials of emerging therapies in ulcerative colitis.
Table 2. Secondary endpoints from phase 3 trials of emerging therapies in ulcerative colitis.
2. Body of review
2.1. Anti-IL-23 agents
The IL-23 axis is an emerging treatment target in IBD (both Crohn’s disease [CD] and UC), with ustekinumab, a human monoclonal antibody targeting the shared p40 subunit of IL-12 and IL-23 already in clinical use in in many countries [Citation6].
IL-23 is a heterodimeric cytokine with a unique p19 subunit and p40 subunit which it shares with IL-12 [Citation13]. IL-23 exerts its effect by binding to the IL-23 receptor and activating intracellular JAKs (predominantly TYK2 and JAK2) [Citation14]. IL-23 is mainly synthesized by dendritic cells and macrophages and is one of the key mediators in the T helper 17 (Th17) cell pathway [Citation15]. These helper cells are characterized by the production of IL-17, IL-21, IL-22, and IL-26. IL-17 producing cells are increased in the serum and mucosa of patients with IBD [Citation16]. Together with T cell receptor activation, IL-6 and transforming growth factor-β (TGF-β) induce the expression of retinoid-related orphan receptor-γt (ROR-γt), a transcription factor that promotes expression of IL-17 and IL-23 receptors on the surface of Th 17 cells. IL-23 can thus establish a positive feedback loop leading to the expansion of Th17 cells. IL-17 also stimulates innate lymphoid cells-3, natural killer T cells, and γδ T cells [Citation14] ().
Figure 1. The biology of interleukin-23 signaling. Abbreviations: IL – interleukin; R – receptor; ROR-γt – retinoid-related orphan receptor- γt; TGF-β – transforming growth factor beta. Created with BioRender.com.
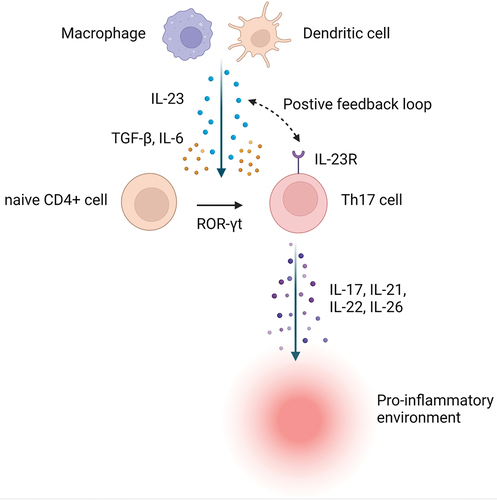
There is considerable cross-regulation between the IL-23 Th 17 and IL-12 Th1 pathway [Citation17,Citation18]. Broadly speaking, IL-12 may mediate more systemic effects, while IL-23 is implicated in mucosal immunology, although these notions remain unsubstantiated in humans [Citation19]. Selective IL-23 inhibitors are approved for psoriasis (guselkumab, risankizumab, tildrakizumab) and psoriatic arthritis (guselkumab) [Citation20–23]. It is uncertain how IL-23-specific monoclonal antibodies targeting the p19 subunit compare to agents targeting both IL-12 and IL-23 through the p40 subunit. Drugs currently in development for UC all target IL-23 in isolation.
2.1.1. Mirikizumab
2.1.1.1. Efficacy
Mirikizumab (LY3074828; Eli Lilly) is a humanized monoclonal antibody against the p19 subunit of IL-23, administered as three intravenous doses (every 4 weeks) during a 12-week induction period followed by subcutaneous maintenance dosing every 4 to 12 weeks. A Phase 2 trial in CD has recently been successfully completed [Citation24], a Phase 3 trial is ongoing (NCT03926130). The drug has not yet been approved for any indication.
The LUCENT-1 study (NCT03518086) assessed the efficacy and safety of mirikizumab as induction therapy in patients with moderate-to-severe UC [Citation25]. In this Phase 3, multicenter, randomized, double-blind, placebo-controlled study, patients with UC were randomized 3:1 to receive mirikizumab 300 mg intravenously every 4 weeks or placebo. Randomization was stratified by biologic failure, baseline corticosteroid use, baseline disease activity by Mayo score, and world region. The study excluded patients who had failed 3 or more biologic therapies or had been exposed to anti-IL-12/23 agents.
The primary endpoint of the induction study was clinical remission at week 12, defined by the modified Mayo score: a rectal bleeding subscore of 0; a stool-frequency subscore of ≤1, with a decrease of ≥1 point from baseline; and a Mayo endoscopy subscore of ≤1. Clinical remission was significantly higher among patients receiving mirikizumab compared to placebo (24.2% [210/868] vs. 13.3% [39/294], P < 0.001). Bio-naïve patients achieved clinical remission in 30.9%, while patients with previous biologic failure achieved clinical remission in 15.2%. Mirikizumab was superior to placebo in achieving endoscopic improvement and histo-endoscopic mucosal improvement (). Results of the maintenance trial are expected to be reported in the very near future.
2.1.1.2. Safety
The overall incidence of adverse events (44.5% [426/958] vs. 46.1% [148/321]) and serious adverse events (2.8% [27/958] vs. 5.3% [17/321]) was comparable between the mirikizumab and placebo group. The most common adverse events included nasopharyngitis, anemia, headache, and worsening of UC. There was no concerning signal for infections (serious, opportunistic, or total), cardiovascular events, or hepatic events. Infusion-related reactions were usually mild and did not lead to cessation of treatment.
2.1.2. Other anti-IL-23 agents
Three additional p19 antibodies, risankizumab, brazikumab, and guselkumab, are being investigated for use in UC, with guselkumab having the first Phase 2 results available.
Risankizumab (BI655066/ABBV066, Abbvie) is a humanized monoclonal antibody against the p19 subunit of IL-23. It has been approved for moderate to severe plaque psoriasis, and has been submitted for approval to regulatory authorities for CD. In UC, a Phase 2/3 induction trial (NCT03398148) and Phase 3 maintenance trial (NCT03398135) are ongoing.
Brazikumab (MEDI2070, previously known as AMG139, Astra Zeneca) is a human monoclonal antibody against the p19 subunit of IL-23. It has not yet been approved for any indication. It was studied in a Phase 2a study in CD [Citation26], with an ongoing Phase 2/3 trial (NCT03759288). In UC, a Phase 2 trial is ongoing (NCT03616821).
Guselkumab (CNTO1959, Janssen) is a human monoclonal antibody against the p19 subunit of IL-23, approved for moderate-to-severe plaque psoriasis. A Phase 2/3 trial is recruiting in CD (NCT03466411, GALAXI). In UC, a Phase 2b randomized, double-blind, placebo-controlled trial demonstrated superiority of guselkumab to placebo for clinical response during induction at week 12 (61.1% pooled for guselkumab at 200 mg and 400 mg doses vs. 27.6% for placebo; P < 0.001) (integrated Phase 2b/3 study; NCT04033445, QUASAR) [Citation27]. The efficacy of the 200 mg and 400 mg doses was comparable. Guselkumab also met key secondary endpoints of clinical remission, symptomatic remission, endoscopic improvement, and histo-endoscopic mucosal improvement. Only the 200 mg dose was superior to placebo for endoscopic normalization.
The efficacy and safety of guselkumab in UC were further studied in a somewhat audacious exploratory Phase 2a trial evaluating combination treatment of guselkumab and golimumab during induction (NCT03662542; VEGA) [Citation28]. Combination therapy was significantly more effective than either drug alone for clinical remission (36.6% for combination, 21.1% for guselkumab alone, 22.2% for golimumab alone) and endoscopic improvement (49.3% for combination, 29.6% for guselkumab alone, 25.0% for golimumab alone). The patients had a short disease duration and were naïve to biologics. Reassuringly, the rates of adverse events were comparable across treatment groups. One patient on combination therapy had serious influenza infection and sepsis. These results open the way to further combination trials which may eventually lead to greater efficacy success than ever seen before.
2.2. Sphingosine-1-phosphate receptor modulators
S1P is a multifunctional molecule, synthesized from sphingosine, an endogenous cellular sphingolipid, through phosphorylation by sphingosine kinases [Citation29,Citation30]. S1P is secreted from cells, thereby establishing a concentration gradient: concentrations are high in the blood and lymph, but low in the interstitial fluid [Citation31]. S1P signals through five subtypes of G-protein coupled receptors (S1PR1-5). The expression of these receptors varies by tissue, and the function of each receptor is highly dependent on the cell type on which it is expressed [Citation32]. Under physiological conditions, the S1P receptor on lymphocytes responds to an S1P gradient, which drives lymphocyte egress from lymphoid tissues into the systemic circulation toward the site of inflammation (). This is mediated by S1PR1, which also has a role in angiogenesis, heart rate regulation [Citation33] and has been associated with fibrosis. S1PR2/3 are involved in regulation of endothelial barrier function, fibrosis, and vasoconstriction. S1PR4 have more limited expression in the hematopoietic and lymphoid tissue, while S1PR5 are mainly expressed in the central nervous system. S1PR4 influence T cell differentiation and modulate cytokine expression, S1PR5 mediate natural killer cell and monocyte trafficking, and maintain endothelial barrier integrity [Citation29].
Figure 2. The biology of sphingosine-1-phosphate signaling. Abbreviations: S1P – sphingosine-1-phosphate; S1PR – sphingosine-1-phosphate receptor. Created with BioRender.com.
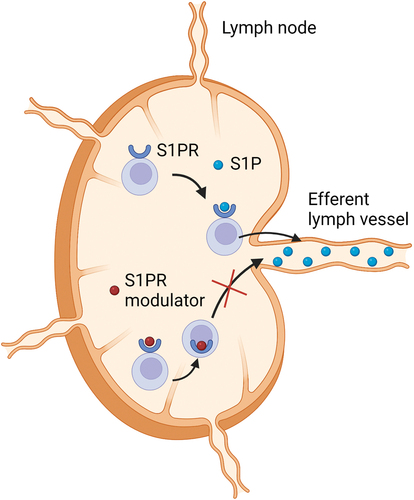
S1P receptor modulators bind to one of the S1PR on activated lymphocytes, which leads to internalization of the receptor and subsequent loss of responsiveness to the S1P gradient [Citation32]. This results in retention of lymphocytes within the lymph nodes and thus inhibition of lymphocyte trafficking to inflammatory sites with potentially preserved immune surveillance as circulating monocytes, natural killer cells, and natural killer T cells were affected only minimally in a study of patients with multiple sclerosis [Citation34]. The majority of patients develop peripheral lymphopenia during treatment with these agents, but this pharmacodynamic effect does not seem to correlate with efficacy.
2.2.1. Ozanimod
2.2.1.1. Efficacy
Ozanimod (RPC-1063) is a modulator of S1PR1 and S1PR5, with a 27-fold selectivity for S1PR1 over S1PR5 and 10,000-fold greater selectivity for S1PR1 in comparison with S1PR2-4 [Citation35]. The drug has been approved for relapsing-remitting multiple sclerosis and UC (Zeposia ®, Bristol Myers Squibb). A Phase 3 study in CD is ongoing (NCT03464097).
Approval of ozanimod for UC was based on the True North study (NCT02435992), which assessed the efficacy and safety of ozanimod as induction and maintenance [Citation36]. In this Phase 3, multicenter, randomized, double-blind, placebo-controlled study, patients with UC were randomized 2:1 to receive oral ozanimod hydrochloride at a dose of 1 mg per day (equivalent to 0.92 mg of ozanimod) or placebo in cohort 1 or to receive open-label ozanimod at the same daily dose in cohort 2 in the 10-week induction period. Patients were assigned to cohort 2 with open-label dosing once the pre-specified number for patients with previous anti-TNF exposure was reached.
The primary endpoint of the induction study was clinical remission at week 10, defined by components of the Mayo score: a rectal bleeding subscore of 0; a stool-frequency subscore of ≤1, with a decrease of ≥1 point from baseline; and an endoscopy subscore of ≤1. Clinical remission was significantly higher among patients receiving ozanimod compared to placebo (18.4% [79/429] vs. 6.0% [13/216], P < 0.001) [Citation36]. Ozanimod was also superior to placebo in achieving endoscopic improvement and mucosal healing (). In a subgroup analysis, stratified by previous anti-TNF exposure, the rate of clinical remission was greater in naïve (22.1% [66/299]) than experienced (10.0% [13/130]) patients.
At week 10, patients with a clinical response (a reduction of ≥3 points and of ≥30% from baseline in the total Mayo score or a reduction of ≥2 points and of ≥35% from baseline in the three-component Mayo score, plus a reduction of ≥1 point in the rectal-bleeding score or an absolute rectal-bleeding score of ≤1 point) to ozanimod underwent re-randomization in a 1:1 ratio, to receive ozanimod at the same dose or placebo for the maintenance period through week 52. At week 52, clinical remission was significantly higher among patients who received ozanimod than among those who received placebo during maintenance (37.0% [85/230] vs. 18.5% [42/227], P < 0.001). Ozanimod was superior to placebo in endoscopic improvement, mucosal healing, maintenance of clinical remission, and durable clinical remission (). In a subgroup analysis, stratified by previous anti-TNF exposure, the rate of clinical remission was greater in naïve (40.9% [63/154]) than experienced (28.9% [22/76]) patients.
2.2.1.2. Safety
The overall incidence of adverse events was comparable between the ozanimod and placebo group during induction (39.9% [318/796] pooled over both ozanimod cohorts vs. 38.0 [82/216] for placebo). During maintenance the incidence of adverse events was higher in the ozanimod group than in the placebo group (49.1% [113/230] vs. 36.6% [83/227]) [Citation36]. However, the rate of serious adverse events was comparable across all groups both in induction and maintenance (ozanimod: 5.0% [40/796] for induction, 5.2% [12/230] for maintenance; placebo: 3.2% [7/216] for induction, 7.9% [18/227] for maintenance). Based on previous associations with S1PR modulation, the following adverse events of special interest were examined in the True North study: bradycardia, cardiac conduction abnormalities (second-degree and higher atrioventricular block), macular edema, cancer, serious or opportunistic infection, pulmonary toxicity, and hepatotoxicity [Citation37,Citation38].
Bradycardia (heart rate <60 beats per minute) was reported more frequently with ozanimod than with placebo during induction (0.6% [5/796] pooled over both ozanimod cohorts vs. 0 for placebo), but not maintenance (no events in either group). No atrioventricular block beyond second-degree Mobitz type 1 was observed; the latter was detected in one patient. Macular edema occurred in three patients receiving ozanimod and resolved with treatment discontinuation. Five malignancies were reported: basal-cell carcinoma in one patient who received ozanimod during the induction period, basal-cell carcinoma and rectal adenocarcinoma in one patient each who received ozanimod during the induction and maintenance periods, and adenocarcinoma of the colon and breast cancer in one patient each who received ozanimod during the induction period and placebo during the maintenance period.
From baseline to week 10, the absolute lymphocyte count decreased by a mean of approximately 54% in ozanimod-treated patients. During the 52-week study, 17 patients had an absolute lymphocyte count of less than 200 cells per mm3, which subsequently increased and remained at a level at or above 200 cells per mm3 during treatment with ozanimod. No patient with a serious or opportunistic infection had an absolute lymphocyte count of less than 200 cells per mm3. Serious infection was observed in less than 2% of the patients in each group during both the induction and maintenance period. Herpes zoster infection occurred in 0.4% (3/796) of ozanimod-treated patients during induction and 2.2% (5/230) during maintenance, this infection did not occur in any patient who did not receive ozanimod. One ozanimod-treated patient had a hypertensive crisis on day 1 of the induction period; the event was moderate and resolved on the same day without treatment interruption. During the maintenance period, hypertensive crisis occurred in one patient each in the ozanimod and the placebo group; neither resulted in treatment discontinuation. Elevated liver aminotransferase levels were more common with ozanimod treatment than with placebo (2.1% [17/796] pooled over both ozanimod induction cohorts vs. 0; 4.8% [11/230] vs. 0.4% [1/227] for placebo). There were no patients with severe liver injury. Progressive multifocal leukoencephalopathy was not observed in this study. This is a rare viral central nervous system infection that has been observed in patients with multiple sclerosis treated with fingolimod (4.75 per100000patient-years) and ozanimod (a single case reported to date) [Citation39]. Long-term safety data from multiple sclerosis studies are reassuring: no high-degree atrioventricular block was observed, malignancy rates remained low with prolonged exposure, and macular edema only developed in patients with underlying risk factors [Citation40].
The safety concerns outlined above have led to recommendations for pre-treatment assessment [Citation41]. All patients should undergo electrocardiography to screen for preexisting (conduction) abnormalities and caution should be applied to patients receiving beta-blockers or calcium channel blockers, although ozanimod is not contraindicated in these patients. Patients with a resting heart rate below 55 beats per minute, Mobitz type I atrioventricular block, a remote history of myocardial infarction or heart failure (New York Heart Association class I/II) should be monitored during 6 hours after the first dose with hourly pulse and blood pressure measurement. To avoid bradycardia, the dose of ozanimod is titrated over 7 days (patients receive color-coded starter packs of pills). Recent myocardial infarction, unstable angina, stroke, transitory ischemic attack (within 6 months), and heart failure (New York Heart Association class III/IV) are contraindications to the use of ozanimod. Patients should also undergo a complete blood count to screen for lymphopenia and perform liver enzyme testing. Ophthalmological evaluation is recommended for patients at higher risk of macular edema (history of uveitis, diabetes, or retinal disease). Vaccination against varicella zoster is recommended in patients who are IgG negative. Concurrent treatment with monoamine oxidase inhibitors is contraindicated due to potential pharmacokinetic interactions.
2.2.2. Etrasimod
Etrasimod (Arena Pharmaceuticals) is a S1PR1, S1PR4, and S1PR5 modulator, which demonstrated superiority over placebo in a Phase 2 study in UC at the 2 mg dose (clinical remission: 33% vs. 8.1%; endoscopic improvement: 41.8% vs. 17.8%), but not at the 1 mg dose [Citation42]. No adverse events, unexpected for an S1P modulator were observed. The Phase 3 studies induction and maintenance completed enrollment in 2021 (NCT03945188, NCT03996369), however the results have not yet been reported. Etrasimod has not yet been approved for any indication.
2.3. Janus kinase inhibitors
JAKs are tyrosine kinases that bind different intracellular cytokine receptors and regulate immune responses, wound healing, and hematopoietic differentiation. The JAK family consists of four intracellular proteins: JAK1, JAK2, JAK3, and TYK2. Their activation requires homo- or heterodimerization with subsequent autophosphorylation [Citation43,Citation44]. Different combinations of JAKs are associated with different cytokine receptors. From a mechanistic viewpoint, JAK1 is the molecule involved in IL-2, IL-4, IL-6, IL-15, and interferon signaling, whereas JAK2 is more involved in signaling of hematopoietic cytokines, prolactin, and growth hormone, which underscores that JAK1 is the most likely therapeutic target of JAK inhibitors in inflammatory bowel disease, although the end effect of selective JAK inhibition is difficult to predict due to cytokine pleiotropism and dose-dependent selectivity [Citation45].
Despite sound in vitro data on the selectivity of JAK inhibitors, its clinical impact is probably dependent not only on the agent itself, but also its dose, cell type, tissue penetration, and individual genetic background [Citation46]. The clinical consequences of JAK-1 selectivity (as claimed for filgotinib and upadacitinib) in IBD remain unknown – both in terms of efficacy and safety. A meta-analysis of JAK inhibition across other immune-mediated diseases in addition to IBD showed a numerically higher rate of herpes zoster with nonselective JAK inhibitors (tofacitinib, baricitinib) compared to selective JAK1 inhibitors (upadacitinib, filgotinib) [Citation47]. Further analyses based on Phase 3 data in IBD are still pending.
2.3.1. Filgotinib
2.3.1.1. Efficacy
Filgotinib is an oral preferential inhibitor of JAK1. It has recently been approved for the treatment of UC in Europe (Jyseleca ®, Galapagos). A Phase 2 trial in CD has been successfully completed [Citation48] and a Phase 3 trial for this indication is ongoing (NCT02914561).
The SELECTION study (NCT02914522) evaluated the efficacy and safety of filgotinib for the treatment of moderate-to-severe UC [Citation49]. In this Phase 2b/3, randomized, double-blind, placebo-controlled study patients with UC were included in two induction studies, based on previous exposure to biologic therapy: induction study A (biologic-naïve) or induction study B (biologic-experienced). They were randomly assigned in a 2:2:1 ratio to receive filgotinib 200 mg, filgotinib 100 mg or placebo once per day for 10 weeks. Patients who had previously received other JAK inhibitors were excluded.
The primary endpoint of the induction study was clinical remission at week 10, defined by components of the Mayo score: a rectal bleeding subscore of 0; a stool-frequency subscore of ≤1, with a decrease of ≥1 point from baseline; and an endoscopy subscore ≤1. Among biologic-naïve patients in induction study A, 26.1% (64/245) in the 200 mg arm (P = 0.016 vs. placebo), 19.1% (53/277) in the 100 mg arm (P = 0.338 vs. placebo), and 15.3% (21/137) in the placebo arm attained clinical remission at week 10. Filgotinib 200 mg, but not 100 mg, was also superior to placebo for endoscopic and histological remission ().
Among biologic-experienced patients in induction study B, 11.5% (30/262) in the 200 mg arm (P = 0.010 vs. placebo), 9.5% (27/285) in the 100 mg arm (P = 0.065 vs. placebo), and 4.2% (6/142) in the placebo arm attained clinical remission at week 10. Neither of the active arms were superior to placebo in achieving endoscopic remission, the 200 mg arm demonstrated superiority for histological remission.
Patients who either had clinical remission or clinical response at week 10 were re-randomized 2:1 at week 11 to either continue their induction filgotinib regimen or to receive placebo. For this re-randomization, patients were stratified based on systemic corticosteroid and immunosuppressant use on day 1, exposure to one versus more than one biologic agent, and participation in induction study A (biologic-naïve patients) versus participation in induction study B (biologic-experienced patients). In total, 664 patients (391 [58.9%] of them from induction study A, 273 [41.1%] from induction study B) entered the maintenance study. At week 58, 37.2% (74/199) of patients in the 200 mg arm had clinical remission, compared to 11.2% (11/98) in the placebo group (P < 0.0001). In the 100 mg arm, 23.8% (41/172) had clinical remission, compared to 13.5% (12/89) in the placebo group (P = 0.042). The treatment effect of filgotinib 200 mg for clinical remission at week 58 was consistent across prespecified subgroups: prior anti-TNF failure (22.7% [17/75] vs. 5.3% [2/38]; P < 0.05), prior vedolizumab failure (27.5% [11/40] vs. 0 [0/21]; P < 0.05), and previous failure of both classes of biologics (25.8% [8/31] vs- 0 [0/19]; P < 0.05). Filgotinib 200 mg was superior to placebo both for endoscopic and histological remission ().
2.3.1.2. Safety
Both during induction and maintenance, the rate of adverse events was similar for the 200 mg arm (53.6% during induction, 66.8% during maintenance), the 100 mg arm (50.4% during induction, 60.3% during maintenance), and placebo (56.3% during induction, 62.2% during maintenance). Serious adverse events occurred in 5.0% (28/562) of patients given filgotinib 100 mg, 4.3% (22/507) of patients given filgotinib 200 mg, and 4.7% (13/279) of patients given placebo during induction. In the maintenance study, serious adverse events were reported in 4.5% (8/179) of patients given filgotinib 100 mg and 7.7% (7/91) of patients in the respective placebo group, by 4.5% (9/202) of patients in the filgotinib 200 mg group, and no patients in the respective placebo group.
The incidence of infections and serious infections, even when adjusted for duration of previous drug exposure, was similar between the filgotinib and placebo groups in both the induction and maintenance studies. Six patients from all studies had herpes zoster infections, none of which were serious or prompted discontinuation of the study drug. One patient who received filgotinib 200 mg in the induction study had pulmonary embolism. No patients who receiving filgotinib had venous thromboses or pulmonary embolisms in the maintenance study. Two patients who received placebo both in the induction study and in the maintenance study had venous thromboses.
Non-melanoma skin cancers occurred in three patients in the induction studies and one patient in the maintenance study, all had previously been treated with thiopurines. Other malignancies were reported in three patients (colon cancer, breast cancer, and malignant melanoma; all in patients who received filgotinib).
In the induction studies, a small increase in serum lipid concentrations was observed in the filgotinib groups, which remained stable during maintenance. The proportion of patients with abnormal creatine kinase increase was higher in the filgotinib groups than in the placebo groups in all three studies, without clinical rhabdomyolysis. No new safety concerns were observed in a 4-year follow-up study of filgotinib in patients with rheumatoid arthritis [Citation50].
Findings from animal studies suggested potential issues with male reproductivity [Citation51]. Two targeted clinical studies are addressing this concern in humans, which both have completed recruitment (NCT03926195, NCT03201445). Full results have not been published as of yet, but a press release reported numerically higher rates of decreases in sperm count with placebo rather than filgotinib. The Food and Drug Administration’s (FDA) black box warning against thromboembolic events based on data for tofacitinib was extended to baricitinib and upadacitinib in 2021 and would probably have included filgotinib if the sponsor had not abandoned the pursuit of approval of the agent for rheumatoid arthritis in the United States [Citation52].
2.3.2. Upadacitinib
2.3.2.1. Efficacy
Upadacitinib (Rinvoq ®, Abbvie) is an oral JAK1 preferential inhibitor. It has been approved in the United States for UC and submitted for regulatory approval in Europe. A Phase 2 trial in CD has been successfully completed [Citation53] and two Phase 3 trials for this indication have completed recruitment (NCT03345836, NCT03345849).
The efficacy and safety of upadacitinib for the treatment of moderate-to-severe UC was evaluated in two Phase 3 induction trials (U-ACCOMPLISH [NCT03653026], U-ACHIEVE [NCT02819635]) [Citation54,Citation55] and one maintenance trial (U-ACHIEVE Maintenance [NCT02819635]) [Citation56]. In the two induction studies, patients with moderate-to-severe UC who had inadequate response, loss of response, or intolerance to aminosalicylates, immunosuppressants, corticosteroids and/or biologics were randomized 2:1 to receive upadacitinib 45 mg daily or placebo for 8 weeks. Randomization was stratified for baseline corticosteroid use, inadequate response to biologics, and adapted Mayo score at baseline (≤7 vs. >7).
The primary endpoint was again clinical remission at week 8, defined by the components of the Mayo score: a rectal bleeding subscore of 0; a stool-frequency subscore of ≤1, and not greater than at baseline; and an endoscopy subscore ≤1. In U-ACCOMPLISH, 33.5% of 341 patients in the upadacitinib group and 4.1% of 174 patients in the placebo group achieved clinical remission (P < 0.001) [Citation54]. Treatment with upadacitinib was also significantly more likely than placebo to result in endoscopic improvement, endoscopic remission, and histologic-endoscopic mucosal improvement (). In U-ACHIEVE, 26.1% of 319 patients in the upadacitinib group and 4.8% of 154 patients in the placebo group achieved clinical remission (P < 0.001). Upadacitinib was superior to placebo in endoscopic, endoscopic remission, and histologic-endoscopic mucosal improvement (). Although upadacitinib was superior to placebo both in patients with and without previous inadequate response to biologics, the difference versus placebo was numerically greater among patients who had not had inadequate response to biologics [Citation57].
At week 8, responders to upadacitinib were re-randomized 1:1:1 at to receive upadacitinib 30 mg (n = 154), 15 mg (n = 148), or placebo (n = 149) through week 52 [Citation56]. Re-randomization was stratified by previous biologic failure status, induction clinical remission status and baseline corticosteroid use. Both the 30 mg dose (51.7%; P < 0.001) and the 15 mg dose (42.3%; P < 0.001) were superior to placebo (12.1%) in achieving clinical remission at week 52. Upadacitinib also met all the secondary endpoints: endoscopic improvement, endoscopic remission and histologic-endoscopic mucosal improvement (). Both doses of upadacitinib were superior to placebo in maintenance of clinical remission (subset of patients in clinical remission at the end of induction) (30 mg: 69.7%; 15 mg: 59.2%; placebo: 22.2%; P < 0.001) and maintenance of endoscopic improvement (subset of patients with endoscopic improvement at the end of induction) (30 mg: 69.5%; 15 mg: 61.6%; placebo: 18.9%; P < 0.001).
2.3.2.2. Safety
With the exception of the U-ACCOMPLISH induction study (52.9% for upadacitinib vs. 39.5% for placebo) the rate of adverse events was comparable between upadacitinib (U-ACHIEVE induction: 56.4%; maintenance: 30 mg 78.6%, 15 mg 77.7%) and placebo (U-ACHIEVE induction: 60.0%; maintenance: 75.8%) [Citation54–56]. The rate of serious adverse events was lower with upadacitinib (U-ACHIEVE induction 2.5%, 30 mg maintenance 5.8%, 15 mg maintenance 6.8%; U-ACCOMPLISH induction 3.2%) than with placebo (U-ACHIEVE induction 5.8%, maintenance 12.8%; U-ACCOMPLISH induction 4.5%).
The rate of serious infections was low and comparable across all groups throughout the whole study. Herpes zoster was only observed in upadacitinib-treated patients with an incidence around 4%. Two venous thromboembolisms occurred in the 30 mg maintenance arm and one in the placebo arm during induction. One major cardiovascular event occurred in the placebo arm, none occurred in the upadacitinib arms. Nonmelanoma skin cancer occurred in two patients receiving upadacitinib 30 mg, but not in other groups. Other malignancies were found at similar rates across all groups (0.7–1.3%). In terms of laboratory abnormalities, elevations in creatine kinase, elevations in liver enzymes, and neutropenia were more commonly recorded with upadacitinib, but this was not the case for anemia and lymphopenia. With low numbers of adverse events of special interest, a potential dose-dependency is difficult to evaluate.
3. Conclusion
A number of drugs belonging to different classes have recently shown efficacy in Phase 3 trials in UC, with some of them already receiving regulatory approval. These include anti-IL-23 antibodies, S1PR modulators, and selective JAK-1 inhibitors. With the increasing number of available therapeutic agents, their positioning in treatment algorithms has become challenging. Of the three classes, IL-23 inhibitors have the most favorable safety profile, while both S1PR modulators and selective JAK inhibitors carry a somewhat higher risk of cardiovascular and infectious adverse events. It remains to be seen whether selective JAK1 inhibition can offset this risk. In the absence of head-to-head studies, comparing efficacy is difficult, but network meta-analyses suggest that upadacitinib could be the single most effective agent. Combinations of anti-TNF agents and anti-IL-23 antibodies are promising to further break the therapeutic ceiling in UC.
4. Expert opinion
The expansion of the therapeutic armamentarium for UC holds both great promise to improve treatment outcomes and great responsibility to select the right drug for the right patient taking into account the activity and severity of the patient’s disease, their comorbidities, prior treatment, and the perceived balance between risks and benefits. In the absence of head-to-head randomized trials [Citation58] or trials stratifying patients by the type and not only number of previously used biologics, temporary conclusions may be drawn from network meta-analyses of randomized controlled trials [Citation59,Citation60] and emerging data from propensity score analyses of real-world cohorts [Citation61,Citation62].
Two network meta-analyses of registration trials in UC have recently been published, providing a benchmark of efficacy and safety for all currently available therapeutic agents for moderate-to-severe UC [Citation59,Citation60]. Both studies identified upadacitinib as the best performing agent for the induction of clinical remission. Infliximab was ranked second and was superior to all other treatment agents except tofacitinib [Citation60]. Upadacitinib also ranked highly for inducing endoscopic improvement (Mayo endoscopic score ≤1): being the best performing agent in one analysis [Citation59] and second to infliximab 10 mg/kg in the other [Citation60]. Focusing on patients with previous anti-TNF exposure, upadacitinib was again ranked highest both for clinical remission and endoscopic improvement [Citation60]. However, upadacitinib also ranked highest in terms of all adverse events in both analyses [Citation59,Citation60], although the rate of serious adverse events or adverse events leading to drug discontinuation was no higher than placebo for any of the drugs [Citation60]. In a more detailed analysis of adverse events focusing on infections, only tofacitinib was found to have a higher risk than placebo. Aside from the usual caveats about indirect comparisons, two additional inherent limitations of these analyses should be borne in mind. Firstly, the included trials span over a period of 15 years and patients included in contemporary trials conceivably had more refractory disease having failed multiple other treatments. Secondly, the definition of clinical remission in recent trials is stricter, mandating a rectal bleeding score of 0, which may have led to an underestimation of effect on clinical remission for ozanimod, filgotinib, upadacitinib, and tofacitinib compared to infliximab, adalimumab, ustekinumab, and vedolizumab.
Balancing efficacy and safety is a continuous challenge in the treatment of UC. JAK inhibitors are particularly problematic in this respect as a black box warning against thromboembolic and major cardiovascular events was issued by the FDA for tofacitinib and subsequently extended to upadacitinib, a selective JAK1 inhibitor, upon authorization in the United States [Citation52]. Enthusiasm for JAK inhibitors may decrease further in light of the recently published ORAL Surveillance study [Citation63]. In patients with rheumatoid arthritis 50 years or older with at least one additional cardiovascular risk factor, treatment with tofacitinib was associated with an increased risk of major cardiovascular events, cancer (excluding non-melanoma skin cancer), and opportunistic infections compared to anti-TNF agents. Due to a signal of increased deaths due to pulmonary embolism in the tofacitinib 10 mg twice daily arm, the dose was switched to 5 mg twice daily mid-trial. Although patients with UC are generally younger than the population in ORAL Surveillance and conceivably have a lower risk of major cardiovascular events, these risks should be discussed with patients upon prescription.
Although more selective JAK inhibition may be associated with a lower risk of herpes zoster [Citation47], the cardiovascular risk profile of these newer agents is expected to emerge during post-marketing surveillance and long-term extension studies of completed Phase 3 trials. No infectious adverse events were associated with lymphopenia, induced by S1PR modulators, although lymphocyte depletion could predispose patients to viral infections. None of these novel oral agents can be used in women planning pregnancy and the potential of filgotinib to cause male reproductive toxicity is under investigation. Of the novel drug classes for UC, IL-23 inhibitors have the most favorable safety profile and this could prove to be a major advantage of this drug class.
Increased selectivity also leads raises questions about efficacy. The exclusion of patients with prior failure of ustekinumab from trials of IL-23 inhibitors and patients with prior failure of tofacitinib from trials of novel JAK inhibitors precludes informed speculation. Intriguingly, p19 inhibition with risankizumab was more effective than nonselective p40 inhibition with ustekinumab in psoriasis [Citation21,Citation64], a substantial proportion of patients with inadequate response to ustekinumab subsequently responded to selective p19 inhibition with guselkumab [Citation20]. Currently, only limited indirect comparisons are possible in IBD, as none of the trials in UC have an active control arm with ustekinumab. In a Phase 2 induction study in CD, the rates of clinical remission and endoscopic response, but not remission, were numerically higher with guselkumab compared to ustekinumab, but the difference did not exceed 10 percentage points [Citation65].
An additional exciting aspect partially addressed by ongoing trials is combination biological therapy or combining biologics with small molecules (NCT03662542, evaluating the efficacy of combined therapy with golimumab and guselkumab versus each of the two drugs in monotherapy). Reports on this approach are currently limited to case series [Citation66–68], a preliminary report on the results of an open-label uncontrolled trial in CD with two agents of known efficacy (adalimumab, vedolizumab) (NCT02764762), and a trial in UC combining a treatment of known efficacy (golimumab) with guselkumab [Citation28]. In real-world clinical practice, combination biological therapy has been attempted either due to uncontrolled luminal disease or quiescent luminal disease with uncontrolled extraintestinal manifestations or concomitant immune-mediated inflammatory disease. Despite largely encouraging results on efficacy, it should be borne in mind that most series were retrospective and criteria for gauging success were not as stringent. Furthermore, 10% of patients in one of the series had an opportunistic infection or an infection necessitating hospitalization [Citation68]. In rheumatology, combination therapy resulted in higher rates of adverse events and marginal gains in efficacy [Citation69]. Ideally, combination biological or small molecule therapy should be undertaken in a controlled trial or at least a thorough and frank discussion with the patient including due consideration of surgical therapy.
Despite considerable advances and new therapeutic agents, the field of IBD is notable for the absence of biomarkers to guide treatment choices, which remain largely empirical. Translational research aiming to address this dilemma and future trials exploring treatment sequencing may help inform clinical practice and break the therapeutic efficacy ceiling.
Article highlights
IL-23 antagonists (mirikizumab, guselkumab, risankizumab, brazikumab), S1PR modulators (ozanimod, etrasimod), and selective JAK inhibitors (filgotinib, upadacitinib) are in late clinical development or have recently received regulatory approval
IL-23 antagonists appear to have a very favorable safety profile
Oral administration of S1PR modulators and JAK inhibitors is convenient and avoids immunogenicity; cardiovascular and infectious adverse events are potential concerns
It remains to be seen whether selective JAK1 inhibitors have a more favorable efficacy/safety profile than pan-JAK inhibitors
Declaration of interest
J Hanzel has received speaker fees from Abbvie, Janssen, and Takeda; consulting fees from Alimentiv Inc. J Grootjans has received speaker fees from Dr. Falk, Janssen, and GSK. G D’Haens has received consultancy and/or speakers’ fees from AbbVie, ActoGeniX, AIM, Allergan, Amgen, Arena Pharmaceuticals, Boehringer Ingelheim, Celgene/Receptos, Celltrion, Cosmo Technologies, Elan Pharmaceuticals, enGene, Falk, Ferring, Galapagos, Genentech, Gilead Sciences, Giuliani SpA, Given Imaging, Gossamer Bio, GSK, Janssen, Lilly, MSD, Neovacs, Novo Nordisk, Otsuka, PDL BioPharma, Pfizer, Progenity, Prometheus Laboratories, Robarts Clinical Trials, Salix, Schering-Plough, Seres/Nestlé, SetPoint, Shire, Takeda, Tillotts, UCB Pharma, Versant, and Vifor Pharma; research grants from AbbVie, Falk, Given Imaging, Janssen, MSD, and PhotoPill; and speaking honoraria from AbbVie, Ferring, MSD, Norgine, Shire, Tillotts, Tramedico, and UCB Pharma. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- Ungaro R, Mehandru S, Allen PB, et al. Ulcerative colitis. Lancet. 2017;389:1756–1770.
- Turner D, Ricciuto A, Lewis A, et al. STRIDE-II: an update on the selecting therapeutic targets in inflammatory bowel disease (STRIDE) initiative of the international organization for the study of IBD (IOIBD): determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology. 2021;160:1570–1583.
- Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476.
- Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710.
- Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376:1723–1736.
- Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381:1201–1214.
- Roda G, Jharap B, Neeraj N, et al. Loss of response to anti-TNFs: definition, epidemiology, and management. Clin Transl Gastroenterol. 2016;7:e135.
- Singh J, Wells G, Christensen R, et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev. 2011; CD008794. 10.1002/14651858.CD008794.pub2.
- Sandborn WJ, Panes J, Sands BE, et al. Venous thromboembolic events in the tofacitinib ulcerative colitis clinical development programme. Aliment Pharmacol Ther. 2019;50:1068–1076.
- Sandborn WJ, Baert F, Danese S, et al. Efficacy and safety of vedolizumab subcutaneous formulation in a randomized trial of patients with ulcerative colitis. Gastroenterology. 2020;158:562–572.
- Schreiber S, Ben-Horin S, Leszczyszyn J, et al. Randomized controlled trial: subcutaneous vs intravenous infliximab CT-P13 maintenance in inflammatory bowel disease. Gastroenterology. 2021;160:2340–2353.
- Bots SJ, Parker CE, Brandse JF, et al. Anti-drug antibody formation against biologic agents in inflammatory bowel disease: a systematic review and meta-analysis. BioDrugs. 2021;35:715–733.
- Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725.
- Moschen AR, Tilg H, Raine T. IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting. Nat Rev Gastroenterol Hepatol. 2019;16:185–196.
- Patel Dhavalkumar D, Kuchroo Vijay K. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity. 2015;43:1040–1051.
- Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70.
- Becker C, Dornhoff H, Neufert C, et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol. 2006;177:2760–2764.
- O’Connor JW, Kamanaka M, Booth CJ, et al. A protective function for interleukin 17A in T cell–mediated intestinal inflammation. Nat Immunol. 2009;10:603–609.
- Uhlig HH, McKenzie BS, Hue S, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–318.
- Langley RG, Tsai TF, Flavin S, et al. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: results of the randomized, double‐blind, phase III NAVIGATE trial. Br J Dermatol. 2018;178:114–123.
- Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650–661.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390:276–288.
- Deodhar A, Helliwell PS, Boehncke W-H, et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395:1115–1125.
- Sands BE, Peyrin-Biroulet L, Kierkus J, et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with crohn’s disease. Gastroenterology. 2021 Epub ahead of print;160:S–37.
- D’Haens G, Kobayashi T, Morris N, et al. OP26 efficacy and safety of mirikizumab as induction therapy in patients with moderately to severely active ulcerative colitis: results from the phase 3 LUCENT-1 study. J Crohn’s Colitis. 2022;16:i028–i029.
- Sands BE, Chen J, Feagan BG, et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, in patients with moderate to severe crohn’s disease: a phase 2a study. Gastroenterology. 2017;153:77–86.
- Dignass A, Rubin D, Bressler B, et al. OP23 the efficacy and safety of guselkumab induction therapy in patients with moderately to severely active ulcerative colitis: phase 2b QUASAR study results through week 12. J Crohn’s Colitis. 2022;16:i025–i026.
- Sands BE, Feagan BG, Sandborn WJ, et al. OP36 efficacy and safety of combination induction therapy with guselkumab and golimumab in participants with moderately-to-severely active ulcerative colitis: results through week 12 of a phase 2a randomized, double-blind, active-controlled, parallel-group, multicenter, proof-of-concept study. J Crohn’s Colitis. 2022;16:i042–i043.
- Wang J, Goren I, Yang B, et al. The sphingosine 1 phosphate/sphingosine 1 phosphate receptor axis: a unique therapeutic target in inflammatory bowel disease. Aliment Pharmacol Ther. 2022;55:277–291.
- Verstockt B, Vetrano S, Salas A, et al. Sphingosine 1-phosphate modulation and immune cell trafficking in inflammatory bowel disease. Nat Rev Gastroenterol Hepatology. 2022; Epub ahead of print.
- Schwab SR, Pereira JP, Matloubian M, et al. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739.
- Nielsen OH, Li Y, Johansson-Lindbom B, et al. Sphingosine-1-phosphate signaling in inflammatory bowel disease. Trends Mol Med. 2017;23:362–374.
- Sanna MG, Liao J, Jo E, et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279:13839–13848.
- Harris S, Tran JQ, Southworth H, et al. Effect of the sphingosine-1-phosphate receptor modulator ozanimod on leukocyte subtypes in relapsing MS. Neurol Neuroimmunol Neuroinflamm. 2020;7:e839.
- Scott FL, Clemons B, Brooks J, et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br J Pharmacol. 2016 Jun;173(11):1778–1792.
- Sandborn WJ, Feagan BG, D’Haens G, et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2021;385:1280–1291.
- Fragoso YD. Multiple sclerosis treatment with fingolimod: profile of non-cardiologic adverse events. Acta Neurol Belg. 2017;117:821–827.
- Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391:1263–1273.
- Sriwastava S, Chaudhary D, Srivastava S, et al. Progressive multifocal leukoencephalopathy and sphingosine 1-phosphate receptor modulators used in multiple sclerosis: an updated review of literature. J Neurol. 2022;269(3): 1678–1687.
- Selmaj KW, Cohen JA, Comi G, et al. Ozanimod in relapsing multiple sclerosis: pooled safety results from the clinical development program. Mult Scler Relat Disord. 2021;51:102844.
- Zeposia (Ozanimod) - summary of product characteristics. European Medicines Agency 2021.https://www.ema.europa.eu/en/documents/product-information/zeposia-epar-product-information_en.pdf
- Sandborn WJ, Peyrin-Biroulet L, Zhang J, et al. Efficacy and safety of etrasimod in a phase 2 randomized trial of patients with ulcerative colitis. Gastroenterology. 2020;158:550–561.
- Danese S, Argollo M, Le Berre C, et al. JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut. 2019;68:1893–1899.
- Salas A, Hernandez-Rocha C, Duijvestein M, et al. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17:323–337.
- De Vries LCS, Wildenberg ME, De Jonge WJ, et al. The future of janus kinase inhibitors in inflammatory bowel disease. J Crohn’s Colitis. 2017;11:885–893.
- Choy EH. Clinical significance of janus kinase inhibitor selectivity. Rheumatology (Oxford). 2019;58(6):953–962.
- Olivera PA, Lasa JS, Bonovas S, et al. Safety of janus kinase inhibitors in patients with inflammatory bowel diseases or other immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology. 2020;158:1554–1573.
- Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet. 2017;389:266–275.
- Feagan BG, Danese S, Loftus EV, et al. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. Lancet. 2021;397:2372–2384.
- Kavanaugh A, Westhovens RR, Winthrop KL, et al. Safety and efficacy of filgotinib: up to 4-year results from an open-label extension study of phase II rheumatoid arthritis programs. J Rheumatol. 2021;48:1230–1238.
- Jyseleca (Filgotinib) summary of product characteristics. European Medicines Agency. 2020.https://www.ema.europa.eu/en/documents/product-information/jyseleca-epar-product-information_en.pdf
- FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. Food and Drug Administration. 2021.https://www.fda.gov/media/151936/download
- Sandborn WJ, Feagan BG, Loftus EV Jr., et al. Efficacy and safety of upadacitinib in a randomized trial of patients with crohn’s disease. Gastroenterology. 2020;158:2123–2138.
- Vermeire S, Danese S, Zhou W, et al. OP23 efficacy and safety of upadacitinib as induction therapy in patients with moderately to severely active ulcerative colitis: results from phase 3 U-ACCOMPLISH study. J Crohn’s Colitis. 2021;15:S021–S022.
- Danese S, Vermeire S, Zhou W, et al. OP24 efficacy and safety of upadacitinib induction therapy in patients with moderately to severely active ulcerative colitis: results from the phase 3 U-ACHIEVE study. J Crohn’s Colitis. 2021;15:S022–S024.
- Panaccione R, Hebuterne X, Lindsay J, et al. Efficacy and safety of upadacitinib maintenance therapy in patients with moderately to severely active ulcerative colitis: results from a randomized phase 3 study. United European Gastroenterol J. 2021;9:1207–1209.
- Vermeire S, Tanida S, Renwei H, et al. Efficacy of upadacitinib induction therapy in patients with moderately to severely active ulcerative colitis by biologic inadequate responder status: results from two randomized phase 3 studies. United European Gastroenterol J. 2021;9:17–18.
- Sands BE, Peyrin-Biroulet L, Loftus EV, et al. Vedolizumab versus adalimumab for moderate-to-severe ulcerative colitis. N Engl J Med. 2019;381:1215–1226.
- Lasa JS, Olivera PA, Danese S, et al. Efficacy and safety of biologics and small molecule drugs for patients with moderate-to-severe ulcerative colitis: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2021;7:161–170. Epub ahead of print
- Burr NE, Gracie DJ, Black CJ, et al. Efficacy of biological therapies and small molecules in moderate to severe ulcerative colitis: systematic review and network meta-analysis. Gut. 2021;gutjnl-2021–326390. Epub ahead of print. DOI:10.1136/gutjnl-2021-326390.
- Dalal RS, Mitri J, Goodrick H, et al. Real-world comparison of tofacitinib vs ustekinumab among bio-exposed patients with ulcerative colitis: a propensity score analysis. Inflamm Bowel Dis. 2021;27:1694–1697.
- Hupe M, Riviere P, Nancey S, et al. Comparative efficacy and safety of vedolizumab and infliximab in ulcerative colitis after failure of a first subcutaneous anti-TNF agent: a multicentre cohort study. Aliment Pharmacol Ther. 2020;51:852–860.
- Ytterberg SR, Bhatt DL, Mikuls TR, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316–326.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551–1560.
- Sandborn W, Chan D, Johanns J, et al. Guselkumab for the Treatment of Crohn's Disease: Induction Results From the Phase 2 GALAXI-1 Study. Gastroenterology. 2022;8 [Epub ahead of print].
- Yang E, Panaccione N, Whitmire N, et al. Efficacy and safety of simultaneous treatment with two biologic medications in refractory Crohn’s disease. Aliment Pharmacol Ther. 2020;51:1031–1038.
- Kwapisz L, Raffals LE, Bruining DH, et al. Combination biologic therapy in inflammatory bowel disease: experience from a tertiary care center. Clin Gastroenterol Hepatol. 2021;19:616–617.
- Goessens L, Colombel JF, Outtier A, et al. Safety and efficacy of combining biologics or small molecules for inflammatory bowel disease or immune-mediated inflammatory diseases: a European retrospective observational study. United European Gastroenterol J. 2021;9:1136–1147.
- Boleto G, Kanagaratnam L, Drame M, et al. Safety of combination therapy with two bDMARDs in patients with rheumatoid arthritis: a systematic review and meta-analysis. Semin Arthritis Rheum. 2019;49:35–42.