
1. Introduction
Asthma is a prevalent, chronic inflammatory respiratory disease defined by airway hyperresponsiveness (AHR), aberrant mucus production, and airway smooth muscle remodeling. Classically, asthma is considered an allergic disease, characterized by infiltrates of ‘type 2’ inflammatory cells including eosinophils, mast cells, basophils, CD4+ T helper 2 (Th2) cells, and group 2 innate lymphoid cells (ILC2s). These cells mediate an allergic inflammatory response via canonical type 2 inflammatory cytokines such as interleukin (IL)-4, IL-5, and IL-13. Increasingly, asthma is recognized as a clinically heterogenous disease – both in terms of phenotypic presentation as well as molecular basis of inflammation – with subsets of steroid-unresponsive patients that have predominantly neutrophilic, rather than eosinophilic, inflammation. Given the paucity of treatment options for those who are refractory to traditional anti-type 2 inflammatory therapies, there is a dire need for therapies targeting alternative pathways that contribute to asthma severity.
Prior preclinical studies demonstrate a potential role for IL-23 in asthma pathogenesis, and observational studies describe the association of serum IL-23 levels with asthma severity in children [Citation1–6]. In mouse models of asthma, IL-23 blockade results in reduced airway inflammation. In the era of targeted molecular therapies, there is intense interest in modulating IL-23 and its related downstream IL-23/CD4+ T helper 17 (Th17) axis as a novel target for asthma therapy. Multiple agents are already approved by the United States Food and Drug Administration (FDA) for the treatment of Th17-mediated diseases such as inflammatory bowel disease and psoriasis (), but they have yet to be proven to be efficacious in asthma and other atopic diseases [Citation7]. Unfortunately, existing animal model data and recent human studies have thus far yielded conflicting and disappointing results. However, a better understanding of the complex interplay between IL-23 and airway inflammation seen in asthma may yet pave the way for future therapies.
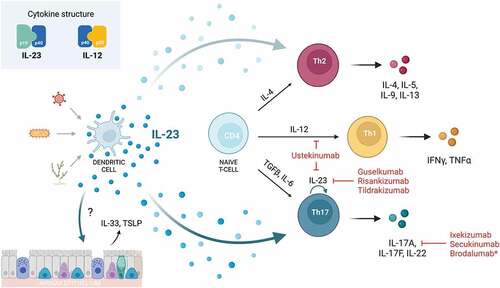
Table 1. FDA-approved monoclonal antibody therapies targeting the IL-23/Th17 axis.
2. Biology of IL-23/IL-23 receptor (IL-23 R)
IL-23 is an IL-12 family cytokine secreted primarily by dendritic cells (DCs), as well as macrophages, B-cells, and endothelial cells [Citation2]. It has a heterodimeric structure composed of an IL-23-specific p19 subunit (IL-23p19) and a p40 subunit shared with IL-12 (IL-12p40) [Citation8]. IL-23 signals through the IL-23R complex, which is composed of the IL-23R alpha subunit and IL-12 receptor beta 1 subunit, shared with the IL-12 receptor complex [Citation9]. IL-23R is expressed on numerous cell types including DCs, macrophages, Th17 cells, γδ T-cells, CD4+ memory T-cells, natural killer cells, and ILCs. Downstream signaling occurs through Janus kinase (JAK) 2 and signal transducer and activator of transcription (STAT) 3 signaling cascades.
Although IL-23 and IL-12 share structural homology, their physiologic functions differ. While IL-12 is the main cytokine involved in ‘type 1’ polarization, driving the differentiation of naive CD4+ T-cells into T helper 1 (Th1) cells and stimulating the production of interferon-gamma, IL-23 has a more convoluted role in the regulation of eosinophilic type 2 and neutrophilic type 3 inflammation [Citation1,Citation10]. Single nucleotide polymorphisms (SNPs) and perturbations in IL-23 homeostasis have been associated with inflammatory bowel disease, ankylosing spondylitis, rheumatoid arthritis, psoriasis, thyroiditis, and asthma [Citation11,Citation12].
3. The IL-23/Th17 Axis
While IL-23 is not essential for the polarization of naïve CD4+ T-cells to Th17 cells, IL-23 plays a key role in the survival and effector function of Th17 cells [Citation13]. Given that activated Th17 cells are responsible for robust neutrophil recruitment and that IL-17A and IL-17F – the predominant cytokines produced by Th17 cells – are elevated in asthmatic individuals, it has been hypothesized that the IL-23/Th17 axis is responsible for neutrophil-dominant inflammation seen in severe, steroid-resistant asthma [Citation14,Citation15]. Blocking this pathway mitigates airway inflammation in animal models, but the specific mechanisms of how the IL-23/Th17 axis contribute to allergic and non-allergic airway inflammation remain unclear [Citation1,Citation16–18]. Given the contribution to both neutrophilic and eosinophilic airway inflammation, there is great interest in harnessing this pathway to develop therapies for severe asthma.
4. Mechanisms of IL-23 in asthma
4.1. IL-23 intensifies both neutrophilic and eosinophilic inflammation
Neutrophil infiltration is seen in steroid-refractory severe asthma and likely contributes to asthma pathology. Neutrophils collected from patients with severe asthma and stimulated with IL-6, IL-21, and IL-23 produce significantly more IL-17A and IL-17F, as well as STAT3 phosphorylation, compared to healthy controls [Citation19].
Although primarily involved in neutrophil recruitment, the IL-23/Th17 axis may also enhance eosinophilic inflammation as well. In an OVA-induced mouse model of asthma, T-cell-specific overexpression of IL-23R results in increased pulmonary eosinophil infiltration compared to wild-type littermate controls [Citation3]. When stimulated ex vivo, T-cells overexpressing IL-23R produce increased levels of Th2 cytokines. Conversely, inhibiting IL-23 signaling in mouse models decreases eosinophil count and Th2 cytokine production in bronchoalveolar lavage fluid (BALF) [Citation20]. In another study, co-transfer of Th17 cells with antigen-specific Th2 cells significantly enhances eosinophil recruitment and Th2-mediated inflammation [Citation1]. Interestingly, in this study, eosinophilic inflammation was seen in response to IL-23 stimulation even in IL-17A deficient mice, suggesting an enhancement of eosinophilic inflammation in an IL-17-independent manner.
4.2. IL-23 amplifies antigen sensitization
Numerous studies demonstrate the ability of IL-23 blockade to mitigate airway inflammation. One study suggests that IL-23 signaling during antigen sensitization may play a key role [Citation16]. In a mouse model of asthma, antibodies against IL-23 were co-administered either during sensitization or challenge with ovalbumin (OVA). Anti-IL-23 given during sensitization decreased eosinophils in BALF and serum OVA-specific IgG, while anti-IL-23 given during challenge exacerbated AHR and had no significant effect on eosinophil infiltration or IgG production. Similarly, mice treated with anti-IL-23 during house dust mite (HDM) sensitization had decreased AHR and airway inflammation, as defined by total inflammatory cells, neutrophils, and eosinophils in BALF. Anti-IL-23 treatment also decreased IL-4, IL-5, IL-17A; antigen-specific IgG1 in BALF; IL-1α and IL-33 from whole lung; and ILC2s in whole lung by flow cytometry [Citation17].
Another study combining allergic sensitization with cigarette smoke exposure in mice found similar results. Anti-IL-23 antibodies administered during sensitization with HDM decreased AHR and the number of ILC2s in whole lung [Citation18]. There was no significant change in Th17 or ILC3 number, which led authors to suggest that type 3 inflammation may not be the primary driver of pathology.
4.3. Interplay of Th2 and Th17 pathways
It is likely that severe, steroid-refractory asthma cannot be easily categorized as ‘eosinophilic’ or ‘neutrophilic.’ Rather, the pathophysiology reflects a complex reciprocal relationship between multiple dysregulated inflammatory pathways. A transcriptional analysis of bronchial biopsies from 51 patients with asthma defined three distinct subgroups based on Th2 and Th17 signatures – Th2-high, Th17-high, and Th2/Th17-low [Citation21]. Despite the independent and seemingly mutually exclusive pattern of gene expression (i.e. there was not a Th2/Th17-high group identified), both Th2- and Th17- high groups were phenotypically associated with eosinophilic inflammation, defying the dichotomy of ‘eosinophilic’ and ‘neutrophilic’ asthma. In this study, blockade of Th2 pathways increased expression of IL-17, proliferation of CD4+ IL-17+ cells, and neutrophil infiltration, while anti-IL-17 use increased IL-4- and IL-13-producing CD4+ T-cells [Citation21]. Reciprocally, treatment of naïve T-cells with IL-13 attenuates Th17 differentiation and IL-17A production [Citation22,Citation23]. Ultimately, the combined blockade of IL-13 and IL-17 may be required to modulate complex inflammation, as seen in a HDM mouse model of asthma [Citation21].
4.4. Putative role for airway epithelial cells
It is perhaps by an independent, epithelial cell-mediated pathway that IL-23 contributes to both eosinophilic and neutrophilic inflammation. Multiple studies have demonstrated that antigen exposure upregulates IL-23 R in epithelial cells [Citation3,Citation17,Citation24]. Given that IL-23 is predominantly secreted by antigen presenting cells, such as DCs, it may act as an upstream amplifier of epithelial alarmins, such as IL-33 and thymic stromal lymphopoietin (TSLP). In a murine model of non-allergic eosinophilic asthma, treatment with recombinant IL-23 (rIL-23) with nonspecific airway irritants results in increased IL-33, TSLP, IL-17+ CD4+ cells, and eosinophils, but not IL-13+ CD4+ cells [Citation24]. Interestingly, in this study, isolated high-dose rIL-23 induces IL-33 and TSLP secretion from epithelial cells and increases ILC2s without the co-administration of airway irritant. In contrast, the use of anti-IL-23 decreases IL-33 and TSLP production [Citation18]. Furthermore, IL-23 blockade in stimulated epithelial cells suppresses IL-13 expression from co-cultured ILC2s, suggesting a potential novel role for epithelial cells as the link between IL-23 and Th2-mediated inflammation () [Citation18].
Figure 1. Mechanisms of IL-23 signaling and IL-23/Th-17 blockade on T-cell-mediated inflammation. This figure illustrates proposed mechanisms by which IL-23 amplifies Th2- and Th17-mediated inflammation. In response to antigen stimulation, antigen presenting cells such as dendritic cells secrete IL-23, which primarily acts on Th17 cells to potentiate Th17 cell proliferation and function, thereby enhancing neutrophilic inflammation. It may also act on airway epithelium to increase secretion of alarmins, IL-33 and TSLP, which act on ILC2s to amplify the Th2 response. This may provide a pathway by which IL-23 increases eosinophilic inflammation, which is seen both clinically and in animal models. Created with BioRender.com.
5. Therapy: Targeting the IL-23/Th17 axis
Both IL-23 and IL-17 blockade are remarkably efficacious in the treatment of Th17-mediated autoimmune diseases. Given the reported role for the IL-23/Th17 axis in the pathophysiology of asthma, such agents have also been explored for asthma; however, results have been conflicting.
5.1. Case reports
Multiple case reports underscore a link between IL-23 blockade and eosinophilic inflammation. Several patients with concurrent chronic psoriasis and asthma achieved resolution of asthma symptoms while on ustekinumab, an anti-IL12p40 antibody [Citation25,Citation26]. On the other hand, a patient with psoriatic arthritis and a remote history of allergic asthma developed a significant increase in peripheral eosinophils and experienced an asthma exacerbation requiring oral steroids shortly after starting guselkumab, an anti-IL-23p19 antibody [Citation27]. Despite significant improvement in psoriatic arthritis, his respiratory symptoms persisted leading to termination of the new agent. In another case report, a woman developed eosinophilic pneumonia that was attributed to initiation of ustekinumab, which resolved with a steroid taper and discontinuation of the antibody therapy [Citation28].
5.2. Clinical trials
Larger clinical trials have also yielded sobering results. Previously, a phase 2b trial with brodalumab, a human anti-IL-17R monoclonal antibody, was discontinued due to interim futility analysis. In this randomized, double-blind, placebo-controlled study, brodalumab failed to demonstrate clinical improvement in moderate-to-severe asthma [Citation29]. In this study, the subjects were not phenotyped at study entry based on their inflammatory phenotype.
Subsequently, in a recent multi-center, randomized, double-blind, placebo-controlled trial in adults with severe asthma, rizankizumab, an anti-IL-23p19 antibody, did not achieve the primary end point of improvement in time to first asthma worsening [Citation30]. In fact, those on risankizumab had a much shorter time to asthma worsening (40 versus 86 days, hazard ratio, 1.46; 95% confidence interval [CI], 1.05 to 2.04; p = 0.03). Sputum transcriptomics demonstrated that genes associated with transcription of Th1 and Th17 cells, including TBX21 and RORC, respectively, were downregulated by risankizumab. Data on GATA3, the gene encoding the transcription factor for Th2 cells, were not included. Unfortunately, the aforementioned case report with guselkumab may suggest that the adverse asthma outcomes in the failed risankizumab trial reflect a class-effect rather than an isolated drug-effect [Citation27]. Of note, a high percentage of those participating in the risankizumab trial were categorized as having allergic asthma (61.9% and 68.8% of the patients in the intervention and placebo arm respectively) [Citation30]. The existing mechanistic data would suggest that individuals with allergic, thereby type 2, asthma would be less likely to benefit from a treatment targeting type 3 and/or type 1 inflammation and more likely to be at risk for an ensuing type 2-skewed exacerbation.
6. Discussion
Patients with asthma often have high levels of IL-23 and IL-17 compared to their heathy controls, and frequently have both neutrophilic and eosinophilic recruitment. Unfortunately, the initial clinical trials targeting IL-23 and IL-17 yielded disappointing results suggesting non-efficacy, at best, and exacerbated disease, at worst. However, there is still hope that an improved understanding of the molecular mechanisms underpinning the IL-23/Th17 axis may result in targeted therapies that benefit a smaller, selective group of well-phenotyped asthma patients. Perhaps, in select patients with severe treatment-resistant disease (e.g. characterized by sputum neutrophilia and peripheral eosinophilia), a dual inhibition of type 2 and type 3 inflammatory responses with combined IL-13 and IL-17 blockade may be necessary to achieve disease remission. Or perhaps, without a current ability to precisely phenotype asthma patients, a complete IL-23 or IL-17 blockade is too heavy-handed. If there is a role for singular Th17 blockade, we suspect the patients most likely to benefit may be those with high sputum neutrophils and lack a history of atopy.
While the IL-23/Th17 axis has been hypothesized to contribute to the pathogenesis of asthma, it is likely not as simple as Th1 versus Th2 versus Th17. The role of IL-23 is likely contextual and may even help regulate the different arms of the cellular response to antigen and play a key role in regulating the balance between Th2 and Th17 inflammation. Given the potential of biologics such as tezepelumab (anti-TSLP antibody), astegolimab (anti-ST2 antibody), and itepekimab (anti-IL-33 antibody) in severe asthma, understanding the role for epithelial cells as the link between IL-23 and Th2-mediated airway inflammation may yet reveal potential uses for IL-23 and IL-17 targeted therapies [Citation31–33]. Ongoing research on underlying immunological pathways and the role of epithelial cells will allow for a more nuanced understanding of interplay between immune cells and their environment in severe asthma patients that is needed to guide therapy.
Declaration of interest
A Wu is a recipient of an NIH Training Award. S Peebles is a recipient of NIH and VA funding. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Wakashin H, Hirose K, Maezawa Y, et al., IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178(10): 1023–1032.
- Lewkowich IP, Lajoie S, Clark JR, et al. Allergen uptake, activation, and IL-23 production by pulmonary myeloid DCs drives airway hyperresponsiveness in asthma-susceptible mice. PLoS ONE. 2008;3(12):e3879.
- Peng J, Yang XO, Chang SH, et al. IL-23 signaling enhances Th2 polarization and regulates allergic airway inflammation. Cell Res. 2010;20(1):62–71.
- Nakajima H, Hirose K. Role of IL-23 and Th17 cells in airway inflammation in asthma. Immune Netw. 2010;10(1):1–4.
- Ciprandi G, Cuppari C, Salpietro AM, et al. Serum IL-23 strongly and inversely correlates with FEV1 in asthmatic children. Int Arch Allergy Immunol. 2012;159(2):183–186.
- Wang N, Brix S, Larsen JM, et al. Innate IL-23/Type 17 immune responses mediate the effect of the 17q21 locus on childhood asthma. Clin Exp Allergy. 2021;51(7):892–901.
- Husein-ElAhmed H, Steinhoff M. Effectiveness of ustekinumab in patients with atopic dermatitis: analysis of real-world evidence. J Dermatolog Treat. 2021;32(1):1–6.
- Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13(5):715–725.
- Parham C, Chirica M, Timans J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168(11):5699–5708.
- Li Y, Hua S. Mechanisms of pathogenesis in allergic asthma: role of interleukin-23: IL-23 and allergic asthma. Respirology. 2014;19(5):663–669.
- Abdollahi E, Tavasolian F, Momtazi-Borojeni AA, et al. Protective role of R381Q (rs11209026) polymorphism in IL-23R gene in immune-mediated diseases: a comprehensive review. J Immunotoxicol. 2016;13(3):286–300.
- Mosayebian A, Ganjalikhani Hakemi M, Meshkat R, et al. Association between interleukin-23 receptor R381Q Gene. Iran J Allergy Asthma Immunol. 2015;14(4):386–391.
- Taherian M, Razavi AR, Izad M, et al. The role of interleukin-23 in stability of in vitro T helper-17 cells. Iran J Allergy Asthma Immunol. 2014;13(2):131–137.
- Al-Ramli W, Préfontaine D, Chouiali F, et al. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol. 2009;123(5):1185–1187.
- Wang Y-H, Wills-Karp M. The potential role of interleukin-17 in severe asthma. Curr Allergy Asthma Rep. 2011;11(5):388–394.
- Masaki K, Suzuki Y, Kagawa S, et al. Dual role of interleukin-23 in epicutaneously-sensitized asthma in mice. Allergol Int. 2014;63(1):13–22.
- Lee HS, Park D-E, Lee J-W, et al. IL-23 secreted by bronchial epithelial cells contributes to allergic sensitization in asthma model: role of IL-23 secreted by bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2017;312(1):L13–L21.
- Lee HS, Park D-E, Lee J-W, et al. Critical role of interleukin-23 in development of asthma promoted by cigarette smoke. J Mol Med (Berl). 2019;97(7):937–949.
- Halwani R, Sultana A, Vazquez-Tello A, et al. Th-17 regulatory cytokines IL-21, IL-23, and IL-6 enhance neutrophil production of IL-17 cytokines during asthma. J Asthma. 2017;54(9):893–904.
- Ogawa R, Suzuki Y, Kagawa S, et al. Distinct effects of endogenous interleukin-23 on eosinophilic airway inflammation in response to different antigens. Allergol Int. 2015;64:S24–9.
- Choy DF, Hart KM, Borthwick LA, et al. Th2 and Th17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med. 2015;7(301):301ra129.
- Newcomb DC, Zhou W, Moore ML, et al. A functional IL-13 receptor is expressed on polarized murine CD4+ Th17 cells and IL-13 signaling attenuates Th17 cytokine production. J Immunol. 2009;182(9):5317–5321.
- Newcomb DC, Boswell MG, Zhou W, et al. Human TH17 cells express a functional IL-13 receptor and IL-13 attenuates IL-17A production. J Allergy Clin Immunol. 2011;127(4):1006–13.e1–4.
- Lee HS, Park D-E, Lee J-W, et al., Role of interleukin-23 in the development of nonallergic eosinophilic inflammation in a murine model of asthma. Exp Mol Med. 2020;52(1): 10–92.
- Torres T, Vilaça S, Velho G, et al. Etanercept-induced asthma in a psoriatic patient resolving with transition to ustekinumab. Eur J Dermatol. 2012;22(5):696–697.
- Amarnani A, Rosenthal KS, Mercado JM, et al. Concurrent treatment of chronic psoriasis and asthma with ustekinumab. J Dermatolog Treat. 2014;25(1):63–66.
- Schmiedeberg K, Rassouli F, von Kempis J, et al. Anti-interleukin-23-directed therapy and the recurrence of severe allergic asthma. Rheumatology. 2022;71(1):63–66
- Kalra SS, Chizinga M, Trillo-Alvarez C, et al. Ustekinumab associated chronic eosinophilic pneumonia. J Asthma. 2021;58(12):1670–1674.
- Busse WW, Holgate S, Kerwin E, et al., Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013;188(11): 1294–1302.
- Brightling CE, Nair P, Cousins DJ, et al., Risankizumab in severe asthma - a phase 2a, placebo-controlled trial. N Engl J Med. 2021;385(18): 1669–1679.
- Menzies-Gow A, Corren J, Bourdin A, et al. Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma. N Engl J Med. 2021;384(19):1800–1809.
- Kelsen SG, Agache IO, Soong W, et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J Allergy Clin Immunol. 2021;148(3):790–798.
- Wechsler ME, Ruddy MK, Pavord ID, et al. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N Engl J Med. 2021;385(18):1656–1668.