
ABSTRACT
Introduction
Autoimmune hemolytic anemia (AIHA) is classified according to the direct antiglobulin test (DAT) and thermal characteristics of the autoantibody into warm and cold forms, and in primary versus secondary depending on the presence of associated conditions.
Areas covered
AIHA displays a multifactorial pathogenesis, including genetic (association with congenital conditions and certain mutations), environmental (drugs, infections, including SARS-CoV-2, pollution, etc.), and miscellaneous factors (solid/hematologic neoplasms, systemic autoimmune diseases, etc.) contributing to tolerance breakdown. Several mechanisms, such as autoantibody production, complement activation, monocyte/macrophage phagocytosis, and bone marrow compensation are implicated in extra-/intravascular hemolysis. Treatment should be differentiated and sequenced according to AIHA type (i.e. steroids followed by rituximab for warm, rituximab alone or in association with bendamustine or fludarabine for cold forms). Several new drugs targeting B-cells/plasma cells, complement, and phagocytosis are in clinical trials. Finally, thrombosis and infections may complicate disease course burdening quality of life and increasing mortality.
Expert opinion
Beyond warm and cold AIHA, a gray-zone still exists including mixed and DAT negative forms representing an unmet need. AIHA management is rapidly changing through an increasing knowledge of the pathogenic mechanisms, the refinement of diagnostic tools, and the development of novel targeted and combination therapies.
1. Introduction
Autoimmune hemolytic anemia (AIHA) is a rare disease with an incidence of 0.8 to 3/100,000 people per year and is caused by an autoimmune attack against erythrocyte antigens [Citation1,Citation2]. While accumulated knowledge exists about disease pathogenesis, the etiology of AIHA is still largely obscure and considered ‘multifactorial.’ Similar to other autoimmune conditions, several risk factors for AIHA development have been described including endogenous and exogenous noxae such as drugs, infections, systemic autoimmune diseases, hematologic and solid neoplasms, and congenital syndromes [Citation3]. Additionally, emotional stressors and environmental pollution may possibly represent under-investigated risk factors for autoimmunity that may deserve dedicated studies in the future. All these elements may contribute to the derangement of immune tolerance and favor the emergence of anti-red cell autoantibodies with different abilities to cause clinically relevant hemolysis. The severity of the latter, the underlying condition, and the therapies used to treat AIHA may all lead to several complications, including infections, thrombosis, and mortality, but also result in an increasingly recognized burden for patients’ quality of life. Recently, treatment of AIHA has been more clearly defined for ‘warm’ (wAIHA, where antibodies are mainly IgG with highest affinity to the antigen at 37°C) and ‘cold’ forms, where cold agglutinins are IgM binding to erythrocytes at 0–4°C), but a huge gray zone of mixed and overlapping conditions still exists and deserves separate consideration from both a pathogenic and diagnostic point of view [Citation4]. Additionally, the advent of novel therapies targeting specific pathogenic steps, further advocates for better disease classification. In this review, we will address AIHA pathogenesis and treatment and try to shed light on the current and future understanding of the ‘causes and consequences’ of anti-red cell autoimmunity.
2. Pathogenic mechanisms
Autoimmunity can arise through several mechanisms, including a cross-reaction between antigens of infectious agents and self-molecules (named ‘molecular mimicry’), a drug-induced modification of erythrocyte membrane by adsorption or binding of drugs, and the production of autoantibodies by the so-called ‘forbidden clones’ during B lymphoproliferative syndromes [Citation1,Citation5]. Additionally, the breakage of ‘self-tolerance’ may be favored by the presence of germinal mutations impairing immune system maturation, such as those of congenital immunodeficiencies, or by the continuous exposure of erythrocyte self-antigens as happens in patients with ineffective erythropoiesis (i.e. congenital anemias and myelodysplastic syndromes) [Citation6]. Overall, AIHA pathogenesis is complex () and mainly involves antibody production by (a) the B-cell compartment and through (b) the alteration of T-lymphocytes homeostasis, and final erythrocyte destruction through (c) the mononuclear phagocytic system and (d) the activation of the complement cascade. Additionally, (e) the efficacy of bone marrow compensatory response is a major determinant of AIHA pathogenesis and severity [Citation7].
Figure 1. Pathogenic mechanisms involved in autoimmune hemolytic anemia (AIHA). Several predisposing conditions are involved in AIHA development including genetic background, presence of underlying immunodeficiencies, infections, solid and hematologic neoplasms. The main immune mechanisms responsible for tolerance breakdown are molecular mimicry, emergence of forbidden clones, and polyclonal B-cell activation. Additionally, drugs and hematologic treatments, such as checkpoint inhibitors (CPI) and hematopoietic stem cell transplant (HSCT) may induce immune dysregulation or generation of neoantigens. The resulting immunologic activation involves antigen presenting cells (APC), T- and B-cells, and cytokines and leads to the production of autoantibodies of several classes. B-cells interact with T helper (Th) cells via CD40 and its ligands but also present cognate antigens, acting as professional APC. This interaction also contributes to generate class-switched autoantibodies. Concerning cytokines, interleukin (IL)-4, IL-6, and IL-10 levels are increased and interferon gamma (IFN-γ) decreased favoring Th2 response and boosting humoral autoimmunity; the IL-2 and IL-12 levels are also increased contributing to Th1 differentiation and cellular autoimmunity. Transforming growth factor beta (TGF-β) is elevated favoring Th17 differentiation and production of IL-17 that decrease T-regulatory (Treg) levels. Once produced, IgG autoantibodies may weakly activate the complement cascade, and mainly destroy erythrocytes via antibody dependent cellular cytotoxicity and phagocytosis primarily in the spleen. IgM autoantibodies strongly activate complement system and cause erythrocyte destruction via C3b opsonization and phagocytosis primarily in the liver. These mechanisms are both called extravascular hemolysis. IgM may lead to terminal complement activation until the formation of the membrane attack complex (MAC) with consequent intravascular hemolysis.
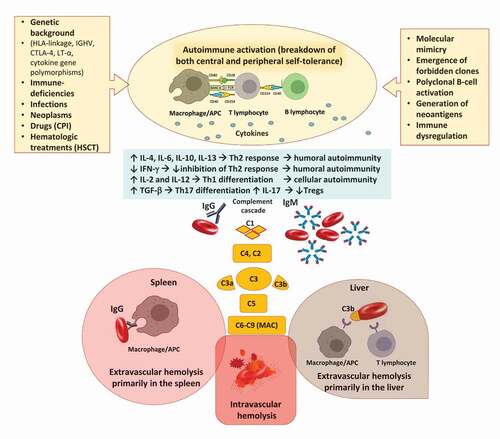
(a) Anti-erythrocyte autoantibodies are mainly produced by B lymphocytes at different stages of maturation. Interestingly, warm IgG autoantibodies are mainly polyclonal and produced by self-reactive nonmalignant lymphocytes residing in bone marrow and spleen, while IgM from CAD is more often monoclonal and sustained by a CD20+ CD5-lymphoid bone marrow infiltrate. This also happens in AIHA secondary to lymphoproliferative disorders, such as chronic lymphocytic leukemia (CLL), where autoantibodies are mainly polyclonal IgG (90%) produced by non-CLL lymphocytes and rarely IgM directly released by the malignant cells [Citation8]. Similar to this mechanism is polyclonal B-cell activation, occurring in different conditions such as infections with EBV, CMV, hepatotropic virus, and HIV.
(b) T-cell compartment is also involved through an imbalance among T helper (Th)1, Th2, Th17, and regulatory T-cells (Tregs), and the production of several cytokines that favor immune tolerance breakage [Citation5]. Results from various studies are sometimes conflicting, highlighting the heterogeneity of immune response in AIHA patients, often tested at different disease stages (onset, remission, and relapse) and under various therapies. Specifically, Th2 response appears overactive, with a consistent increase of interleukin (IL)-4, IL-6, and IL-10 in AIHA patients versus healthy controls [Citation9,Citation10], and a decrease of Interferon (IFN)-γ, promoting humoral autoimmunity. Additionally, IL-10 has anti-inflammatory and regulatory properties and its dysregulation likely contributes to loss of self-tolerance to RBC autoantigens and autoantibody production. Cellular immunity is also involved, with elevated activity of cytotoxic CD8 + T lymphocytes, natural killer cells, activated macrophages, and increased levels of IL-2 and IL-12 that promote Th1 differentiation [Citation7]. Transforming growth factor (TGF)-β is another regulatory cytokine whose levels were found to be elevated in AIHA. TGF-β favors the differentiation of Th17, which produce IL-17 that amplifies the pro-inflammatory and autoimmune responses and decreases T-regs [Citation11]. Finally, follicular T-helper cells, which contribute to generate memory B cells and long-lived plasma cells, have also been implied in autoantibodies production in AIHA [Citation7].
(c) Hemolysis mediated by the monocytic/macrophage system is typical of IgG wAIHA and mainly occurs extra-vascularly in the spleen [Citation12]. The mechanism includes the recognition of the Fc fragment of IgG by cells of the monocyte-macrophage system with consequent erythrocyte phagocytosis and antibody-dependent cellular cytotoxicity (ADCC). Additionally, other immune cells such as natural killer (NK) and neutrophils that express receptors for the IgG Fc fragment may also mediate an ADCC. Notably, the spleen is also a lymphatic organ able to produce autoantibodies. The monocytic/macrophage system is also involved in complement-mediated (C3b) extravascular hemolysis, mainly occurring in lymphoid organs and liver [Citation5,Citation6].
(d) Complement system is activated through the classical pathway [Citation13], mainly by pentameric IgM (mainly cold reactive, rarely warm), but also by abundant monomeric IgG, also depending on the subclasses (i.e. IgG3 more than IgG1; IgG2 is a weak activator; no evidence for IgG4). This results in the phagocytosis of C3b-coated cells (extravascular hemolysis) or, to a lesser extent, in the terminal complement activation, with formation of the membrane attack complex and intravascular hemolysis.
(e) Hematopoietic bone marrow compensates anemia by increasing erythropoiesis as mirrored by the increased number of reticulocytes (immature RBCs with nuclear residue) in the peripheral blood. Reticulocytosis indicates an efficient bone marrow compensation, while reticulocytopenia, present in up to 20% of adults and 40% of children, correlates with greater clinical severity and slower recovery [Citation14,Citation15] and may be due to autoimmunity against bone marrow precursors [Citation7].
Finally, it should be noted that anti-RBC autoantibodies are not necessarily pathogenic since they can be detected in healthy individuals (including blood donors); the lack of pathogenicity may be due to different Ig subclasses, avidity, ability to fix complement, and thermal amplitude (i.e. cold autoantibodies reacting at 4°C are less likely to induce clinically significant hemolysis) [Citation1,Citation2].
3. Diagnosis and classification of the different types of AIHA
The first step is a thorough medical history and physical examination for signs and symptoms of acute (symptomatic anemia possibly accompanied by abdominal pain and hemoglobinuria) or chronic hemolysis (milder fatigue, jaundice, gallstones, splenomegaly, peripheral cold-induced cyanosis, etc.); furthermore, recent infections, drug exposure, chronic diseases, tumors, and transplant should be evaluated. Laboratory workup shows anemia of various degrees with elevated reticulocytes, increased indirect bilirubin and LDH (more typical of intravascular hemolysis), and decreased haptoglobin [Citation1,Citation2,Citation16]. The direct antiglobulin test (DAT) is the mainstay of AIHA diagnosis and should be performed with monospecific antisera (anti-IgG, -IgA, -IgM, anti-complement, -C), allowing the classification of AIHA into wAIHA (DAT positive with IgG or IgG+C at low titer, <64), and cold forms (DAT positive with anti-C only and presence of cold agglutinins usually of high titer). The latter are further classified in cold agglutinin diseases (CAD), where monoclonal IgM is sustained by a clonal lymphoproliferative bone marrow disease similar to low-grade non-Hodgkin lymphoma, termed ‘CA-associated lymphoproliferative bone marrow disorder’ [Citation17–19], and cold agglutinin syndrome (CAS), occurring secondary to infections, such as Mycoplasma pneumoniae, EBV, CMV, and COVID-19, or malignancies (aggressive B-cell lymphoma). Rarer forms include mixed AIHA [Citation20], where DAT is positive for IgG and C and cold agglutinins with high thermal amplitude are detected. Additionally, some Authors suggest that the titer of cold agglutinins, along with positive autoagglutination at 20°C, may be helpful to distinguish mixed AIHA from IgG+C wAIHA [Citation2,Citation4]. The very rare paroxysmal cold hemoglobinuria (PCH) should also be considered, the latter is sustained by a biphasic IgG hemolysin called Donath-Landsteiner antibody [Citation4] that binds to erythrocytes at low temperatures but activates complement at 37°C resulting in intravascular hemolysis. The disease is nowadays mainly temporary, self-limiting, and occurring in children after viral infections [Citation4]. Other atypical cases include warm IgA driven AIHA, the very severe and potentially fatal warm IgM cases, characterized by huge intravascular hemolysis due to massive complement activation at body temperature, and DAT negative AIHA [Citation1,Citation21]. DAT may be falsely negative due to a low number of erythrocyte-bound autoantibodies or to low-affinity ones. In these cases, more sensitive second-level tests may be performed in reference centers. However, DAT remains negative in up to 10% of AIHAs and the diagnosis is based on the exclusion of other hemolytic conditions. Specifically, other acquired forms should be ruled out, including mechanical and toxic noxae (i.e. anamnesis for intravascular devices, prosthetic valves, angiomas, radiation, and drugs), paroxysmal nocturnal hemoglobinuria (high-resolution cytofluorimetry), thrombotic microangiopathies (platelets values, coagulation parameters, blood smear for schistocytes), and infections (i.e. malaria); subsequently, congenital hemolytic anemias (i.e. RBC membrane and enzyme defects) should be considered through the evaluation of family history, blood smear, osmotic fragility tests, and enzyme activities [Citation3,Citation7]. Thereafter, response to steroid therapy allows the ex juvantibus diagnosis of DAT negative AIHA.
In all patients, the clinical evaluation is completed by the investigation of underlying alloantibodies in patient’s serum (indirect agglutinin test, IAT), reported in about one-third of AIHA patients [Citation22]. The latter may cause severe hemolytic reactions in the case of transfusion. In complex cases, allo- and autoantibody may be distinguished by immune-absorbance techniques and extended RBC genotyping [Citation3]. Additionally, autoantibodies may be eluted from the washed patient’ RBCs to determine the class, specificity, titer, and thermal range. As regards specificity, in wAIHA autoantibodies are mainly directed against RBC membrane proteins including Band 3, glycophorin A, and Rh system, while in CAD the polysaccharide regions of glycoproteins, such as I/i or Pr antigen, are the most common target.
Bone marrow evaluation is useful to evaluate compensatory erythroid hyperplasia, features suggestive of underlying bone marrow failure or lymphoma, and the presence and type of lymphoid infiltrate that may be differently targeted with available therapy. It is advised in CAD at diagnosis, in relapsed/refractory cases of wAIHA, and in cases of suspected underlying hematologic diseases [Citation2,Citation4]. Similarly, whole-body CT scan, serology for infections and autoimmune diseases, and testing of anti-phospholipid antibodies are recommended upon specific clinical suspects. Finally, the determination of endogenous erythropoietin (EPO) levels is suggested in patients with inadequate reticulocytosis, particularly if relapsing and heavily treated [Citation7].
4. Conditions associated with AIHA development and secondary forms
Slightly more than 50% of AIHA cases are secondary to associated conditions, i.e. chronic lymphocytic leukemia, systemic lupus erythematosus (SLE), common variable immune deficiency, or other immunologic or lymphoproliferative disorders [Citation8,Citation23,Citation24]. Furthermore, several endogenous and exogenous noxae may increase the risk of autoimmunity as detailed thereafter.
4.1. Endogenous noxae associated with AIHA development
4.1.1. Genetics and congenital conditions associated with AIHA
Several studies highlighted the association of AIHA with alterations of genes involved in antigen recognition, T- and B-cell maturation and proliferation, and cytokine homeostasis. The formers include specific HLA loci (HLA-B8, BW6, DR, and DQ) [Citation25,Citation26] and stereotyped rearrangements of genes encoding the variable region of the immunoglobulin heavy and light chains (IGHV4-34, IGHV3, and IGKV3-20) [Citation27–30]. Additionally, the G polymorphism of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4, a negative regulator of T-cell responses) and the AG configuration of lymphotoxin-α (LT-α) gene showed a higher frequency among patients with AIHA [Citation31,Citation32]. A stronger genetic predisposition was recently demonstrated in pediatric patients with Evans syndrome, where AIHA occurs concomitantly with autoimmune thrombocytopenia and/or neutropenia. By next-generation sequencing, a genetic lesion was detected in 65% of cases, mainly involving genes related to primary immunodeficiencies (TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS). These pathogenic hints have prognostic and therapeutic importance, since mutated patients diagnosed with primary immunodeficiencies are more prone to catastrophic infections after immunosuppression [Citation33] and may be candidates for new biological drugs used in these conditions. Mutations typical of primary immunodeficiencies include those of KMT2D and CARD11, which have been described in 69% and 31% of CAD patients, respectively [Citation34], and are the hallmark of Kabuki syndrome. The latter is a congenital disorder characterized by malformations, immune-deficiency, and autoimmune phenomena, including AIHA in 4% of cases. Other primary immunodeficiencies include common variable immunodeficiency (CVID) [Citation35], IgA deficiency [Citation36], and autoimmune lymphoproliferative syndromes (ALPS) [Citation37]. Finally, AIHA may develop in patients with congenital anemia, including beta-thalassemia (up to 6%), sickle cell disease, and hereditary spherocytosis, where erythropoiesis is overstimulated and largely ineffective [Citation38]. With more sensitive methods, such as mitogen-stimulated DAT (MS-DAT), anti-erythrocyte autoantibodies were demonstrated in up to 61% of patients with hereditary spherocytosis, although with doubtful pathogenic significance [Citation39].
4.1.2. AIHA in the context of connective tissue diseases and other autoimmune conditions
AIHA belongs to the criteria for the diagnosis of SLE and may represent a mild manifestation of the disease or be its leading feature, with overt hemolysis and treatment requirements. The frequency of AIHA was about 14% in children and 3% in adults with SLE, again underlining the likely contribution of a more immature immune system in the former [Citation40]. Other associated autoimmune diseases include inflammatory bowel diseases (complicated by AIHA in about 4% of cases), Sjogren's syndrome (in about 2.8% of cases), autoimmune liver disorders (in 1.4% of cases), and case reports of systemic sclerosis, scleroderma, CREST syndrome, and thyroid autoimmune disorders. Although such associations do not necessarily imply a causative relationship, AIHA severity is correlated with the degree of organ involvement and disease activity of the underlying autoimmune condition [Citation3].
4.1.3. AIHA associated with neoplasms
Concerning solid neoplasms, AIHA is highly associated with ovarian teratoma and thymoma (up to 9% of cases), mainly occurs during disease progression, and tends to ameliorate after tumor resection, thymectomy, and/or chemotherapy [Citation41]. An association has also been reported with carcinomas [Citation42]. Very recently, a Japanese group described ectopic band 3 expression and erythrocyte membrane-bound IgG (by flow cytometry) in 97% of 50 colorectal cancer patients, with a DAT positivity in 10 of them. The presence of anti-band three autoantibodies was confirmed in patients’ sera and mouse experiments suggested that ectopic band 3 expression occurs in tumor cells under hypoxic conditions [Citation43]. Regarding hematological diseases, AIHA is frequently associated with lymphoproliferative disorders, such as CLL (5–10% of cases), non-Hodgkin lymphoma (NHL, 2–3%), and rarely HL (0.2%), which may precede or follow the hemolytic disease [Citation3,Citation8]. AIHA prevalence increases in specific NHL subtypes, being maximal in angioimmunoblastic T-cell lymphoma [Citation44] and in marginal zone lymphoma [Citation45]. Notably, CAD is associated with the above-mentioned CAD-associated lymphoproliferative disease that may be difficult to distinguish from other NHLs but usually presents with a 10–15% isolated bone marrow infiltrate [Citation18]. Similar to solid cancers, CLL/NHL treatment usually induces AIHA recovery. As described for congenital anemias, the positivity of anti-erythrocyte autoantibodies by MS-DAT is frequent in CLL [Citation46], but even in myelodysplastic syndromes, and in primary myelofibrosis [Citation47,Citation48], again with only a few cases developing clinical hemolysis. Finally, AIHA may rarely complicate multicentric Castleman disease and large granular lymphocyte (LGL) lymphoproliferative disorders and should be included in the differential diagnosis of anemia in this setting [Citation49].
4.2. Exogenous noxae associated with AIHA development
4.2.1. Infections associated with AIHA and the role of microbiome
AIHA may follow several infections, particularly Parvovirus B19, in up to 20% of cases, hepatotropic virus infections, including HCV, HBV, and HAV, and more rarely HIV [Citation3]. In particular, HCV-related AIHA was mainly sustained by cold agglutinins (CAS) and had a mortality rate as high as 56% in a large study [Citation50]. The risk of AIHA in HCV patients consistently increased from 2.8- to 11.6-fold in cases treated with interferon [Citation51]. Additionally, AIHA may develop in up to 3% of patients with infectious mononucleosis, within 1–2 weeks of onset, and more frequently as a CAS [Citation3]. Regarding bacteria, CAS may follow Mycoplasma pneumoniae infection [Citation52], as well as tuberculosis, and usually recovers after anti-tuberculosis treatment [Citation53]. More rare associations include AIHA complicating brucellosis or undulant fever, and the above mentioned PCH classically preceded by syphilis and virus infections [Citation3].
Finally, alterations of the microbiome (i.e. the ensemble of microorganisms colonizing various districts of the body) are emergingly described through next-generation sequencing (NGS) techniques and have been associated with the development of autoimmune diseases, including autoimmune cytopenias [Citation54]. A disrupted microbiome composition may lead to autoimmunity through molecular mimicry, chronic inflammation, alteration of epithelial barriers, and dissequestration of self-antigens. On the other hand, the microbiome perturbations may also be an effect of the autoimmune disease and its treatment. For instance, in immune thrombocytopenia and chronic idiopathic neutropenia, a peculiar microbiota composition has been identified, possibly predictive of response to therapy, and future studies will possibly clarify its role in other conditions including AIHA.
4.2.2. Drugs associated with AIHA
Drug-induced AIHAs [Citation1,Citation23,Citation55–57] are generally categorized according to the pathogenic mechanism in forms due to (1) drug-dependent antibodies that activate an immune response only while the drug is present and (2) drug-independent antibodies. The first encompasses hapten-mediated antibodies, recognizing a mixed epitope composed of erythrocyte parts and drug non-covalently bound to RBC, and includes penicillin (DAT positive for IgG only) and ceftriaxone (DAT positive for IgG, IgG + C, or C only). Drug-independent antibodies induce AIHA via adsorption, immune dysregulation, or unknown mechanisms and may occur after cladribine, methyldopa, and fludarabine [Citation2,Citation3]. The latter, a purine analogue used in the CLL treatment, further induces an imbalance between Th17 and T-regs [Citation7], that is restored by the association of cyclophosphamide and rituximab that kill autoreactive T-cells and markedly reduce AIHA [Citation58]. Concerning novel oral B-cell receptor inhibitors, increased incidence of autoimmune hepatitis, colitis, and pneumonitis has been reported after the PI3K inhibitor idelalisib, and a single case report of AIHA post- the BCL-2 inhibitor venetoclax has been published. Contrarily, Bruton’s tyrosine kinase inhibitor ibrutinib seems safe and favored the resolution of primary and secondary AIHAs, possibly through the inhibition of autoantibodies producing B-cells and restoration of T-cell homeostasis [Citation59–61]. Finally, novel immune-activating anti-cancer drugs whose therapeutic effect is based on enhancing host immune response against tumor cells may favor AIHA development. Immune checkpoint inhibitors (CPIs) that reactivate T-cell mediated immunosurveillance are the main example. They inhibit programmed death receptor 1 and its ligand (PD1 and PDL1) or CTLA-4 and may evoke severe autoimmune reactions, including fulminant AIHA [Citation62,Citation63]. Recent studies reported a total of 82 cases mainly occurring after nivolumab, followed by pembrolizumab, ipilimumab, and atezolizumab. All cases displayed severe hemolysis, with transfusion requirement (80%), high prevalence of DAT negativity (38%), relapse in about 50% of patients, and mortality as high as 17% [Citation62,Citation63]. A last mention deserves the occurrence of cytopenias in about 30% of patients with acute lymphoblastic leukemia and NHL treated with chimeric antigen T-cell infusions; these cases may be life-threatening and steroid-refractory and are likely to be immune mediated [Citation64].
4.2.3. Transplant associated AIHA
AIHA complicates 5–10% of liver and/or intestinal transplantation and 2.5% of pancreas transplants [Citation2]. In this context, it is worth mentioning the passenger lymphocyte syndrome that is sustained by immune competent lymphocytes transferred with the donor organ [Citation65]. Onset is between 3- and 24-days post-transplant, the risk is proportional to the burden of transplanted lymphocytes, ranging from 9 to 70% of cases (kidney < liver < heart-lung transplants) [Citation66], and hemolysis is generally transient. Regarding hematopoietic stem cell transplant (HSCT), 2–4% of patients may experience AIHA after 3–10 months, and mortality increases with infections [Citation67,Citation68]. Some risk factors for AIHA developing post HSCT include unrelated donor, HLA-mismatch, graft-versus-host disease (GVHD), cord blood as HSC source, age <15 years, CMV reactivation, alemtuzumab use, and nonmalignant diagnosis pre-HSCT. The passenger lymphocyte syndrome may also occur after HSCT and has been linked to HLA mismatches, use of peripheral blood as stem cell source, cyclosporine alone for GVHD prophylaxis, reduced-intensity conditioning, and female donors, while cord blood use appears protective. Careful transfusion procedures are warranted in transplanted patients, particularly in mismatched cases [Citation69].
4.2.4. Possible effect of emotional stress on the development of autoimmunity
Several hematologic patients, including those with autoimmune conditions, report that their diagnosis had been preceded by a variety of traumas/stressors either physical (accidents, infections, etc.) or psychological (bereavement, dismissal, divorce, etc.). Their contribution to disease development is difficult to establish, but it has been speculated that they may interact with the immune system by altering the adrenergic/glucocorticoid axis thus interfering with disease course [Citation70]. Some evidence come from pediatric studies evaluating the long-term effects of childhood traumatic stress, including increased prevalence of altered inflammatory markers and higher risk of ischemic heart disease [Citation71,Citation72]. A recent analysis of 15,357 adults enrolled in the Adverse Childhood Experiences (ACEs) Study from 1995 to 1997 in California, and followed-up until 2005, evaluated the effect of physical, emotional, or sexual abuse on the frequency of hospitalizations for autoimmune diseases. The Authors found that 64% of patients reported at least one ACE and that the occurrence of 2 or more events conferred a 70% increased risk for hospitalization for autoimmune diseases [Citation70]. Although only nine AIHAs were reported in this study, this remains an open field for further investigation.
4.2.5. Possible association of pollution with autoimmunity
Several epidemiology studies linked air pollution (a heterogeneous mixture including carbon monoxide, nitrates, sulfur dioxide, ozone, lead, toxic by-product of tobacco smoke and particulate matter) to the occurrence of autoimmune diseases through various mechanisms including systemic inflammation, oxidative stress, epigenetic modifications, and chronic airway damage [Citation73]. In this view, air pollutants have been shown to bind to the aryl hydrocarbon receptor (AHR) in the lung to regulate Th17 and Tregs balance. Furthermore, pollutants trigger inducible bronchus associated with lymphoid tissue, resulting in the production of pro-inflammatory cytokines. The latter stimulates B- and dendritic cells thus favoring the production of antibodies and self-reactive T lymphocytes. Clinical examples are the effect of smoking and occupational exposure to silica on rheumatoid arthritis and SLE development, and the association of inhalation of chemical solvents, herbicides, and silica with scleroderma and ANCA associated vasculitis [Citation74]. Although a direct effect on AIHA development has not been reported yet, air pollutants have been recently shown to affect erythropoiesis, and particulate matter (PM2.5) and nitrogen dioxide (NO2) levels have been associated with lower hemoglobin in older Americans and were also related to higher C-reactive protein levels as markers of systemic inflammation [Citation75].
5. Standard therapy
Based on the above-mentioned differences in physiopathology, the treatment of warm and cold AIHA forms differs in the choice of the type and sequence of immunosuppressive agents [1; 2,4,6]. In both conditions, the utility of supportive measures, including transfusions, nutrient supplementation, erythropoiesis stimulating agents, and anticoagulant and anti-infective prophylaxis should be considered. Finally, secondary forms may necessitate treatment for the underlying disease, such as chemo-immunotherapy for lymphomas and solid tumors, intravenous immunoglobulin (IVIG) in primary immunodeficiencies, cytotoxic immunosuppressants in systemic autoimmune diseases ().
Table 1. Standard therapies for warm AIHA (wAIHA) and cold agglutinin disease (CAD).
5.1. Primary warm-AIHA
Predniso(lo)ne 60–100 mg or 1 mg/kg body weight per day for 2–3 weeks is the recommended first-line therapy, followed by a slow tapering in 3–6 months. About 80% respond initially, but only 30–40% experience sustained remission after 1 year [Citation76]. Rituximab addition in the first line, which prolonged response duration in two prospective studies [Citation77], should be considered in severe settings (i.e. Hb<8 g/dL, presence of atypical AIHA or Evans syndrome). In steroid refractory patients, second-line rituximab is the preferred choice, after re-assessment for secondary AIHA forms and bone marrow evaluation, with response rates of 70–80% in about 3–6 weeks. The conventional dose is 375 mg/m2 weekly for 4 weeks, but a fixed low dose of 100 mg weekly for 4 weeks yielded equal efficacy [Citation78]. Splenectomy is currently recommended in patients unresponsive to/relapsing after rituximab, with responses of about 70%. The risk of severe infection and thrombosis should be taken into account and prevented (see the dedicated section) [Citation79]. Further options include azathioprine, cyclophosphamide, cyclosporine, mycophenolate mofetil, or bortezomib, basing mostly on small retrospective series and case reports. For emergencies, high-dose intravenous methylprednisolone and IVIG may be appropriate, and some reports or retrospective series of plasma exchange, emergency splenectomy, or partial splenic embolization have been published. Finally, limited evidence regards the use of bortezomib plus dexamethasone, high-dose cyclophosphamide, or stem cell transplantation in ultra-refractory cases [Citation2].
5.2. Cold agglutinin disease
In a proportion of patients with mild anemia or compensated hemolysis without circulatory symptoms and fatigue, non-pharmacologic management with thermal protection is advised [Citation2,Citation4,Citation80]. In symptomatic cases, glucocorticoids are effective only at unacceptably high doses, so that rituximab has become the preferred first-line therapy with response rates of 45–60%, mainly partial. A majority of responders relapse within 12–15 months, and repeated rituximab courses have a fair chance to succeed. The addition of fludarabine has yielded higher responses, but with more toxicity, while the association with bendamustine seems safer and even more effective. The overall response exceeded 70% and deepened overtime, with an estimated median response duration of >88 months [Citation81]. For refractory patients, bortezomib monotherapy (1 cycle of 1.3 mg/sm iv for total 4 biweekly doses) may be considered, with responses in 32% in a small prospective study [Citation82]. Finally, a recent case series of 10 CAD patients (primary or secondary) treated with ibrutinib, described responses in all of them, suggesting the need of future investigation [Citation83]. Complement inhibitors represent a promising approach that will be addressed in the following paragraph.
5.3. Other AIHAs
In mixed AIHA cases [Citation20], which are generally more severe and refractory/relapsing, clinical-laboratory features (monospecific DAT, cold agglutinin titers, autoagglutination, presence of cold agglutinin symptoms) should be evaluated at each relapse to assess the prevailing form (warm versus cold). Glucocorticoids should be given generously, and rituximab considered as an early second line, particularly in the presence of CAD features, while splenectomy is discouraged [Citation4].
The very rare PCH is usually self-remitting and may be handled with steroids. Recently, the efficacy of a single dose of the complement C5 inhibitor eculizumab in a child was reported [Citation84]. Treatment for drug-induced hemolytic anemia mainly consists of discontinuation of suspected medications, and transfusions and steroids were required in severe cases such as those occurring post-CPIs. For the latter, early rituximab may be required given the high rate of steroid-refractory cases.
5.4. Supportive measures
Transfusions must not be denied in cases of life-threatening anemia (i.e. Hb<6 g/dL or conditions of increased O2 requests), but over-transfusion should be avoided. If time permits, an extended phenotyping should be performed with respect to Rh subgroups, Kell, MNS, Kidd, S/s, and Duffy antigens to avoid alloimmunization that may occur in up to one-third of cases [Citation2,Citation7,Citation22]. Genotyping can be considered but will often be too time-consuming. In CAD, compatible erythrocytes by crossmatching at 37°C are usually available and may be safely transfused provided adequate warming of the patient and the extremity chosen for infusion. Vitamin (B12, folic acid, vitamin D) and iron supplementation should be tailored on specific deficiencies/need of osteoporosis prevention. Anticoagulant and anti-infective prophylaxis should also be considered. Finally, recombinant erythropoietin (i.e. epoetin alpha 40,000 UI/week subcutaneously) may improve hemoglobin in w-AIHA as well as CAD and can be used as a supportive strategy in patients with inadequate reticulocytosis [Citation85]. Plasma exchange (with the use of albumin substitution instead of plasma to avoid complement supplementation) may be successfully used in refractory life-threatening cases, while specific immunosuppressive therapy is instituted [Citation2,Citation6].
6. New drugs
Novel treatments mainly target autoantibody production by the B-cell/plasma-cell compartment or the final erythrocyte breakdown by either complement or mononuclear phagocyte system () [Citation6]. The formers include monoclonal antibodies mainly used in secondary AIHA, such as ofatumumab (anti-CD20), alemtuzumab (anti-CD52), and daratumumab (anti-CD38). The latter targets long-lived plasma cells that do not express CD20 and may cause rituximab refractoriness. Isatuximab, another anti-CD38 MoAb is under investigation in wAIHA in phase 1 study [NCT04661033]. Oral B-cell receptor inhibitors parsaclisib [NCT03538041 and NCT05073458], ibrutinib [NCT03827603], and rilzabrutinib [NCT05002777] are also being studied in clinical trials in wAIHA and CAD with promising results.
Table 2. New drugs in warm AIHA (wAIHA) and cold agglutinin disease (CAD).
Complement modulation is the most promising drug under study for CAD: sutimlimab, a monoclonal antibody against complement protein C1s, demonstrated short time to response, rapid normalization of hemolysis, and good safety profile in a phase 3 trial [Citation86]; pegcetacoplan, a pegylated peptide that inhibits C3 [Citation6] also showed good activity in CAD and wAIHA with IgG+C DAT positivity. In the latter, a novel C1q inhibitor ANX005 is also being developed [NCT04691570]. Notably, complement inhibitors do not eliminate antibody production and will probably have to continue indefinitely. However, the very short time to response may be particularly helpful in severely anemic patients and acute crises. The reticuloendothelial system may be targeted by inhibiting the spleen tyrosine kinase with fostamatinib, which also inhibits the B-cell receptor downstream pathway and is now in phase 2 studies in wAIHA [NCT02612558]. Finally, the safety/efficacy of several inhibitors of the neonatal Fc receptor (FcRn) such as intravenous nipocalimab [NCT03075878] and subcutaneous RVT-1401 [NCT04253236], are under investigation. The FcRn, which is homologous to the MHC Class I receptor family, is widely expressed, and mediates IgG recycling conferring to the antibodies their long half-life. Blocking FcRn increases IgG clearance including that of pathogenic IgG autoantibodies.
7. Consequences of AIHA and its treatment
The detrimental events that may complicate AIHA clinical course include disease relapse, the above-mentioned association with other autoimmune cytopenias, the occurrence of thrombosis and infections, and the rare evolution into other lymphoid or myeloid hematologic conditions. All these may have an impact on patients’ mortality and overall quality of life and are discussed thereafter [Citation3].
7.1. Risk of AIHA relapse
The severity of anemia at onset constituted the major determinant of disease relapse in two recent large multicenter studies [Citation15,Citation87]. Relapse risk augmented with Hb decrease, by about 7% increased risk per each gram of reduction of Hb. Concerning AIHA type, CAD, mixed and atypical forms were at higher risk of relapse and required multi-treatment, while wAIHA had a more benign course. Overall, complement activation seems to be an additional risk factor for relapse, particularly after rituximab.
7.2. Evans syndrome (ES)
The association of immune thrombocytopenia and/or neutropenia to AIHA has a significant impact on prognosis. Particularly, relapse rate exceeded 70% in these patients, and more than 50% required ≥3 therapy lines in a recent analysis [Citation88]. If cumulated with Hb < 8 g/dL or forms other than WAIHA, the presence of ES was associated wit 3-/4-folds higher than other cases [Citation87]. ES patients had also a higher incidence of infections (33%) and thrombotic complications (21%) as compared with primary AIHA cases, correlating with the number of therapy lines and resulting in a mortality rate approaching that of low-grade hematologic malignancies, despite young age.
7.3. Risk of thrombosis
Thrombotic events occur in about 15–20% of patients [Citation17,Citation89,Citation90] and were mainly observed in patients with very severe anemia (i.e. Hb levels <6 g/dL) at onset, intravascular hemolysis (LDH > 1.5 x upper limit of normality ULN), previous splenectomy, and presence of ES [Citation87,Citation91]. The pathogenesis is still unclear and likely includes the contribution of nitric oxide depletion by free Hb, the release of erythrocyte microparticles [Citation92], and the close interplay among autoimmunity/autoinflammation, complement aactivation,and the coagulation cascade. Moreover, the presence of underlying diseases, such as anti-phospholipid syndrome, active infections, and neoplasms may favor the occurrence of thrombotic complications [Citation91,Citation93]. On the whole, the use of anticoagulant prophylaxis should be considered in hyper-hemolytic, infected AIHA patients, provided safe platelet counts. A comprehensive evaluation of all risk factors is advised, including thrombophilia screening (particularly lupus anticoagulant, LAC) in selected cases and before splenectomy [Citation2,Citation88,Citation91]. Finally, an ongoing prospective randomized study will evaluate the efficacy of prolonged oral anticoagulation with apixaban in preventing thrombosis during the 12-weeks after AIHA diagnosis [NCT05089227].
7.4. Risk of infections
Infections may be both a trigger for AIHA development and relapse and be caused by immunosuppressive treatment in a vicious circle. They complicate about 20% of AIHA cases, with a frequency increase along with the number of therapy lines. Interestingly, beyond splenectomy, which is associated with a known risk of sepsis by capsulated bacteria, repeated steroid therapy at each relapse seemed to have a prominent role in recent studies [Citation87,Citation88,Citation94]. In thrombosis, underlying diseases such as primary immunodeficiencies and malignancies and their therapies may lead to increased infectious risk deserving special attention. On the whole, testing HBV/HCV status, quantiferon for tuberculosis, is highly advised in patient candidates to immunosuppression with high-dose steroids, rituximab, or cytotoxic drugs to prompt adequate prophylaxis. Cotrimoxazole is advisable in patients receiving >25 mg day prednisone (or equivalent) for >4 weeks, anti-viral agents (i.e. lamivudine) and surveillance in those at risk for HBV reactivation, referral to an infective disease specialist for anti-tubercular treatment if tuberculosis is suspected. HCV positive subjects should be referred for possible eradication. Vaccinations against capsulated bacteria (Meningococcus, Haemophilus, and Pneumococcus) are mandatory before splenectomy [Citation95].
7.5. Steroid induced side effects
In a recent survey, it has been calculated that AIHA patients received a cumulative dose of 8.2 ± 2.1 kg (mean± standard error) in a mean follow-up of 12 ± 3.5 months [Citation94]. This may be associated with well known but sometimes under recognized side effects including arterial hypertension, diabetes, gastrointestinal dysfunction, neurological disorders, and osteoporosis. Concerning the latter, assessment of bone density scan and supplementation with calcium and vitamin D may aid prompt recognition/prevention of harmful spontaneous fractures. Additionally, vitamin D levels were found reduced in AIHA patients versus controls, particularly in those who had received more therapy lines; furthermore, the addition of vitamin D exerted a dose-dependent inhibition on in vitro production of anti-erythrocyte antibodies [Citation96]. These findings further suggest preventive administration of vitamin D in patients with AIHA.
7.6. AIHA evolving into other hematologic diseases
Various reports described the possible evolution of AIHA into clonal hematologic diseases and bone marrow failure. In particular, lymphoproliferative diseases may become clinically overt in patients followed for AIHA, more frequently as the evolution of an underlying misdiagnosed lymphoid indolent infiltrate into aggressive forms, particularly in CAD [Citation17]. Concerning myeloid diseases (from idiopathic cytopenia/dysplasia of unknown significance to myelodysplastic syndromes), we previously described the presence of hypercellularity, dyserythropoiesis, and increased reticulin fibrosis in about one-third of AIHA cases, associated with inadequate reticulocytopenia and chronic/relapsing course. Furthermore, we reported two cases of AIHA and ES with anti-erythroblast antibodies, which showed a clear-cut bone marrow erythrocyte precursor hyperplasia at diagnosis but evolved into idiopathic cytopenia/dysplasia of uncertain significance (ICUS/IDUS) and aplastic anemia (AA) after several years [Citation97]. It may be hypothesized that refractory/relapsing AIHAs may shift from a predominant ‘peripheral’ pattern toward a ‘central’ autoimmunity over time, with a likely contribution of chronic inflammation, aging, and accumulation of somatic mutations [Citation6]. In this sense, small clones of paroxysmal nocturnal hemoglobinuria have been described in AIHA patients, possibly evolving to clinically overt disease [Citation98].
7.7. Mortality of AIHA
In a large retrospective series, mortality of AIHA patients was around 10–20% and mainly associated with very severe anemia. Patients presenting with Hb <6 g/dL had an overall mortality of 24% versus 18% of those with Hb >6 g/dL. Age, multi-treatment, comorbidities, and the occurrence of complications, further impacted patients outcome [Citation99,Citation100]. Specifically, mortality risk was predicted by ES, acute renal failure, and infections (hazard risk, HR 8, 95% CI 2.5–26; HR 6.3, 1.4–29; HR 4.8, 1.5–15, respectively) [Citation15,Citation87]. In a series of 13 very severe multitreated primary AIHA mortality reached 57%, despite transfusions, steroid boli, IVIG, rituximab, EPO, and plasma-exchange [Citation101]. More recently, Lafarge et al. described a mortality rate of up to 30% in 44 AIHA patients admitted to the intensive care unit for severe anemia [Citation102]. In CAD, a recent large multicenter study reported a disease-related mortality of 11% and a median estimated survival of 16 years from diagnosis (5-year survival 83%) [Citation17]. Finally, secondary AIHAs may also impact on disease severity and prognosis and higher mortality was observed in AIHA secondary to lymphoma and cancer [Citation2].
7.8. Quality of life in AIHA
Clinicians are used to evaluate patients’ quality of life (QoL) by simply asking the patients to communicate the perception of health status and disease impact on daily living in their own words. However, communication skills of both the patient and the caregiver are highly heterogeneous and impede an objective evaluation of quality of life, and the use of structured surveys is helpful. The UK group recently administered a semi-structured interview to 12 adult patients with AIHA and found that the disease had a significant impact on QoL for most patients [Citation103]. On a 0–10 scale – where 10 was the ‘worst I’ve ever felt’ – 77% of subjects scored 5–10, and 31% scored 9 or 10. Patients described that the worst aspects of AIHA included the impact of investigation or treatment, symptoms (mainly fatigue), and anxiety over outcome. Multiple QoL domains were affected, including daily living, working, leisure, and even sleep. Finally, patients and their partners’ emotional wellbeing were adversely affected. Available questionnaires are being actively used in the afore mentioned clinical trials to assess the beneficial impact of novel drugs on QoL [Citation86]. However, an effort to design disease-specific surveys will be expected in the next future.
8. COVID-19 infection and AIHA
8.1. AIHA associated with SARS-CoV-2 infection
In the last two years, SARS-CoV-2 has become one of the major triggers for the development and exacerbations of autoimmune cytopenias, including AIHA and Evans syndrome [Citation104–107]. The latter may in turn increase the need of immunosuppressive treatment and, consequently, the frailty of this patient population. Pathogenically, molecular mimicry among SARS-CoV-2 antigens and red cells’ epitopes seems the prominent mechanism [Citation108], followed by the over-inflammation triggered by the virus that may result in cross activation of the many arms of the immune system (humoral and cellular immunity, complement, coagulation cascade, etc.) against self-antigens in an ‘innocent bystander’ fashion. The latter may also favor the oxidative stress of erythrocytes and platelets with exposure of phosphatidyl serine and consequent clearance by the mononuclear phagocyte system [Citation109,Citation110]. COVID-19-associated AIHA also displays some epidemiologic peculiarities. In fact, a higher incidence of ES was noted, while it usually complicates only one-fifth of AIHAs [Citation15], and mainly in adults that are more frequently infected by the virus. Additionally, a higher prevalence of cold-reactive complement-activating autoantibodies (up to 50%) was found as compared to primary AIHA [Citation111]. From a therapeutic point of view, SARS-CoV-2 associated AIHA and ES were mainly handled with a combination of steroids and intravenous immunoglobulins, irrespective of AIHA type. Rituximab was administered in only one case displaying cold reactive autoantibodies [Citation104]. This drug represents a concern during septic states, including SARS-CoV-2 infection, and has been associated with a more severe COVID19 in rheumatic disorders [Citation112]. Supportive treatments, including rEPO and IVIG may be also considered. Finally, the thrombotic risk of AIHA adds to that of COVID19 thus advocating for anticoagulant prophylaxis with low molecular weight heparin in these patients, provided a safe platelet count [Citation113].
8.2. AIHA associated with SARS-CoV-2 vaccine
Several reports highlighted the possible impaired immune response to SARS-CoV-2 vaccines in patients requiring immunosuppression, including autoimmune cytopenias. Specifically, recent reports showed that hematological patients treated with monoclonal antibodies (such as rituximab, daratumumab, and the anti-B cell early maturation antigen belantamab) do not mount humoral response to vaccines. Additionally, it has been shown that steroids reduce both humoral and T cell response to COVID19 vaccine in hematologic patients and in those with autoimmune diseases [Citation114]. On the other hand, SARS-CoV-2 vaccine may trigger autoimmune phenomena by activating ADCC as well as the classical complement cascade. In the literature, various AIHA, both warm and cold, ITP, and Evans syndromes developing/reactivating after SARS-CoV-2 vaccine have been reported [Citation115–118], irrespective of vaccine type and dose. All patients have been successfully managed with steroids, adjustment of ongoing treatment, or supportive therapy [Citation118]. Interestingly, two large cohort studies showed that post-vaccine ITP has an incidence inferior to that expected for primary ITP in the general population (0.8 versus 3.3 cases per 100,000 adults per year) [Citation119,Citation120]. Overall, it is suggested to perform vaccinations as far as possible from rituximab treatment and at the lowest steroid dose and to strictly monitor Hb and hemolytic markers for possible exacerbations since they are manageable if promptly recognized.
9. Conclusions
In conclusion, this review highlights the multifaceted nature of AIHA including a multifactorial etiology, encompassing genetic predispositions and environmental exposures leading to tolerance breakage. The distinction between cold and warm forms by DAT with monospecific antisera is pivotal since they show diverse hemolytic patterns (i.e. IgG mediated extravascular hemolysis in the spleen versus complement mediated hemolysis in the liver or intravascularly) and should be treated differently. Furthermore, a prompt recognition/exclusion of the associated conditions (infections, drugs, tumors, hematologic conditions, systemic autoimmune diseases, congenital immunodeficiencies, etc.) is fundamental, since they may complicate AIHA diagnosis, and may require specific management (i.e. CT scan, bone marrow evaluation, genetic testing). Treatment of wAIHA mainly relies on steroids as the first line, followed by rituximab in refractory/relapsing cases. Splenectomy is generally deferred to the three line in young low-comorbid patients. Asymptomatic CAD patients may be handled with cold temperatures avoidance, while those subjects displaying moderate-to-severe anemia or disabling peripheral symptoms require frontline rituximab (since steroids are effective only at high unacceptable doses). For the rarer mixed and atypical AIHA forms, which may be more severe and difficult to diagnose, there is great uncertainty and a close interaction with the laboratory physicians is pivotal. The role of bone marrow compensation and the efficacy of recombinant erythropoietin in patients with inadequate erythropoiesis should be always considered. Several novel drugs are being studied in clinical trials and will target the different pathogenic steps of AIHA that may dynamically change during the clinical course. The latter may be affected by several complications including the occurrence of thrombosis and infections, and the rare evolution into other lymphoid or myeloid hematologic conditions. Finally, SARS-CoV-2 and its vaccines represent novel triggers of AIHA development/exacerbations, including warm and cold forms, as well as cases of Evans syndrome with several diagnostic and therapeutic pitfalls.
10. Expert opinion
Anti-erythrocyte autoimmunity has long been recognized as the most suitable scholastic example to teach autoimmunity, since it involves different classes of antibodies with variable thermal characteristics, complement, cytokines, and cellular immune effectors, such as T-lymphocytes and monocyte-macrophages. Additionally, AIHA is unusual among autoimmune diseases in that the target autoantigens are well characterized (i.e. band 3, glycophorin A, and Rh system in wAIHA and I/i system in CAD). Notwithstanding the simplicity of the cell targeted by the immune attack, and the easiness of monitoring the degree of the immune activation (as compared to other more complex systemic autoimmune diseases), anti-erythrocyte autoimmunity highlights the complex interaction of several immune effectors and the existence of gray zones that elude classification. For example, complement activation is the well-established key mechanisms in CAD; however, the disease is undoubtedly due to an antibody (mainly IgM, usually monoclonal). Thus, the therapeutic strategy would better benefit from a combination of B-cell/plasma cell-directed drugs and complement inhibition. Additionally, thermal characteristics are not trivial: while an IgM reacting only at cold temperature will result in minimal/negligible clinical consequences, the very rare IgM with thermal range close to body temperature may be the most severe and potentially fatal forms. Paradoxically, these cases have not been included in CAD trials, even if they would probably benefit from early complement inhibition. On the other hand, in wAIHA, it is assumed that complement plays a secondary role, and most clinical trials are focused on targeting B-cells/plasma cells, inhibiting ADCC, or removing pathologic antibodies via the Fc-Rn. This strategy may exclude the severe wAIHA with DAT positivity for IgG and C, where complement inhibition may have a definite reason to be performed. Finally, the strict and scholastic distinction between wAIHA and CAD leaves an unmet need for mixed forms, which are generally more severe and relapsing than other AIHAs, and have no therapeutic options after steroids and rituximab failure. Altogether, these considerations underline the need to identify not only the main mechanism involved in erythrocyte destruction but also all the complex interactions of the different immune effectors. This is also pivotal for a disregarded mechanism, i.e. bone marrow compensation, which may be optimal in early disease, but progressively decreasing with patient’s age, disease duration/relapses, and presence of anti-erythroblast autoimmunity. Furthermore, in clinical practice, strict classifications may be difficult to apply, and tests’ positivities may vary overtime and according to the clinical setting (host immunity, previous treatments, presence of triggers). For instance, a high dose steroid treatment received for the first AIHA episode may abate/mask the ‘warm’ part of a mixed AIHA at relapse. Additionally, during polyclonal activation, as that occurring during infections, B-cell populations may release autoantibodies of different Ig subclasses. This may also occur during primary IgM immune responses that may be immature and less specific. Finally, the presence of a complement-activating trigger, such as SARS-CoV-2, may favor complement deposition on red blood cells and further interfere with DAT findings.
Beyond the diagnostic issues, the study of the so-called risk factors and predisposing conditions is gaining importance. The latter include modifiable environmental triggers that affect the general population, such as air pollutants and other stressors, that might allow the establishment of preventive measures in the future. In this view, a fascinating field is the analysis of the microbiome and its interactions with disease development and treatment. Dysbiosis may in fact represent both a modifiable risk factor for AIHA onset and relapses, as well as a trigger for infectious complications following immunosuppression. In the near future, the availability of NGS techniques will improve our ability to analyze these microorganisms with a likely impact on how we manage several diseases, including AIHA. Broader, molecular techniques will allow the definition of the genomic landscape of this disease, such as the presence of germinal and somatic mutations that may predispose to autoimmunity through an altered antigen recognition, an imbalance of B/T populations and of cytokine milieu, and the emergence of clonal entities. The latter may include ‘clonal myelopoiesis/lymphopoiesis of indeterminate potential’ that may be the herald of autoimmunity in hematology. The same techniques will possibly help to clarify why some patients are healed, some relapse and become chronic, and some other evolve into neoplastic conditions or bone marrow failure.
Therefore, the management of AIHA will significantly change through a deepening knowledge of the various pathogenic mechanisms, to the refinement of diagnostic tools and disease classification, and to the development of novel targeted and combination therapies.
Article highlights
AIHA is a rare and heterogeneous disease with an incidence of 0.8-3/100,000 people year.
AIHA recognizes several pathogenic mechanisms including autoantibody production, complement activation, monocyte/macrophage phagocytosis, and bone marrow compensation.
Standard therapies should be differentiated between warm AIHA (steroids followed by rituximab) and cold AIHA (rituximab alone or in association with bendamustine or fludarabine as the first line).
Several new drugs targeting B-cells/plasma cells (B-cell receptor inhibitors, anti-CD38 monoclonal antibodies), complement (anti-C1s, C1q, C3 monoclonal antibodies), and phagocytosis (spleen tyrosine kinase inhibitor and anti-neonatal Fc receptor monoclonal antibodies) are under study.
Several complications, including thrombosis and infections, may worsen disease course burdening quality of life and increasing mortality.
Declaration of interest
B Fattizzo received consultancy for Amgen, Alexion, Annexon, Apellis, Momenta, Novartis, and Sobi. W Barcellini received consultancy for Agios, Alexion, Annexon, Sanofi, Novartis, and Sobi. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
B Fattizzo and W Barcellini wrote the manuscript and revised it for important intellectual content. All authors agree for the final version of the manuscript to be published.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Petz LD, Garratty G. Immune Hemolytic Anemias. 2nd ed. Philadelphia: Churchill Livingstone; 2004.
- Jäger U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the first international consensus meeting. Blood Rev. 2020May; 41:100648. https://doi.org/10.1016/j.blre.2019.100648. Epub 2019 Dec 5. PMID: 31839434
- Barcellini W, Giannotta J, Fattizzo B. Autoimmune hemolytic anemia in adults: primary risk factors and diagnostic procedures. Expert Rev Hematol. 2020Jun;13(6):585–597. Epub 2020 Apr 29. PMID: 32274943.
- Berentsen S, Barcellini W. Autoimmune hemolytic anemias. N Engl J Med. 2021 Oct 7;385(15):1407–1419. PMID: 34614331.
- Barcellini W, Fattizzo B. The changing landscape of autoimmune hemolytic anemia. Front Immunol. 2020 Jun 3;11:946. https://doi.org/10.3389/fimmu.2020.00946. PMID: 32655543; PMCID: PMC7325906.
- Barcellini W, Zaninoni A, Giannotta JA, et al. New insights in autoimmune hemolytic anemia: from pathogenesis to therapy stage 1. J Clin Med. 2020 Nov 27;9(12):3859. PMID: 33261023; PMCID: PMC7759854.
- Fattizzo B, Barcellini W. Autoimmune cytopenias in chronic lymphocytic leukemia: focus on molecular aspects. Front Oncol. 2020 Jan 10;9:1435. DOI:https://doi.org/10.3389/fonc.2019.01435. PMID: 31998632; PMCID: PMC6967408.
- Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology. 7th ed. Philadelphia: Saunders; 2012.
- Barcellini W, Clerici G, Montesano R, et al. In vitro quantification of anti-red blood cell antibody production in idiopathic autoimmune haemolytic anaemia: effect of mitogen and cytokine stimulation. Br J Haematol. 2000;111(2):452–460.
- Toriani-Terenzi C, Fagiolo E. IL-10 and the cytokine network in the pathogenesis of human autoimmune hemolytic anemia. Ann N Y Acad Sci. 2005;1051(1):29–44.
- Xu L, Zhang T, Liu Z, et al. Critical role of Th17 cells in development of autoimmune hemolytic anemia. Exp Hematol. 2012;40(12):994–1004.
- Gallagher MT, Branch DR, Mison A, et al. Evaluation of reticuloendothelial function in autoimmune hemolytic anemia using an in vitro assay of monocyte-macrophage interaction with erythrocytes. Exp Hematol. 1983Jan; 111: 82–89 PMID: 6832239.
- Berentsen S. Role of complement in autoimmune hemolytic anemia. Transfus Med Hemother. 2015Sep;42(5):303–310. Epub 2015 Sep 7. PMID: 26696798; PMCID: PMC4678321.
- Conley CL, Lippman SM, Ness PM, et al. Autoimmune hemolytic anemia with reticulocytopenia and erythroid marrow. N Engl J Med. 1982 Feb 4;306(5):281–286. PMID: 7054700.
- Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014 Nov 6;124(19):2930–2936. Epub 2014 Sep 16. PMID: 25232059.
- Fattizzo B, Giannotta JA, Serpenti F, et al. Difficult cases of autoimmune hemolytic anemia: a challenge for the internal medicine specialist. J Clin Med. 2020 Nov 27;9(12):3858. PMID: 33261016; PMCID: PMC7760866.
- Berentsen S, Barcellini W, D’Sa S, et al., Cold agglutinin disease revisited: a multinational, observational study of 232 patients. Blood. 136(4): 480–488. 2020.
- Randen U, Trøen G, Tierens A, et al. Primary cold agglutinin-associated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica. 2014;99(3):497–504.
- Małecka A, Delabie J, stlie I, et al. Cold agglutinin-associated B-cell lymphoproliferative disease shows highly recurrent gains of chromosome 3 and 12 or 18. Blood Adv. 2020;4(6):993–996.
- Shulman IA, Branch DR, Nelson JM, et al. Autoimmune hemolytic anemia with both cold and warm autoantibodies. JAMA. 1985 Mar 22-29;253(12):1746–1748. PMID: 3974053.
- Sokol RJ, Booker DJ, Stamps R, et al., Direct coombs test-negative autoimmune hemolytic anemia and low-affinity IgG class antibodies. Immunohematology. 1997;13(4):115–118.
- Branch DR, Petz LD. Detecting alloantibodies in patients with autoantibodies. Transfusion. 1999 Jan;39:6–10. PMID: 9920160
- Hill QA, Stamps R, Massey E, et al., Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. 2017;177(2):208–220.
- Azizi G, Tavakol M, Rafiemanesh H, et al. Autoimmunity in a cohort of 471 patients with primary antibody deficiencies. Expert Rev Clin Immunol. 2017;13(11):1099–1106.
- Abdel-Khalik A, Paton L, White AG, et al. Human leucocyte antigens A, B, C, and DRW in idiopathic “warm” autoimmune haemolytic anaemia. Br Med J. 1980;280(6216):760–761.
- Wang-Rodriguez J, Rearden A. Reduced frequency of HLA-DQ6 in individuals with a positive direct antiglobulin test. Transfusion. 1996;36(11–12):979–984.
- Silberstein LE, Jefferies LC, Goldman J, et al. Variable region gene analysis of pathologic human autoantibodies to the related i and I red blood cell antigens. Blood. 1991;78(9):2372–2386.
- Potter KN, Hobby P, Klijn S, et al. Evidence for involvement of a hydrophobic patch in framework region 1 of human V4-34-encoded Igs in recognition of the red blood cell I antigen. J Immunol. 2002;169(7):3777–3782.
- Jefferies LC, Carchidi CM, Silberstein LE. Naturally occurring anti-i/I cold agglutinins may be encoded by different VH3 genes as well as the VH4.21 gene segment. J Clin Invest. 1993;92(6):2821–2833.
- Malecka A, Trøen G, Tierens A, et al. Immunoglobulin heavy and light chain gene features are correlated with primary cold agglutinin disease onset and activity. Haematologica. 2016;101(9):e361–4.
- Pavkovic M, Georgievski B, Cevreska L, et al. CTLA-4 exon 1 polymorphism in patients with autoimmune blood disorders. Am J Hematol. 2003;72(2):14714–14719.
- D’Abronzo LS, Barros MM, Bordin JO, et al. Analysis of polymorphisms of TNF-a, LT-a, IL-10, IL-12 and CTLA-4 in patients with warm autoimmune haemolytic anaemia. Int J Lab Hematol. 2012;34(4):356–361.
- Hadjadj J, Aladjidi N, Fernandes H, et al., members of the French reference center for pediatric autoimmune cytopenia (CEREVANCE). Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. 2019;134(1):9–21.
- Malecka A, Trøen G, Tierens A, et al. Frequent somatic mutations of KMT 2D (MLL 2) and CARD 11 genes in primary cold agglutinin disease. Br J Haematol. 2018;183(5):838–842.
- Feuille EJ, Anooshiravani N, Sullivan KE, et al. Autoimmune cytopenias and associated conditions in CVID: a report from the USIDNET registry. J Clin Immunol. 2018;38(1):28–34.
- Odineal DD, Gershwin ME. The epidemiology and clinical manifestations of autoimmunity in selective IgA deficiency. Clin Rev Allergy Immunol. 2019;58(1):107–133.
- Oliveira JB. The expanding spectrum of the autoimmune lymphoproliferative syndromes. Curr Opin Pediatr. 2013;25(6):722–729.
- Motta I, Giannotta J, Ferraresi M, et al. Autoimmune hemolytic anemia as a complication of congenital anemias. a case series and review of the literature. J Clin Med. 2021 Aug 2;10(15):3439. PMID: 34362222; PMCID: PMC8347040.
- Zaninoni A, Vercellati C, Imperiali FG, et al. Detection of red blood cell antibodies in mitogen-stimulated cultures from patients with hereditary spherocytosis. Transfusion. 2015;55(12):2930–2938.
- Gormezano NW, Kern D, Pereira OL, et al. Autoimmune hemolytic anemia in systemic lupus erythematosus at diagnosis: differences between pediatric and adult patients. Lupus. 2017;26(4):426–430.
- Nenova IS, Valcheva MY, Beleva EA, et al. Autoimmune phenomena in patients with solid tumors. Folia Med (Plovdiv). 2016;58(3):195–199.
- Sokol RJ, Booker DJ, Stamps R. Erythrocyte autoantibodies, autoimmune haemolysis, and carcinoma. J Clin Pathol. 1994 Apr;47(4):340–343. PMID: 8027372; PMCID: PMC501938.
- Kitao A, Kawamoto S, Kurata K, et al. Band 3 ectopic expression in colorectal cancer induces an increase in erythrocyte membrane-bound IgG and may cause immune-related anemia. Int J Hematol. 2020 May;111(5):657–666. Epub 2020 Jan 30. PMID: 31997080
- Crickx E, Poullot E, Moulis G, et al. Clinical spectrum, evolution, and management of autoimmune cytopenias associated with angioimmunoblastic T-cell lymphoma. Eur J Haematol. 2019;103(1):35–42.
- Dasanu CA, Bockorny B, Grabska J, et al. Prevalence and pattern of autoimmune conditions in patients with marginal zone lymphoma: a single institution experience. Conn Med. 2015;79(4):197–200.
- Barcellini W, Montesano R, Clerici G, et al. In vitro production of anti-RBC antibodies and cytokines in chronic lymphocytic leukemia. Am J Hematol. 2002 Nov;713:177–183. PMID: 12410572.
- Zaninoni A, Imperiali FG, Cattaneo A, et al. Detection of erythroblast antibodies in mitogen-stimulated bone marrow cultures from patients with myelodysplastic syndromes. Transfusion. 2016;56(8):2037–2041.
- Barcellini W, Iurlo A, Radice T, et al. Increased prevalence of autoimmune phenomena in myelofibrosis: relationship with clinical and morphological characteristics, and with immunoregulatory cytokine patterns. Leuk Res. 2013;37(11):1509–1515.
- Barcellini W, Giannotta JA, Fattizzo B. Autoimmune complications in hematologic neoplasms. Cancers (Basel). 2021 Mar 26;13(7):1532. PMID: 33810369; PMCID: PMC8037071.
- Ramos-Casals M, García-Carrasco M, López-Medrano F, et al. Severe autoimmune cytopenias in treatment-naive hepatitis C virus infection: clinical description of 35 cases. Medicine (Baltimore). 2003;82(2):87–96.
- Chiao EY, Engels EA, Kramer JR, et al. Risk of immune thrombocytopenic purpura and autoimmune hemolytic anemia among 120 908 US veterans with hepatitis C virus infection. Arch Intern Med. 2009;169(4):357–363.
- Stein B, DeCredico N, Hillman L. Evaluation of the Direct Antiglobulin Test (DAT) in the setting of mycoplasma pneumoniae Infection. JAMA. 2018;319(13):1377–1378.
- Ramagopalan SV, Goldacre R, Skingsley A, et al. Associations between selected immune-mediated diseases and tuberculosis: record-linkage studies. BMC Med. 2013;11(1):97.
- Fattizzo B, Cavallaro F, Folino F, et al. Recent insights into the role of the microbiome in malignant and benign hematologic diseases. Crit Rev Oncol Hematol. 2021Apr; 160: 103289. Epub 2021 Mar 2. PMID: 33667659
- Garratty G. Drug-induced immune hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2009;2009(1):73–79. PMID: 20008184.
- Habibi B. Drug induced red blood cell autoantibodies co-developed with drug specific antibodies causing haemolytic anaemias. Br J Haematol. 1985Sep;61(1):139–143. PMID: 2864949.
- Mueller-Eckhardt C, Salama A. Drug-induced immune cytopenias: a unifying pathogenetic concept with special emphasis on the role of drug metabolites. Transfus Med Rev. 1990;4(1):69–77.
- Fischer K, Bahlo J, Fink AM, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208–215.
- Autore F, Pasquale R, Innocenti I, et al. Autoimmune hemolytic anemia in chronic lymphocytic leukemia: a comprehensive review. Cancers (Basel). 2021 Nov 19;13(22):5804. PMID: 34830959; PMCID: PMC8616265.
- Moreno C. Autoimmune cytopenia and CLL ride together. Blood. 2021 Jun 24;137(25):3464–3465. PMID: 34165542.
- Vitale C, Salvetti C, Griggio V, et al. Preexisting and treatment-emergent autoimmune cytopenias in patients with CLL treated with targeted drugs. Blood. 2021 Jun 24;137(25):3507–3517. PMID: 33651883.
- Tanios GE, Doley PB, Munker R. Autoimmune hemolytic anemia associated with the use of immune checkpoint inhibitors for cancer: 68 cases from the food and drug administration database and review. Eur J Haematol. 2019;102(2):157–162.
- Leaf RK, Ferreri C, Rangachari D, et al. Clinical and laboratory features of autoimmune hemolytic anemia associated with immune checkpoint inhibitors. Am J Hematol. 2019;94(5):563–574.
- Schubert ML, Schmitt M, Wang L, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 2021;32(1):34–48.
- Hows J, Beddow K, Gordon-Smith E, et al. Donor-derived red blood cell antibodies and immune hemolysis after allogeneic bone marrow transplantation. Blood. 1986Jan; 67(1): 177–181. PMID: 3079641.
- Skeate R, Singh C, Cooley S, et al. Hemolytic anemia due to passenger lymphocyte syndrome in solid malignancy patients treated with allogeneic natural killer cell products. Transfusion. 2013;53(2):419–423.
- Barcellini W, Fattizzo B, Zaninoni A. Management of refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: current perspectives. J Blood Med. 2019;10:265–278.
- González-Vicent M, Sanz J, Fuster JL, et al. Autoimmune hemolytic anemia (AIHA) following allogeneic hematopoietic stem cell transplantation (HSCT): a retrospective analysis and a proposal of treatment on behalf of the Grupo Español De Trasplante De Medula Osea En Niños (GETMON) and the Grupo Español de Trasplante Hematopoyetico (GETH). Transfus Med Rev. 2018;S0887-7963:30164–30165.
- Kruizinga MD, VT MJD, Bekker V, et al., Risk factors, treatment, and immune dysregulation in autoimmune cytopenia after allogeneic hematopoietic stem cell transplantation in pediatric patients. Biol Blood Marrow Transplant. 24(4): 772–778. 2018.
- Dube SR, Fairweather D, Pearson WS, et al. Cumulative childhood stress and autoimmune diseases in adults. Psychosom Med. 2009Feb;712:243–250. Epub 2009 Feb 2. PMID: 19188532; PMCID: PMC3318917
- Felitti VJ, Anda RF, Nordenberg D, et al. The relationship of adult health status to childhood abuse and household dysfunction. Am J Prev Med. 1998;14(4):245–258.
- Danese A, Pariante CM, Caspi A, et al. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Nat Acad Sci. 2007;104(4):1319–1324.
- Zhao CN, Xu Z, Wu GC, et al. Emerging role of air pollution in autoimmune diseases. Autoimmun Rev. 2019Jun;186:607–614. Epub 2019 Apr 5. PMID: 30959217.
- Farhat SC, Silva CA, Orione MA, et al. Air pollution in autoimmune rheumatic diseases: a review. Autoimmun Rev. 2011Nov;111:14–21. Epub 2011 Jul 6. PMID: 21763467.
- Honda T, Pun VC, Manjourides J, et al. Anemia prevalence and hemoglobin levels are associated with long-term exposure to air pollution in an older population. Environ Int. 2017 Apr;101:125–132. Epub 2017 Jan 31. PMID: 28153527; PMCID: PMC5361751.
- Barcellini W, Fattizzo B. How I treat warm autoimmune hemolytic anemia. Blood. 2021;137(10):1283–1294.
- Michel M, Terriou L, Roudot-Thoraval F, et al., A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm autoimmune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 92(1): 23–27. 2017.
- Murakhovskaya I. Rituximab use in warm and cold autoimmune hemolytic anemia. J Clin Med. 2020;9(12):4034.
- Patel NY, Chilsen AM, Mathiason MA, et al. Outcomes and complications after splenectomy for hematologic disorders. Am J Surg. 2012;204(6):1014–1019.
- Berentsen S. How I treat cold agglutinin disease. Blood. 2021;137(10):1295–1303.
- Berentsen S, Randen U, Oksman M, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017 Jul 27;130(4):537–541. Epub 2017 May 22. PMID: 28533306.
- Pasquale R, Giannotta JA, Barcellini W, et al. Bortezomib in autoimmune hemolytic anemia and beyond. Ther Adv Hematol. 2021 Nov 12;12:20406207211046428. PMID: 34795889; PMCID: PMC8593301.
- Jalink M, Berentsen S, Castillo JJ, et al. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood. 2021 Nov 18;138(20):2002–2005. PMID: 34293088.
- Lau-Braunhut SA, Stone H, Collins G, et al. Paroxysmal cold hemoglobinuria successfully treated with complement inhibition. Blood Adv. 2019 Nov 26;3(22):3575–3578. PMID: 31738828; PMCID: PMC6880888.
- Fattizzo B, Michel M, Zaninoni A, et al. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: a multicenter international study. Haematologica. 2021 Feb 1;106(2):622–625. PMID: 32354865; PMCID: PMC7849557.
- Röth A, Barcellini W, D’Sa S, et al. Sutimlimab in cold agglutinin disease. N Engl J Med. 2021 Apr 8;384(14):1323–1334. PMID: 33826820.
- Barcellini W, Zaninoni A, Fattizzo B, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am J Hematol. 2018Sep;939:E243–6. Epub 2018 Aug 25. PMID: 29981267
- Fattizzo B, Michel M, Giannotta JA, et al. Evans syndrome in adults: an observational multicenter study. Blood Adv. 2021 Dec 28;5(24):5468–5478. PMID: 34592758; PMCID: PMC8714709.
- Audia S, Bach B, Samson M, et al. Venous thromboembolic events during warm autoimmune hemolytic anemia. PLoS One. 2018;13(11):e0207218.
- Broome CM, Cunningham JM, Mullins M, et al. Increased risk of thrombotic events in cold agglutinin disease: a 10-year retrospective analysis. Res Pract Thromb Haemost. 2020 Apr 9;4(4):628–635. PMID: 32548562; PMCID: PMC7292660.
- Fattizzo B, Bortolotti M, Giannotta JA, et al. Intravascular hemolysis and multitreatment predict thrombosis in patients with autoimmune hemolytic anemia. J Thromb Haemost. 2022 May 12; Epub ahead of print. PMID: 35555857.
- Barcellini W, Zaninoni A, Giannotta JA, et al. Circulating extracellular vesicles and cytokines in congenital and acquired hemolytic anemias. Am J Hematol. 2021 Apr 1;96(4):E129–32. Epub 2021 Feb 9. PMID: 33491786.
- Samuelson Bannow BT, Lee A, Khorana AA, et al. Management of cancer-associated thrombosis in patients with thrombocytopenia: guidance from the SSC of the ISTH. J Thromb Haemost. 2018;16(6):1246–1249.
- Giannotta JA, Fattizzo B, Barcellini W. Infectious complications in a cohort of autoimmune hemolytic anemia patients. Annual meeting of the European Hematology Association. Hemasphere 2020. Abstract n. PB2401.
- Giannotta JA, Fattizzo B, Cavallaro F, et al. Infectious complications in autoimmune hemolytic anemia. J Clin Med. 2021 Jan 5;10(1):164. PMID: 33466516; PMCID: PMC7796467.
- Fattizzo B, Zaninoni A, Giannotta JA, et al. Reduced 25-OH vitamin D in patients with autoimmune cytopenias, clinical correlations and literature review. Autoimmun Rev. 2016Jul;157:770–775. Epub 2016 Mar 14. PMID: 26988993
- Barcellini W, Fattizzo B, Zaninoni A, et al. Clinical evolution of autoimmune cytopenias to idiopathic cytopenias/dysplasias of uncertain significance (ICUS/IDUS) and bone marrow failure syndromes. Am J Hematol. 2017;92(3):E26–9.
- Fattizzo B, Giannotta J, Zaninoni A, et al. Small paroxysmal nocturnal hemoglobinuria clones in autoimmune hemolytic anemia: clinical implications and different cytokine patterns in positive and negative patients. Front Immunol. 2020 Jun 4;11:1006. PMID: 32582157; PMCID: PMC7287021.
- Barcellini W, Fattizzo B, Cortelezzi A. Autoimmune hemolytic anemia, autoimmune neutropenia and aplastic anemia in the elderly. Eur J Intern Med. 2018;58:77–83.
- Rattarittamrong E, Eiamprapai P, Tantiworawit A, et al. Clinical characteristics and long-term outcomes of warm-type autoimmune hemolytic anemia. Hematology. 2016;21(6):368–374.
- Fattizzo B, Zaninoni A, Nesa F, et al. Lessons from very severe, refractory, and fatal primary autoimmune hemolytic anemias. Am J Hematol. 2015;90(8):E149–51.
- Lafarge A, Bertinchamp R, Pichereau C, et al., Prognosis of autoimmune hemolytic anemia in critically ill patients. Ann Hematol. 98(3): 589–594. 2019.
- Hill QA, Horan M, Charlton A, et al. Developing the evidence base for the management of autoimmune haemolytic anaemia (AIHA): the UK experience. Br J Haematol. 2021Jan;1922:e54–7. Epub 2020 Nov 20. PMID: 33216965
- Fattizzo B. Evans syndrome in the SARS-CoV-2 era: “springing up like mushrooms.” Blood Transfus. 2021 Dec 1; Epub ahead of print. PMID: 34967726.
- Osti N, Ceolan J, Piccoli P, et al. Acute haemolysis by cold antibody during SARS-CoV-2 infection in a patient with Evans syndrome: a case report and literature review. Blood Transfus. 2021;20(2):168–172.
- Barcellini W, Giannotta JA, Fattizzo B. Are patients with autoimmune cytopenias at higher risk of COVID-19 pneumonia? The experience of a reference center in Northern Italy and review of the literature. Front Immunol. 2021;11:609198.
- Haberman R, Axelrad J, Chen A, et al. Covid-19 in immune-mediated inflammatory diseases. Case series from New York. N Engl J Med. 2020;383(1):85–88.
- Angileri F, Légaré S, Marino Gammazza A, et al. Is molecular mimicry the culprit in the autoimmune haemolytic anaemia affecting patients with COVID-19? Br J Haematol. 2020;190(2):e92–3.
- Chauhan AJ, Wiffen LJ, Brown TP. COVID-19: a collision of complement, coagulation and inflammatory pathways. J Thromb Haemost. 2020;18(9):2110–2117.
- Kubánková M, Hohberger B, Hoffmanns J, et al. Physical phenotype of blood cells is altered in COVID-19. bioRxiv. 2021;120:2838–2847.
- Jacobs J, Eichbaum Q. COVID −19 associated with severe autoimmune hemolytic anemia. Transfusion. 2021;61(2):635–640.
- Avouac J, Drumez E, Hachulla E, et al. COVID-19 outcomes in patients with inflammatory rheumatic and musculoskeletal diseases treated with rituximab: a cohort study. Lancet Rheumatol. 2021;10:1016/S2665-9913(21)00059–X. [Epub ahead of print.
- Fattizzo B. Evans syndrome and infections: a dangerous cocktail to manage with caution. Blood Transfus. 2021;19(1):5–8.
- Ehmsen S, Asmussen A, Jeppesen SS, et al. Antibody and T cell immune responses following mRNA COVID-19 vaccination in patients with cancer. Cancer Cell. 2021;39(8):1034–1036.
- Murdych TM. A case of severe autoimmune hemolytic anemia after a receipt of a first dose of SARS-CoV-2 vaccine. Int J Lab Hematol. 2021;44(1):e10–e12. Epub ahead of print.
- Brito S, Ferreira N, Mateus S, et al. A case of autoimmune hemolytic anemia following COVID-19 messenger ribonucleic acid vaccination. Cureus. 2021;13(5):e15035.
- Pérez-Lamas L, Moreno-Jiménez G, Tenorio-Núñez MC, et al. Hemolytic crisis due to Covid-19 vaccination in a woman with cold agglutinin disease. Am J Hematol. 2021;96(8):e288–91.
- Fattizzo B, Giannotta JA, Cecchi N, et al. SARS-CoV −2 vaccination in patients with autoimmune cytopenias: the experience of a reference center. Am J Hematol. 2021;96(11):e413–6.
- Simpson CR, Shi T, Vasileiou E, et al. First-dose ChAdOx1 and BNT162b2 COVID-19 vaccines and thrombocytopenic, thromboembolic and hemorrhagic events in Scotland. Nat Med. 2021;27(7):1290–1297.
- Welsh KJ, Baumblatt J, Chege W, et al. Thrombocytopenia including immune thrombocytopenia after receipt of mRNA COVID-19 vaccines reported to the Vaccine Adverse Event Reporting System (VAERS). Vaccine. 2021;39(25):3329–3332.