
ABSTRACT
Introduction
Chimeric antigen receptor (CAR) T-cells have emerged as a ground-breaking therapy for the treatment of hematological malignancies due to their capacity for rapid tumor-specific killing and long-lasting tumor immunity. However, the same success has not been observed in patients with solid tumors. Largely, this is due to the additional challenges imposed by safe and uniform target selection, inefficient CAR T-cell access to sites of disease and the presence of a hostile immunosuppressive tumor microenvironment.
Areas covered
Literature was reviewed on the PubMed database from the first description of a CAR by Kuwana, Kurosawa and colleagues in December 1987 through to the present day. This literature indicates that in order to tackle solid tumors, CAR T-cells can be further engineered with additional armoring strategies that facilitate trafficking to and infiltration of malignant lesions together with reversal of suppressive immune checkpoints that operate within solid tumor lesions.
Expert opinion
In this review, we describe a number of recent advances in CAR T-cell technology that set out to combat the problems imposed by solid tumors including tumor recruitment, infiltration, immunosuppression, metabolic compromise, and hypoxia.
1. Introduction
Chimeric antigen receptors (CARs) are synthetic fusion molecules which are genetically delivered to immune cells to reprogram their specificity against one or more native cell surface-associated target(s). CARs consist of a modular fusion of a targeting moiety (most commonly antibody-derived), a variable spacer/hinge domain, a transmembrane element, and a functional signaling domain. Greatest success has been achieved using second-generation designs, originally invented by Finney et al. more than 25 years ago [Citation1], and which contain a fused co-stimulatory and activation domain. Since 2017, six second-generation CAR T-cell therapies targeted against either CD19 [Citation2–7] or BCMA [Citation8–10] have been approved for use in patients with hematological malignancies. However, the same clinical success has not yet been achieved for patients with solid tumors. Compared to hematological malignancies, solid tumors present a much greater number of obstacles including the lack of tumor-specific targets, poorly accessible disease burden, and an immunologically hostile tumor microenvironment (TME). Despite the substantial inter- and intra-tumoral heterogeneity that is observed in solid tumors, more than 100 candidate CAR T-cell targets have been identified pre-clinically [Citation11], of which more than 30 have been the subject of clinical investigation [Citation12]. Nevertheless, clinical efficacy of these approaches remains limited.
To achieve adequate and durable anti-tumor responses in patients with solid tumors, CAR T-cells must overcome a series of specific hurdles (). Tumor recruitment represents the first challenge, in contrast to hematological malignancies. Second, CAR T-cells must extravasate through the highly disorganized tumor vasculature into malignant tissue. Third, CAR T-cells must penetrate the high interstitial pressure within the stroma which encapsulates the tumor. Tumor stroma provides a protective matrix that is composed of immune cells, cancer-associated fibroblasts (CAFs), endothelium, and extracellular matrix (ECM) molecules. It shields tumor cell islands from immune surveillance mechanisms through the combined effects of a physical barrier and an array of immunosuppressive molecules and cells, including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T-cells (Tregs) [Citation13]. Operating within the TME, CAR T-cells must also endure the hypoxic, pro-oxidative, and acidic environment while sustaining their functional activity through serial encounter with tumor cells.
Figure 1. Steps required for successful CAR T-cell immunotherapy of solid tumors. CAR T-cells must successfully negotiate the indicated steps in order to elicit effective anti-tumor activity.
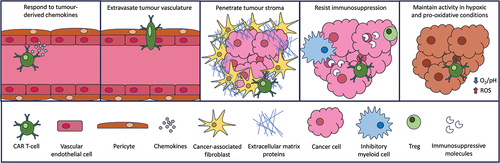
In this review, we explore different approaches that may be taken to facilitate each of these steps in order to enhance CAR T-cell efficacy for solid tumors. To achieve this, we have reviewed the literature contained in the PubMed database from December 1987, when the first CAR was described by Kuwana et al. [Citation14], through to the present day.
2. Body of review
2.1. CAR T-cell recruitment to solid tumors
The first requirement for successful CAR T-cell immunotherapy of solid tumors is their efficient recruitment to sites of disease. However, tumors frequently downregulate the expression of T-cell recruiting chemokines [Citation15]. Furthermore, there is often a mismatch between the chemokines secreted by the TME and the chemokine receptors expressed by effector T-cells, with a particular tendency for tumors to produce factors that preferentially recruit inhibitory immune cells [Citation16]. Clinical imaging studies have demonstrated that even if CAR T-cells do access tumor sites, they often preferentially accumulate at the periphery of such lesions, mimicking an immune excluded phenotype [Citation17]. In selected cases, intra-tumoral delivery may be used to overcome this [Citation18,Citation19] particularly in tumors located in the central nervous system [Citation20]. However, such a strategy is generally not feasible for disseminated disease. To circumvent this issue, investigators have explored the armoring of CAR T-cells to express a chemokine receptor with specificity for cognate ligands that are overproduced within the TME. Several examples of this approach are described below.
The CXCR2 chemokine receptor is typically expressed by neutrophils and MDSCs, but very rarely by T-cells. CXCR2 binds seven chemokines, namely CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8, also known as interleukin (IL)-8. Several tumor types have been shown to secrete CXCR2 ligands, notably IL-8 [Citation21,Citation22]. To harness this, Kershaw et al. genetically engineered primary human T-cells to express CXCR2. As a result, T-cells acquired the ability to undergo chemotaxis in response to CXCL1, which correlated with T-cell interferon (IFN)-γ production [Citation23]. More recently, CXCR2 has been co-expressed with αvβ6-, CD70-, or Glypican (GPC)3-specific CARs and resulted in enhanced CAR T-cell migration in tumor models in which cognate chemokines are produced [Citation21,Citation24,Citation25]. CAR T-cells have also been transduced to express the alternative IL-8 receptor, CXCR1 [Citation21,Citation24]. Although no differences were observed in CAR T-cell phenotype, tumor cell lysis, or cytokine production in vitro, expression of either CXCR1 or CXCR2 was associated with enhanced T-cell infiltration into the tumor, granzyme B (GZMB) production, T-cell persistence, and expansion in a number of xenograft mouse models [Citation21]. Although CXCR1-expressing CAR T-cells demonstrated enhanced anti-tumor efficacy as compared to controls, CXCR2-expressing counterparts achieved superior disease control likely due to the capacity of this receptor to bind a greater diversity of chemokines [Citation24]. Moreover, CXCR2 armoring has been shown to enhance CAR T-cell safety as a result of more rapid clearance of the infused cells from healthy organs, accompanied by enhanced ingress into sites of disease [Citation21].
The CCL2 chemokine is also secreted by a variety of solid tumors, including neuroblastoma and mesothelioma [Citation26,Citation27]. Accordingly, anti-GD2- and anti-mesothelin CAR T-cells have been engineered to co-express the cognate chemokine receptor, CCR2b. This resulted in augmented migration toward CCL2 gradients and improved GD2- and mesothelin-specific CAR T-cell tumor infiltration, expansion, and disease control in mice bearing either SK-N-SH, SK-N-AS (both neuroblastoma) or M108 (mesothelioma) tumors, respectively [Citation26,Citation27].
A number of solid tumors produce a chemokine known as Fractalkine/CX3CL1 [Citation28]. Taking advantage of this, Siddiqui et al. engineered T-cells to express CX3CR1. Although no CAR was used in this study, they did demonstrate enhanced in vivo trafficking of these T-cells to tumors which produced Fractalkine/CX3CL1 [Citation28].
In a further example of this approach, gene expression was compared between Panc02 tumor-infiltrating antigen-specific T-cells and matched splenocyte controls [Citation29]. This revealed a distinct upregulation of both CCL1 in the tumor and cognate receptor CCR8 in successfully infiltrated T-cells. Correspondingly, CCR8-overexpressing EpCAM- and mesothelin-directed CAR T-cells mediated efficient migration toward CCL1 gradients conferring heightened tumor infiltration and reduced tumor burden in Panc02-bearing C57Bl/6 mice, particularly when combined with transforming growth factor β receptor II (TGFβRII) dominant negative receptor armoring of the T-cells.
Gene expression analysis of lung, colorectal, and hepatocellular cancer samples derived from The Cancer Genome Atlas (TCGA) revealed elevated CCL20 within tumors as compared to normal lung tissue [Citation30]. To harness this, an epidermal growth factor receptor (EGFR)-targeted CAR was co-expressed with the unique CCL20 receptor, CCR6. As a result, enhanced tumor infiltration, cytokine production, reduced tumor burden, and improved survival were observed in NPG mice bearing either A549/CCL20 or H23 lung cancer xenografts [Citation30].
A screen of the Panc02 pancreatic carcinoma cell line identified CXCL16 as the most highly expressed chemokine. CXCL16 was similarly found to be upregulated by primary pancreatic tumors and myeloid stromal cells [Citation31]. To extend CAR T-cell chemokine responsiveness to CXCL16, the cognate receptor for CXCL16, CXCR6, was transduced into mesothelin- or EpCAM-specific CAR T-cells. This facilitated CAR T-cell penetration of organoids and enhanced tumor infiltration in Panc02/SUIT-2-bearing mice, resulting in improved anti-tumor responses [Citation31].
In addition to the induction of more effective CAR T-cell tumor trafficking, transgenic chemokine receptor expression can also competitively inhibit the recruitment of suppressive immune cells such as MDSCs through chemokine sequestering. Illustrating this, CXCR4-expressing claudin18.2-specific CAR T-cells were found to limit MDSC recruitment to the tumor via this mechanism [Citation32]. Similarly, CXCR2 armoring of CAR T-cells reduces the recruitment of immunosuppressive myeloid cells to the TME due to competition for CXCR2 ligands [Citation33].
Engineering of CAR T-cells to secrete chemokines has demonstrated additional benefits in tumor recruitment. CCR7 is constitutively expressed by naïve T-cells and dendritic cells and drives their recruitment to lymphoid organs. Formation and maintenance of the T-cell zone in lymphatic organs is dependent on the combination of IL-7 and CCL19 [Citation34]. To exploit this, Adachi et al. have armored CAR T-cells to produce IL-7 and CCL19 to facilitate recruitment of both T-cells and dendritic cells to tumor deposits [Citation34]. This approach is now under evaluation in the clinic in patients with GPC3- or mesothelin-expressing solid tumors (NCT03198546). Two examples of impressive tumor control in man have recently been described using this approach [Citation35]. More recently, enhanced anti-tumor activity has been reported using claudin 18.2-specific CAR T-cells when armored with IL-7 and CCL21, which also binds CCR7 [Citation36]. Although CAR T-cell expression of CCR7 ligands had no effect on cytokine production or cytotoxicity in vitro, expression was associated with enhanced T-cell and dendritic cell tumor infiltration and hence overall tumor control in mice engrafted with a variety of tumors [Citation34,Citation36]. However, many effector T-cells do not express CCR7 and consequently the CXCR3 axis has also been exploited to recruit effector T-cells to the tumor. CXCR3 binds CXCL9, CXCL10, and CXCL11 but most potently to CXCL11. While CXCL11 expression by mesothelin-specific CAR T cells showed no benefit, combining CAR T-cells with CXCL11-generating oncolytic vaccinia virus improved T-cell tumor infiltration and overall anti-tumor responses in NSG mice bearing EMMeso tumors [Citation37]. Boosting both dendritic cell and T-cell recruitment to the tumor through expression of CCR7 or CXCR3 ligands may also contribute to epitope spreading to complement direct CAR T-cell-mediated anti-tumor effects.
In conclusion, the armoring of CAR T-cells with appropriate chemokines and/or chemokine receptors is a potentially valuable approach to increase the efficiency of delivery of these cells to the TME. Several Phase I clinical trials are now evaluating the safety and efficacy of this approach, including CXCR2+ anti-CD70 CAR T-cells (NCT05353530), CXCR4+ anti-BCMA CAR T-cells (NCT04727008), CXCR5+ anti-EGFR CAR T-cells (NCT04153799, NCT05060796), CCR4+ anti-CD30 CAR T-cells (NCT03602157), and anti-GPC3 CAR T-cells armored with IL-7 and CCL19 (NCT03198546). A potential limitation of the co-expression of a chemokine receptor in CAR T-cells is the fact that some tumors express low levels of chemokines and that chemokine profiles can fluctuate between different tumor types. Optimal CAR T-cell recruitment may therefore be delivered by a tumor tailored approach or using a combination of various chemokine receptors which can respond to the most common tumor-associated chemokines. Alternatively, approaches to inhibit downstream actions of protein kinase A have been shown to increase CAR T-cell migration into tumors [Citation38].
2.2. CAR T-cell infiltration of solid tumors
2.2.1. Targeting of tumor-associated vasculature
In principle, CAR T-cell efficacy against solid tumors can also be improved through increased access via the vasculature. Tumor cells stimulate the formation of new blood vessels to ensure adequate oxygen and nutrient delivery to the TME and sufficient removal of metabolic waste. However, the tumor-associated vascular network is irregular with abnormalities in shape, diameter, and inconsistent pericyte coverage [Citation39]. In addition, blood flow is suboptimal, generating a hypoxic and acidic environment which is detrimental to infiltrating immune cells. The aberrant tumor vasculature limits CAR T-cell adherence and extravasation, providing a rationale to deploy interventions that can normalize this and thereby improve CAR T-cell access. Indeed, targeting of the tumor vasculature may simultaneously promote tumor starvation, hindrance of metastasis, and facilitation of immune cell infiltration into the tumor.
Several approaches have been evaluated for their ability to enhance CAR T-cell delivery to tumors via vascular disruption. Combrestatin A-4 phosphate (CA4P) is one such agent, acting on endothelial cells within the TME via the inhibition of microtubule polymerization. When combined with HER2- or mesothelin-directed CAR T cells, it resulted in a reduction in CD31+ blood vessel density while increasing vascular permeability [Citation40]. As a result, improved CAR T-cell tumor infiltration and anti-tumor efficacy were observed [Citation40]. Tumor cells secrete vascular endothelial growth factor (VEGF), which binds its cognate receptor (VEGFR) on endothelial cells to stimulate angiogenesis or lymphangiogenesis. Expression of the VEGFR is highly expressed by the proliferating tumor-associated vasculature and thus represents an attractive target. Consequently, VEGF blockade has been used to reduce tumor vascularization in animal models [Citation41,Citation42], enabling better CAR T-cell infiltration. Illustrating this, the combination of anti-EGFR variant III (vIII) CAR T-cells with the specific VEGF-A IgG1 monoclonal antibody, bevacizumab, induced direct apoptosis of vascular endothelial cells, permitting improved CAR T-cell access and infiltration within the tumor [Citation43,Citation44]. This was associated with increased CAR T-cell cytokine production (IL-2, IFN-γ), reduced Treg infiltration, and enhanced survival in vivo in murine models of glioblastoma [Citation44]. Similarly, small-molecule VEGF inhibitors, sorafenib and sunitinib, augmented CAR T-cell therapy by delaying tumor growth, improving CAR T-cell tumor infiltration and cytokine production [Citation45,Citation46]. However, both of these agents are likely to target additional receptor tyrosine kinases, in particular, since sorafenib also altered CAR T cell activation directly [Citation45]. Thus, the extent to which the observed benefits were due to altered tumor vasculature is not fully clear.
In an alternative approach, CAR T-cells themselves may be directed against target molecules that undergo selective upregulation on tumor-associated endothelial cells. Both in vitro and in vivo studies have demonstrated the utility of VEGFR-2-targeted CAR T-cells including efficient elimination of CD31+ tumor-associated blood vessels, improved CAR T-cell tumor infiltration, activation, and anti-tumor response [Citation47–60]. To further potentiate efficacy, combinatorial strategies have been pursued. For example, tumor-specific T-cells were transduced with a VEGFR-2-directed CAR, which facilitated tumor entry and improved tumor killing through epitope spreading [Citation51,Citation52]. Alternatively, VEGFR-2-specific CAR T-cells were designed to co-express NFAT-inducible IL-12, which further boosted CAR T-cell infiltration, persistence, IFN-γ production, and anti-tumor responses [Citation50]. VEGFR-2-specific CAR T-cells have also been assessed in metastatic cancer patients in a Phase I/II clinical trial (NCT01218867) [Citation61]. However, this trial was suspended due to a lack of objective responses. Lanitis et al. similarly found limited efficacy of VEGFR-2-targeted CAR T-cells due to compensatory tumor-derived VEGF-A upregulation [Citation56]. Increased VEGF-A competitively inhibited CAR T-cell target binding, but the combination of CAR T-cells with VEGF-A blockade was found to restore efficacy [Citation56]. In a similar fashion, the angiogenesis inhibitor TNP-470 was found to have an additive effect when combined with VEGFR-2-specific CAR T-cells [Citation48]. The higher affinity VEGFR-1 receptor has also been targeted using CAR T cells resulting in the efficient lysis of endothelial cells, reduced tube formation and improved overall anti-tumor responses in mice engrafted with A549 lung tumor xenografts [Citation62]. Cancer patients with pleural or peritoneal metastases are currently being recruited to a Phase I clinical trial (NCT05477927) in which CAR T-cells are simultaneously co-targeted against both VEGFR-1 and PD-L1. However, as yet, no clinical outcome data have been reported. Although VEGFR-1- and VEGFR-2-specific CAR T-cells have demonstrated efficacy in vitro and have been tested in human trials, targeting of multiple VEGFR receptor types may be a preferred option. Illustrating this, CAR T-cells were engineered to recognize both angiogenic VEGFR-2 and lymphangiogenic VEGFR-3 receptors using the VEGF-C fragment as the CAR targeting moiety [Citation63]. This strategy allowed for simultaneous targeting of both blood and lymphatic endothelial cells, resulting in improved overall anti-tumor responses and survival in MDA-MB-231- and HCC1806 tumor-bearing mice [Citation63].
Aside from VEGFR, several other tumor-associated vascular markers have been targeted using CAR T-cells. Angiogenic endothelial cells, CAFs, TAMs, and tumor cells commonly express integrin ⍺vβ3 [Citation64]. CAR T-cell targeting of ⍺vβ3 resulted in delayed tumor growth, improved survival, and was associated with excessive bleeding which was confined to tumor tissues with no damage observed in the normal vasculature [Citation65–68]. Fibronectin is a key component of the ECM and is expressed by endothelial cells or tumor cells with a role in cell adhesion, growth, and migration through interaction with integrins. CAR T-cell targeting of oncofoetal fibronectin splice variants, extra domain (ED)A or EDB, was found to reduce the number of CD31+ tumor blood vessels, increase CAR T-cell tumor infiltration and subsequent activation [Citation69–72]. CD13 is also overexpressed within proliferating endothelial cells and can be targeted using the NGR motif of the tissue factor (TF) protein, resulting in local thrombosis upon engagement. GD2-specific CAR T-cells were transduced to express a soluble TF mutant that is produced only upon NFAT activation, thereby imparting activation-specific secretion [Citation73]. Infusion into mice engrafted with the Kelly neuroblastoma tumor xenograft led to improved CAR T-cell infiltration and tumor control [Citation73]. Prostate-specific membrane antigen (PSMA) is also frequently found within the tumor endothelium [Citation74] but is absent from the normal vasculature making it a further candidate for targeting. In keeping with this, CAR targeting of PSMA was found to reduce tumor-associated blood vessels [Citation75]. In another example, Robinson et al. identified CLEC14A as a marker of tumor-associated vascular cells [Citation76] and developed a CLEC14A-targeting CAR [Citation77]. The CAR was infused into RIP-Tag2 mice which are susceptible to spontaneous insulinoma formation. As a result, increased vascular apoptosis, reduced tumor vessel density, and overall tumor burden were observed but without any impact on wound healing [Citation77].
To identify further tumor vasculature-associated markers, St Croix et al. compared the transcriptomes of blood vessels taken from normal and tumor-associated colorectal tissue samples [Citation78]. Cancer-associated genes were assigned tumor endothelial marker (TEM) designations. Although TEM1, now referred to as CD248 or endosialin, was originally assumed to be overexpressed by tumor endothelial cells, it has since been recognized as a marker of pericytes and CAFs [Citation79–85]. TEM1-targeting CAR T-cells were found to reduce TEM1+ tumor blood vessels, limit tumor progression, and reduce metastasis in vivo in a number of tumor models [Citation85]. These findings demonstrate that targeting tumor-associated pericytes represents yet another approach to control tumors using vascular targeted CAR T-cells.
Although expression of most tumor vasculature-associated markers is limited in normal vascular tissue, off-tumor effects have been observed. For example, TEM8 was found to be enriched within the tumor vasculature with only low-level expression in healthy vasculature [Citation78]. CAR T-cell targeting of TEM8 reduced the number of CD31+ tumor blood vessels, decreased tumor burden, and improved survival in vivo [Citation86] but led to on-target, off-tumor toxicity [Citation87].
Alternatively, investigators have sought to enhance the ability of T-cells to adhere to tumor-associated endothelium. This strategy is predicated on the fact that vascular adhesion is essential for efficient T-cell diapedesis into the tumor deposits. While tumor-associated endothelium may downregulate adhesion molecules including VCAM-1, ICAM-1, and ICAM-2, integrin αvβ3 is frequently upregulated at this site [Citation64]. GPI-KISS31 is an αvβ3 ligand fused to CD31. When expressed in primary human T-cells [Citation88], it successfully mediated their adhesion to αvβ3 integrin, facilitating subsequent migration across the endothelium of well-vascularized LLC-1 lung cancer xenografts [Citation88]. However, tumor-specificity and utility of this system in CAR T-cells remain to be tested.
In conclusion, CAR T-cell targeting of the tumor-associated vasculature offers several advantages over tumor-associated targets. First, tumor-associated endothelial cells are in direct contact with circulating CAR T-cells unlike malignant cells which are protected by the stroma. Second, vascular endothelial cells are genetically stable and therefore much less susceptible to mutation and subsequent antigen escape. Third, tumor endothelial markers are largely conserved and broadly expressed in a variety of tumors. Finally, destroying or impeding the tumor vasculature may also hinder nutrient provision to tumor cells supplied by the targeted vessels and limit metastasis. However, due to the ability of the tumors to upregulate VEGF-A production in response to targeting of VEGFR-2, it may be necessary to target all three VEGFR receptors in concert or to simultaneously neutralize VEGF-A to resist this countermeasure. Moreover, although a benefit is observed when targeting VEGFR receptors with CAR T-cells, responses are often not durable. This provides a rationale to combine VEGFR-specific with tumor antigen-specific CAR T-cells. Furthermore, while most tumor vasculature-associated markers are reasonably specific, expression can be observed at low levels within the normal vasculature or during physiological angiogenesis and thus off-tumor toxicity should be monitored for with caution. Options that may mitigate such risk include the targeting of the tumor vasculature using a chimeric costimulatory receptor (CCR) or engineering the regulated expression of the CAR under the control of activation-specific markers such as NFAT or a mechanism that is activated by tumor-associated hypoxia [Citation89].
2.2.2. Disruption of tumor-associated stroma
In solid tumors such as pancreatic cancer, stroma can account for up to 90% of the total tumor mass [Citation90]. In addition to the vasculature, other components of the stroma can also hinder successful CAR T-cell tumor infiltration. Mesenchymal cells such as CAFs produce the majority of ECM proteins within the TME and exert potent immunosuppressive effects [Citation91,Citation92]. These are mediated in part through inhibitory cytokines such as IL-6 and TGF-β that limit CAR T-cell function, in addition to CXCL12 and IL-8 which recruit inhibitory immune cells [Citation93,Citation94]. Targeting CAFs and the ECM offers key advantages including shared target expression across a spectrum of different tumor types and genetic stability. Consequently, approaches that target tumor-associated stroma may in principle be applicable to multiple solid cancers while reducing the possibility of antigen escape through mutation. Furthermore, targeting these elements simultaneously relieves the physical barrier and a key source of immunosuppression that operates within the TME.
The ECM represents a diverse network of components including fibrous proteins, proteoglycans, glycoproteins, and glycosaminoglycans that shield the tumor from immune attack. Collagen forms a major part of the ECM, comprising up to 90% of the total ECM protein content [Citation95]. T-cell proliferation and infiltration are both reduced in tumors with a high collagen density [Citation96]. Moreover, collagen fragments generated as a result of enhanced matrix metalloproteinase activity within the TME exert immunosuppressive effects on local T-cells [Citation97]. Another significant ECM component is the glycosaminoglycan, hyaluronan, which impedes CAR T-cell efficacy by limiting cell motility, cytotoxicity, and cytokine production [Citation98]. Hyaluronan is synthesized by hyaluronan synthase, which correlates with poor prognosis in cancer patients [Citation98]. Mesothelin- or GPC3-targeted CAR T-cells engineered to co-express the hyaluronidase enzyme, PH20, demonstrated improved efficacy with increased T-cell infiltration, proliferation and efficacy in BGC823, MKN28 (both gastric cancer derived), or Huh7-HSA2 hepatoma xenografts [Citation98,Citation99]. Heparan sulfate proteoglycans also form part of the complex ECM network and can be degraded by heparanase. Caruana et al. demonstrated that heparanase is downregulated in cultured CAR T-cells and further depleted following chronic activation [Citation100]. GD2-specific CAR T-cells engineered to co-express heparanase degraded the ECM resulting in increased T-cell infiltration and anti-tumor responses in neuroblastoma xenograft models [Citation100].
Matrix metalloproteases (MMPs) contribute to the repair of tissue damage and fibrosis, but also efficiently degrade many tumor-associated ECM components. Macrophages are a major source of MMPs, the secretion of which is enhanced by CD147. To exploit this, a HER2-specific CAR was engineered in which the transmembrane and intracellular domains were derived from CD147. Macrophages engineered to express this CAR were found to increase expression of several MMPs but had no additional effect on in vitro tumor growth when compared to unmodified macrophages [Citation101]. Infusion of such CAR macrophages into HER2+ 4T1 orthotopic tumor-bearing mice resulted in increased T-cell tumor infiltration, cytokine secretion (IFN-γ, IL-12), and tumor control, accompanied by reduced ECM-associated collagen and tumor-infiltrating inhibitory myeloid cells [Citation101].
Although several studies have demonstrated a benefit of directly targeting ECM proteins, many strategies are directed against a single protein and thus other ECM components can still maintain a locally immunosuppressive environment. Additionally, ECM proteins are not specific to the TME and therefore toxicity may be observed. For these reasons, the direct targeting of CAFs themselves may represent a preferred approach.
Several approaches have been described to target CAFs in the content of CAR T-cell immunotherapy of solid tumors. Small-molecule inhibitors such as the platelet-derived growth factor receptor beta (PDGFRB) inhibitor, nintedanib, provide one such strategy. When co-administered with mesothelin-specific CAR-NK cells, this combination was found to limit CAF growth and CAF-derived IL-6 release [Citation102]. Moreover, anti-tumor responses in mice engrafted with Capan2 pancreatic tumor xenografts were also enhanced [Citation102].
The most well-documented cell surface CAF target is fibroblast activation protein (FAP). FAP is a serine protease involved in ECM remodeling and is expressed by almost all solid tumors making FAP an attractive stromal target [Citation103]. Furthermore, only low to moderate expression is observed in healthy tissue, minimizing risk of on-target, off-tumor toxicity [Citation103]. Patients receiving FAP-targeting F19 monoclonal antibodies or humanized sibrotuzumab tolerated treatment well without evidence of toxicity, but unfortunately with limited anti-tumor efficacy [Citation104–106]. FAP-targeting CAR T-cells were found to efficiently kill CAFs, reduce ECM expression, increase T-cell tumor infiltration, activation, and subsequently delay tumor growth in a range of animal models [Citation93,Citation107–118]. CAF depletion was also associated with a reduction in CXCL12 and, as a result, limited MDSC recruitment to the TME [Citation114,Citation115]. Although most pre-clinical studies did not report any toxicity, Roberts et al. and Tran et al. identified toxic effects of targeting FAP in mice including loss of skeletal muscle mass and diminished hematopoiesis due to targeting FAP-expressing stromal cells in the muscles and bone marrow [Citation109,Citation119]. Nevertheless, the potential therapeutic benefit of targeting FAP using CAR T-cells was investigated in a Phase I trial of patients with pleural mesothelioma (NCT01722149) [Citation120,Citation121]. Despite the intrapleural administration of a subtherapeutic dose, systemic CAR T-cell expansion was observed without adverse impact [Citation120]. To enhance safety, fluorescein-specific universal CAR T-cells were engineered which only become activated in the presence of fluorescein-conjugated OncoFAP, a high affinity organic FAP binder [Citation122]. This strategy has also been pursued using a second universal CAR system known as UniCAR T-cells which have specificity for a cryptic nuclear autoantigen, La/SS-B. The corresponding epitope was fused to a FAP-specific scFv, meaning that FAP-dependent CAR activation only occurs when the binder fragment is also present [Citation123]. Using both approaches, CAR activation is therefore more tightly controlled, dependent on targeting module infusion.
Since FAP is predominantly expressed on CAFs, simultaneous targeting of tumor cells may be required for optimal efficacy. Accordingly, the combination of FAP-specific CAR T-cells with CAR T-cells directed against tumor-associated antigens EphA2 [Citation107], Nectin-4 [Citation112], BCMA [Citation93], Mesothelin [Citation113,Citation115], or Claudin 18.2 [Citation114] have all been used to achieve additive therapeutic effects. Co-targeting of FAP- and Nectin-4 was explored in a Phase I clinical trial in patients with Nectin-4-expressing solid tumors (NCT03932565). However, no clinical outcome data have been released as yet. Anti-tumor responses have also been enhanced by combination of FAP-targeting with other activating agents such as IL-7, CCL19, and PD-1 blockade in pre-clinical studies [Citation112,Citation113,Citation115]. Alternatively, mesothelin-specific CAR T-cells have been engineered to secrete anti-FAP/anti-CD3 bispecific T-cell engagers to simultaneously recruit CAR T-cells to FAP+ CAFs and mesothelin+ tumor cells [Citation124].
In addition to FAP, a number of novel CAF markers have been identified. As mentioned above, TEM1 is expressed by CAFs and tumor-associated pericytes [Citation79–85]. TEM1-directed CAR T-cells resulted in CAF cell death, disruption of the tumor vasculature, tumor necrosis, and reduced metastasis [Citation85]. Moreover, CD70 has been identified as a marker of both solid tumor cells and tumor-promoting CAFs, providing additional opportunities for co-targeting [Citation125]. One of the 14 renal cell carcinoma patients treated in the COBALT clinical trial with allogeneic CD70-specific CAR T-cells has achieved a complete response that remains stable beyond 18 months [Citation126].
Despite the benefit observed when CARs are directed against different ECM components, efficacy is often limited due to expression of alternative ECM proteins and the risk of off-tumor toxicity. However, CAF-associated FAP is present in most solid tumors with negligible expression in normal tissue. Despite some instances of toxicity, targeting FAP is generally well tolerated in patients, but with limited anti-tumor activity. FAP-targeting can be limited to the TME by instead generating a FAP-targeting CCR or by expressing a TME-inducible CAR and complementing with tumor antigen-targeting.
2.3. Tumor-associated immunosuppression
Following successful tumor infiltration, CAR T-cells must resist the highly immunosuppressive TME and retain capacity for serial tumor cell killing. However, this ability is limited by CAR T-cell exhaustion. During exhaustion, T-cells undergo terminal differentiation, upregulate inhibitory immune checkpoint proteins, and undergo altered transcriptomic and epigenetic programming (). This includes a reduction in TCF1-driven gene expression and a transition into a more TOX-, TOX2- and NR4A-associated transcriptomic profile [Citation127–135]. Following persistent antigen exposure within the TME, exhausted CAR T-cells enter a hyporesponsive state with significantly diminished effector cell function in terms of cytotoxicity, proliferation, and cytokine production. This is further potentiated by immunosuppressive mechanisms including secretion of TGF-β, adenosine, IL-10, Indoleamine-pyrrole 2,3-dioxygenase (IDO), and prostaglandin E2 (PGE2) or through expression of immune checkpoint ligands, such as programmed death-ligand 1 (PD-L1). However, several CAR T-cell armoring strategies have been developed to combat these mechanisms of exhaustion.
Figure 2. Comparison between activated CAR T-cells with sustained effector function and dysfunctional, exhausted CAR T-cells.
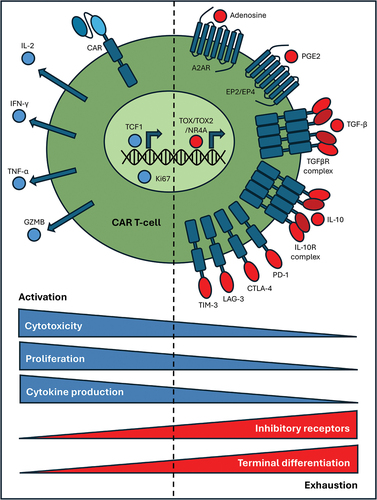
2.3.1. Immune checkpoints
Chronic T-cell activation leads to the increased cell surface expression of inhibitory immune checkpoint proteins including cytotoxic T-lymphocyte associated protein 4 (CTLA-4), programmed cell death protein 1 (PD-1), lymphocyte-activation gene 3 (LAG-3), and T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3). Clinical intervention using immune checkpoint blockade has revolutionized the treatment of highly immunogenic tumors, such as metastatic melanoma, through neutralization of these immune checkpoints and subsequent T-cell reinvigoration. CAR T-cells are similarly susceptible to upregulation of immune checkpoints in response to antigen-specific stimulation, which renders them hypofunctional and thus limits the extent of anti-tumor responses [Citation136–138]. Jacobson et al. pooled patient data from different CAR T-cell clinical trials and reported that patients who were refractory to treatment frequently incurred elevated tumor expression of the PD-1 ligand, PD-L1 [Citation139]. Correspondingly, PD-L1 has been found to directly inhibit CAR T-cell function [Citation140,Citation141]. Additional clinical data also support the notion that non-responding patients have higher levels of PD-1, PD-L1, LAG-3, and TIM-3 [Citation142–144]. Consequently, immune checkpoint blockade has been explored as an avenue to rescue CAR T-cell function.
Inhibition of the PD-1/PD-L1 signaling axis represents the most common immune checkpoint intervention used in combination with CAR T-cells. The combination of PD-1 neutralizing antibodies with CAR T-cells targeted against a host of different tumor-associated antigens, including HER2 [Citation136,Citation145–148], GD2 [Citation149], mesothelin [Citation138,Citation140], PSMA [Citation150], FAP [Citation116], EGFR [Citation151], and ROR1 [Citation152], have demonstrated enhanced pre-clinical anti-tumor activity. Studies demonstrated an increase in CAR T-cell activation, proliferation, cytokine production, tumor infiltration, and anti-tumor responses in vivo. Several clinical trials combining the PD-1-targeting agents pembrolizumab, nivolumab, or tislelizumab with CAR T-cells highlight that the combination has been well-tolerated () [Citation156–159,Citation170–174]. In the context of CD19-specific CAR T-cell treatment of lymphoma patients, anti-PD-1 therapy has induced responses in some lymphoma patients who fail CAR T-cell treatment [Citation158]. However, it is generally acknowledged that combined treatment with CD19 CAR T-cells and anti-PD1 drugs has not achieved significant additional clinical impact in patients reported thus far [Citation141]. Notably, recent data suggest that CAR affinity may influence susceptibility to PD-1 mediated inhibition [Citation175].
Table 1. Strategies undergoing clinical investigation to improve tumor recruitment, infiltration, or promote CAR T-cell function against solid tumors.
PD-L1-blocking antibodies can similarly restore activation of previously hypofunctional CAR T-cells targeted against CD19 [Citation176], HER2 [Citation177], mesothelin [Citation117,Citation178], PSCA [Citation176] and FAP [Citation117]. More specifically, PD-L1 blockade conferred resistance to type 2 tumor-associated macrophage (TAM)-mediated suppression and additionally resulted in loss of TAM populations through IFN-γ signaling [Citation176]. Similarly, CD22-specific CAR T-cells have been engineered to secrete anti-PD-L1 antibodies and efficacy is currently being explored in a Phase I clinical trial (NCT04556669). However, the timing of anti-PD-1/PD-L1 treatment appears to be crucial in the sense that early immune checkpoint blockade may compromise efficacy, whereas ongoing delayed treatment may favor CAR T-cell re-expansion [Citation141,Citation159]. Combined use of PD-1 blockade and CAR T-cell immunotherapy in the context of solid tumors has also proven disappointing to date [Citation156,Citation160], although 2 complete metabolic responses were reported in a series of 18 mesothelioma patients who received intrapleural mesothelin-specific CAR T-cells in combination with pembrolizumab [Citation157].
It is well known that systemic PD-1/PD-L1 blockade can result in autoimmune disease. To reduce the incidence of such toxicities, CAR T-cells have been engineered to directly secrete anti-PD-1/PD-L1 antibodies, thereby restricting these drugs to local tumor-infiltrating immune cells. This approach was found to limit expression of inhibitory immune checkpoints, increase CAR T-cell proliferation, cytokine production, and tumor infiltration when using CAR T-cells targeted against CD19 [Citation179–181], CD20 [Citation182], mesothelin [Citation183], EGFR [Citation184], MUC-16 [Citation180], IL-13 Rα2 [Citation151], carcinoembryonic antigen (CEA) [Citation185], ROR1 [Citation152], CAIX [Citation186,Citation187] or CD44v6 [Citation188]. Li et al. demonstrated that CD19-specific CAR T-cells which directly secrete anti-PD-1 antibodies had superior efficacy when infused into mice bearing H292-CD19 tumors as compared to anti-PD-1 antibody co-treatment in terms of IFN-γ production, tumor immune cell infiltration and overall survival [Citation179]. This benefit may stem from the rescue of tumor-infiltrated immune cells from exhaustion and subsequent epitope spreading. To further enhance specificity of PD-1/PD-L1 blockade to the TME, an anti-PD-1 scFv gene has been placed downstream of an NFAT-based promoter system to achieve antigen-specific activation-dependent expression [Citation152].
Rather than limiting PD-1/PD-L1-mediated immunosuppression through antibody blockade, PD-1 sensitivity can be transcriptionally reduced in CAR T-cells themselves. Engineering CAR T-cells to co-express siRNA/shRNA achieved substantial PD-1 knockdown, which resulted in enhanced proliferation, cytokine production (IFN-γ, TNF-α), and cytotoxicity [Citation140,Citation189–192]. However, anti-tumor responses were often limited due to a sustained low level of PD-1 expression which maintained sensitivity to PD-L1. As a result, CRISPR/Cas9 was implemented to knockout PDCD1 in CAR T-cells and thereby completely abrogate PD-1 signaling. Although most studies have demonstrated a benefit of this approach [Citation137,Citation193–196], this has not been observed consistently [Citation197,Citation198]. Agarwal et al. suggest that PD-1 is essential for CD28 signaling with deletion nullifying the benefit of CTLA4 knockout in CAR T-cells [Citation197]. Furthermore, Wei et al. have reported that PD-1 silencing may inhibit CAR T-cell responses by limiting proliferation [Citation199]. Consequently, rather than deleting PDCD1 altogether, other approaches have implemented co-expression of the PD-1 dominant negative receptor (DNR) which lacks the PD-1 intracellular signaling domain and thus facilitates PD-L1 binding but does not initiate downstream signaling. Although CAR T-cells maintain a low level of PD-1 signaling through endogenous PD-1, expression of the PD-1 DNR improved mesothelin-specific CAR T-cell expansion, cytokine production, and cytotoxicity both in vitro and in MSTO-MSLN tumor-bearing mice [Citation140]. The PD-1 DNR was further enhanced by the addition of the intracellular tail of CD28, thereby coupling the binding of PD-L1 to the delivery of a costimulatory signal. Expression of this PD-1/CD28 switch receptor in CAR T-cells resulted in improved cytokine production, activation, persistence, tumor infiltration, and anti-tumor responses in vivo [Citation200–202]. When co-expressed in SS1-directed CAR T-cells, superior efficacy was observed in EMMESO-bearing mice when compared to co-treatment with PD-1 neutralizing antibodies [Citation200]. In a similar fashion, the fusion of a PD-L1-binding scFv and CD28 intracellular domain has demonstrated enhanced efficacy in mesothelin-specific CAR T-cells in which CAR T-cell PD-L1 can engage the switch receptor and promote a favorable phenotype through the CD70/CD27 axis [Citation203]. The PD-1/CD28 switch receptor has since undergone evaluation in two Phase I clinical trials (NCT03258047, NCT04684459) [Citation165,Citation166].
In addition to PD-1-based chimeric switch receptors, PD-1/PD-L1-targeting CARs have been described [Citation69,Citation204–208]. PD-1 targeting has also been combined with tumor antigen targeting whereby anti-GPC3 and anti-PD-1 scFv molecules were fused together to allow CAR T-cell activation in the presence of either GPC3 or PD-1 [Citation209]. Unfortunately, however, a Phase I clinical trial testing the efficacy of a PD-L1-targeting CAR in patients with PD-L1+ non-small cell lung cancer resulted in significant pulmonary toxicity which led to early termination of the trial (NCT03330834) [Citation168]. PD-1/PD-L1 expression is not confined to the tumor and hence on-target, off-tumor toxicity is a risk of this approach.
Despite the potential promise that PD-1 inhibition offers to CAR T-cell immunotherapy, PD-L1 is not consistently expressed by all tumors and other T-cell immune checkpoints can compensate for the loss of PD-1-mediated immunosuppression. Furthermore, T-cells also demonstrate a high level of heterogeneity in terms of immune checkpoint expression. For example, PD-1 or TIM-3 blockade improved EGFRvIII-specific CAR T-cell anti-tumor responses to a greater extent than CTLA-4 inhibition in D270 tumor-bearing mice [Citation151]. By contrast, CTLA-4 blockade achieved superior impact when combined with IL-13 Rα2-directed CAR T-cells which correlated with distinct checkpoint expression by the CAR T-cells [Citation151]. Thus, optimal CAR T-cell function may require simultaneous inhibition of multiple immunosuppressive molecules or selective and context-dependent disablement of specific immune checkpoints.
Targeting alternative immunosuppressive markers can also relieve CAR T-cell immunosuppression in certain contexts. This includes the use of CTLA-4/TIM-3 inhibitors, CTLA-4/Fas/LAG-3 deletion, and an ‘AND’ switch CAR which co-expresses a first-generation anti-CD19 CAR with a CTLA-4/4-1BB switch receptor [Citation151,Citation197,Citation198,Citation210,Citation211]. However, the benefit of CTLA-4-targeting siRNA or shRNA was limited in the absence of CD80 co-expression or dual CTLA-4/PD-1 knockdown and further demonstrates the need in some circumstances to target multiple immune checkpoints simultaneously [Citation189,Citation212]. Indeed, triple knockdown of PD1, LAG-3, and TIM-3 using specific shRNA sequences in HER2-specific CAR T-cells improved CAR T-cell tumor infiltration in addition to both cytokine and chemokine production in SKOV3-bearing mice, when compared to single (PD-1) and double (PD-1/LAG-3) gene knockdowns [Citation190]. Moreover, the combination of a truncated, signaling-deficient TIM-3 mutant with the TGFβRII dominant negative receptor or combining a PD-1 inhibitor with an adenosine A2AR inhibitor also had additive effects [Citation146,Citation213].
While immune checkpoint inhibition has the potential to promote enhanced CAR T-cell function, it may also promote autoimmunity. It is therefore desirable to restrict immune checkpoint blockade to the TME, where tumor-infiltrating lymphocytes can regain tumor targeting potential which can contribute to epitope spreading. While complete PDCD1 knockout or PD-1 knockdown in CAR T-cells can be inhibitory, expression of dominant negative receptors and chimeric switch receptors can simultaneously deliver co-stimulation while avoiding systemic PD-1 inhibition. Simultaneous targeting of more than one immune checkpoint may also be required in some instances to enable optimal efficacy of this approach.
2.3.2. Transforming growth factor-β
Transforming growth factor beta (TGF-β) is secreted by stromal cells and inhibitory immune cells within the TME. This pleiotropic cytokine limits proliferation, cytotoxicity, and differentiation of Th1, Th2, and Th17 T-cell subsets and promotes the generation of suppressive Tregs. Similarly, TGF-β signaling has been found to dampen CAR T-cell cytotoxicity and cytokine production [Citation213–223]. Since TGF-β represents one of the major immunosuppressive factors in the TME, considerable effort has been expended into the development of solutions to antagonize TGF-β in order to reinvigorate CAR T-cells.
Co-administration of TGF-β inhibitors with CAR T-cell therapy was found to reduce phosphorylation of SMAD2 (pSMAD2), the initiating step of TGF-β downstream signaling, and hence limited the immunosuppressive effects of TGF-β. This resulted in increased CAR T-cell cytokine production, activation, proliferation, overall anti-tumor response, and reduced exhaustion [Citation223,Citation224]. Inhibition of the TGFβR complex in CAR T-cells displayed similar benefits [Citation221,Citation225]. Similar results have been achieved via the secretion of TGF-β traps, including soluble TGFβRII or bispecific proteins which simultaneously neutralize both TGF-β and PD-1 [Citation226,Citation227]. Alternatively, TGF-β-targeted CAR T-cells and TGF-β-inhibitory adenoviruses have been developed, both of which were found to improve T-cell activation, cytokine production, expansion, and cytotoxicity [Citation228–230]. Adaptor CAR T-cells have also been directed against latency-associated peptide (LAP) which can be found bound to precursor TGF-β [Citation231]. Targeting TGF-β or LAP directly may also have the additional benefit of rescuing tumor-infiltrating lymphocytes (TILs) from exhaustion and favoring epitope spreading. Conceptually however, TGF-β inhibition could also lower the threshold for onset of autoimmunity and should ideally be confined to the tumor site.
CAR T-cells have also been engineered to express cell surface TGF-β traps. These include the TGFβRII DNR truncated mutant which maintains TGF-β binding through expression of the extracellular domain but is dysfunctional due to deletion of the essential intracellular signaling domain [Citation232,Citation233]. Expression of this TGFβRII DNR by Epstein Barr virus- or gp100-specific T-cells counteracted the inhibitory effects of TGF-β with reduced pSMAD2 and overall improved CAR T-cell function [Citation226,Citation234,Citation235]. Similar findings were obtained in CAR T-cells, accompanied by enhanced CAR T-cell stemness, tumor infiltration, and anti-tumor responses [Citation29,Citation214,Citation217,Citation218,Citation220,Citation230,Citation236–239]. TGFβRII DNR+ CAR T-cells outperformed their counterparts which secreted soluble TGFβRII or soluble TGFβRII-IgG2a Fc TGF-β traps [Citation226]. This pre-clinical success led to in-human clinical trials, whereby the TGFβRII DNR was co-expressed in PSMA- (NCT03089203, NCT04227275), HER2- (NCT00889954), or BCMA- (NCT05976555) targeted CAR T-cells and infused into cancer patients [Citation169]. Unfortunately, however, the PSMA trial was stopped because two patients died due to neurotoxicity [Citation240].
Although CAR T-cell expression of TGF-β traps improves CAR T-cell function, efficacy can be potentiated by harnessing TGF-β to deliver an activating stimulus. First-generation CAR T-cells were generally found to be susceptible to TGF-β-mediated suppression [Citation241]. The same was true for second-generation CAR T-cells containing a 4-1BB costimulatory domain. However, second-generation CAR T-cells expressing a CD28 costimulatory domain were found to be largely resistant to the effects of TGF-β through their ability to bind Lck [Citation216,Citation241]. In the absence of CD28 Lck binding motifs, CAR T-cells were re-sensitized to TGF-β, but became impervious once again upon addition of IL-2, IL-7 or IL-15 [Citation216]. The use of TGFβRII/IL-7 R or TGFβRII/4-1BB switch receptors, composed of the TGFβRII extracellular domain and either IL-7 R or 4-1BB intracellular domains, has been shown to further enhance CAR T-cell proliferation, cytokine production, tumor infiltration, and survival compared to truncated TGFβRII alone [Citation220,Citation242,Citation243].
An alternative strategy to counteract TGF-β mediated immunosuppression entails the targeting of downstream signaling components. SMAD7 is a negative regulator of the TGF-β signaling cascade. When overexpressed, it has been shown to increase cytokine production, stemness, proliferation, tumor infiltration, anti-tumor responses and to reduce exhaustion, Tregs, and TGFβR expression [Citation218,Citation219].
Insufficient inhibition of the TGF-β signaling axis may render T-cells prone to low level TGF-β-mediated immunosuppression. To address this, CAR T-cells have also been subjected to TGFBR2 knockout using CRISPR/Cas9-mediated knockout. Improved CAR T-cell cytokine production, cytotoxicity, stemness, and proliferation were observed, accompanied by resistance to TGF-β immunosuppression [Citation215,Citation222].
A further approach whereby CAR T-cell insensitivity to TGF-β may be achieved entails the use of γδ T-cells as a vehicle for CAR expression. When these cells are expanded in the presence of TGF-β, intrinsic anti-tumor activity is paradoxically enhanced while sensitivity to immunosuppressive effects of TGF-β is nullified [Citation244].
2.3.3. Adenosine
Adenosine is crucial for nucleic acid synthesis and energy metabolism but contributes to immunosuppression when secreted into the extracellular space during inflammation and hypoxia. Adenosine originates from the dephosphorylation of ATP through the activity of ectonucleases, CD39 and CD73 [Citation245–248]. Binding of adenosine to the adenosine receptor, A2AR, on T-cells results in the activation of protein kinase A (PKA) which interferes with NFκB signaling and therefore optimal T-cell activation. Accordingly, A2AR signaling has been found to limit cytokine production, proliferation, and cytotoxicity in CAR T-cells [Citation146,Citation249–254]. Upon activation or PD-1 blockade, CAR T-cells have been demonstrated to upregulate expression of A2AR as an immune checkpoint mechanism to avoid overactivation [Citation146,Citation249,Citation251,Citation253,Citation254]. Several strategies have therefore been developed to enable CAR T-cells to resist adenosine-mediated suppression. For instance, addition of A2AR inhibitors reverses the suppressive effects of adenosine through increased CAR T-cell cytokine production, proliferation, and cytotoxicity [Citation146,Citation249,Citation253,Citation254]. Similarly, CD19-specific CAR T-cells designed to deliver liposome-encapsulated A2AR inhibitors to both TILs and CAR T-cells alike boosted T-cell persistence and anti-tumor responses in SKOV3.CD19-bearing mice [Citation255].
Despite improved anti-tumor responses, these agents may only achieve transient and incomplete A2AR inhibition. Consequently, direct modification of A2AR expression in CAR T-cells may be more favorable. This has been achieved through expression of A2AR-targeting shRNA or complete knockout of the gene encoding A2AR, ADORA2A. Constitutive expression of A2AR-targeting shRNA by CAR T-cells was associated with reduced A2AR expression and increased CAR efficacy in terms of cytokine production and tumor control in both in vitro and in vivo studies [Citation146,Citation250,Citation252,Citation253]. However, CRISPR/Cas9-mediated knockout of ADORA2A had superior effects compared to the use of A2AR inhibitors and targeted shRNAs [Citation250]. Giuffrida et al. generated both heterozygous ADORA2A± and homozygous knockout ADORA2A-/- CAR T-cells and revealed that partial expression of the A2AR is insufficient to rescue T-cells from adenosine-mediated suppression [Citation250]. Furthermore, CAR T-cells expressing A2AR-targeted shRNAs were associated with a more differentiated phenotype and reduced persistence compared to their ADORA2AKO counterparts [Citation250].
Aside from adenosine receptor modulation, altered adenosine metabolism has also shown benefit in CAR T-cells. An in silico screen of T-cell metabolic regulators identified the importance of adenosine deaminase (ADA) in transcriptomic expression and cytokine production [Citation256]. ADA is an enzyme that irreversibly deaminates adenosine to produce inosine, a nucleoside involved in promoting stemness and reducing terminal differentiation in CAR T-cells [Citation257]. ADA overexpression (ADAOE) was found to protect CAR T-cells from the suppressive effects of adenosine through improved cytokine production and cytotoxicity as compared to ADA wild type and ADA knockout versions [Citation256]. In addition, ADAOE CAR T-cells were less differentiated and exhausted [Citation256,Citation258]. Furthermore, infusion of ADAOE CAR T-cells was associated with a reduced Treg population within the tumor and improved tumor responses in vivo [Citation256,Citation258]. The benefit of ADA overexpression was enhanced through conjugation to a collagen-binding domain to increase recruitment and retention in the TME [Citation258]. ADA exists in two different isoforms with isoform ADA1 having a greater adenosine degradation rate than the ADA2 isoform [Citation259]. However, ADA1 is not readily secreted and is largely intracellular [Citation259]. In contrast, the lower activity ADA2 isoform is naturally secreted [Citation259]. As a result, an ADA2 mutant with enhanced activity was developed and introduced into CAR T-cells which increased the adenosine degradation rate [Citation259]. Although there was a reduction in adenosine, the impact of this ADA2 mutant on CAR T-cell anti-tumor responses remains to be explored.
Inhibition of the adenosine pathway is potentially only beneficial for tumors where there is accumulation of adenosine. Several tumors have an adenosine-enriched TME, but the extent to which this is cancer type- or patient-specific remains to be determined. There is clear evidence that a reduction in A2AR expression and/or an increase in ADA limits adenosine-mediated suppression in CAR T-cells. However, while targeting the CD39/CD73 axis may reduce adenosine accumulation, compensatory mechanisms of ATP dephosphorylation into adenosine may occur through the NAD+/CD38 axis. Overexpression of ADA by CAR T-cells is simultaneously beneficial to local TILs, but owing to expression of A2AR, cells may retain some sensitivity to adenosine. Interruption of A2AR expression therefore represents the preferred method to resist adenosine-mediated suppression.
2.3.4. Other immunosuppressive molecules
Prostaglandin E2 (PGE2) represents another abundant immunosuppressive factor within the TME. PGE2 can limit T-cell proliferation through PKA activation, which can be rescued using PGE2 receptor (EP2, EP4) inhibitors [Citation260]. Indeed, EP2/EP4 inhibition improved mesothelin-specific CAR T-cell cytokine production (IL-2, IFN-γ) and cytotoxicity against the AsPC-1 tumor cell line [Citation260]. As both adenosine and PGE2 activate PKA, interfering with PKA directly may simultaneously reduce signaling by both immunosuppressive mediators. To achieve this, mesothelin-specific CAR T-cells were engineered to express a small peptide, RIAD, which interferes with PKA localization at the immune synapse. This was associated with improvements in cytokine production (IFN-γ), cytotoxicity, and resistance to both adenosine and PGE2-mediated suppression [Citation38]. Additionally, co-expression of RIAD by anti-mesothelin CAR T-cells resulted in superior tumor control and enhanced CAR T-cell persistence in mice engrafted with EMmeso, AE17meso, or PDA4662 tumors [Citation38].
IL-10 is an anti-inflammatory cytokine typically secreted by Tregs to inhibit T-cell activation. To counteract Treg-mediated suppression, anti-CEA CAR T-cells were co-administered with IL-10 blockade which was found to rescue CAR T-cell proliferation, activation, and anti-tumor cytotoxicity [Citation261]. In addition to Tregs, activated T-cells, including CD28-based CAR T-cells, also secrete IL-10 to avoid prolonged activation [Citation262]. However, this may be controlled by replacing the CD28 costimulatory domain for OX40 [Citation262]. Although IL-10 is generally considered to be suppressive to T-cells, it can be beneficial in limiting exhaustion. In fact, expanding CAR T-cells with IL-10 has been found to improve anti-tumor cytotoxicity [Citation263] and engineering CAR T-cells to directly secrete IL-10 has improved CAR T-cell effector function, proliferation, stemness, and anti-tumor efficacy [Citation264].
Indoleamine-pyrrole 2,3-dioxygenase 1 (IDO1) promotes the catalysis of tryptophan into kynurenine and is highly expressed by myeloid cells and various cancers which can induce T-cell immunosuppression through tryptophan depletion. IDO1 is present at high levels within the TME but can be further induced upon exposure to CAR T-cell-derived IFN-γ [Citation265]. It inhibits CAR T-cell cytotoxicity, cytokine production, expansion, and induces a senescent phenotype [Citation265–267]. As a result, CD19-directed CAR T-cells lose responsivity against IDO1-overexpressing Raji cells which is restored upon IDO1 inhibition [Citation266]. In a similar fashion, IDO1 downregulation in colorectal cancer cell lines using targeted miRNAs improves CAR T-cell cytokine production, proliferation, and anti-tumor activity [Citation265].
Finally, other immunosuppressive cytokines found in the TME may be also harnessed locally to boost CAR T-cell function. For example, colony-stimulating factor (CSF)-1 is over-produced by a range of solid tumors and promotes the formation of immunosuppressive M2 polarized macrophages [Citation268]. However, co-expression of the c-fms encoded receptor in CAR T-cells enabled their chemotactic migration in response to CSF-1 gradients, accompanied by the co-stimulation of proliferation and cytokine release [Citation269]. Similarly, IL-4 is overproduced in the TME of a number of solid tumors and also contributes to the development of immunosuppressive myeloid cells [Citation270]. This can similarly be harnessed to foster local CAR T-cell proliferation by co-expression of a chimeric cytokine receptor in which the IL-4 receptor α ectodomain is fused to the shared β chain employed by the receptors for IL-2 and IL-15 [Citation271].
2.3.5. Exhaustion
TME-mediated immunosuppression contributes to CAR T-cell exhaustion, but armoring strategies can be implemented to resist individual suppressive mechanisms. However, reducing CAR T-cell susceptibility to exhaustion as a whole through altered transcriptional programming may have superior outcomes. Transcription factors TOX, TOX2 and the three NR4A variants (NR4A1, NR4A2, NR4A3) all contribute to the indication of a dysfunctional CAR T-cell phenotype [Citation135]. Methylation profiling of anti-CD19 CAR T-cells isolated from patients with acute lymphoblastic leukemia post-infusion revealed important epigenetic changes [Citation272]. Specifically, the TCF7 locus (encoding TCF1) was found to be hypermethylated resulting in repression of TCF1; yet the TOX locus remained hypomethylated suggesting a dynamic shift in chromatin accessibility which associates with exhaustion. Correspondingly, exhausted PD-1hi TIM-3hi CAR T-cells express a high level of TOX, TOX2, and NR4A1–3 [Citation132,Citation135].
Owing to the importance of TOX, TOX2 and NR4A in maintaining CAR T-cell dysfunction, inhibiting the expression of these transcription factors has been explored as an avenue to limit exhaustion. Although TOX2 is found to have a role in central memory T-cell development, TOX knockdown is associated with a less differentiated and exhausted phenotype [Citation273]. Given this indication, a complete double knockout of both TOX and TOX2 or triple knockout of NR4A1, NR4A2, and NR4A3 in CD19-reactive CAR T-cells resulted in reversal of the exhausted phenotype accompanied by a notable reduction in inhibitory receptor expression (PD-1, TIM-3, LAG-3), an increase in cytokine secretion and enhanced cytotoxicity with improved survival when infused into tumor-bearing mice [Citation132,Citation135]. The importance of NR4A in exhaustion has also been exploited by placing pro-inflammatory cytokines under the control of an NR4A promoter so that upon exhaustion, stimulatory cytokines will be secreted to improve CAR T-cell persistence and activation [Citation274].
2.4. Hypoxia and oxidative stress
Due to their high proliferative and metabolic activity, solid tumors often rapidly outgrow their blood supply. Despite the accelerated angiogenesis that is frequently observed in solid tumors, oxygenation levels can dip as low as 0.02–2% O2 resulting in regions of hypoxia within the TME. Furthermore, this inadequate oxygen supply generates an acidified environment with redox imbalances. Together with acidosis and oxidative stress, hypoxia induces T-cell anergy and thus limits anti-tumor responses. Similarly, this environment impairs CAR T-cell expansion and activity [Citation275–278]. However, CAR T-cells can be modified to resist such a hostile environment.
HIF-1α is a master regulator of hypoxia and accumulates at low oxygen levels. The HIF-1 complex, consisting of HIF-1α and HIF-1β, binds to hypoxia response elements (HRE) to promote gene expression. HIF-1α contains oxygen-dependent degradation (ODD) domains, restricting protein expression to conditions of hypoxia. Tumors significantly upregulate expression of HIF-1α to survive in the hypoxic TME in which high levels of HIF-1α confer a poorer prognosis in cancer patients [Citation279]. To counteract this, Meymandi et al. combined mesothelin-specific CAR T-cells with the HIF-1α inhibitor PX-478 [Citation279]. Surprisingly, however, PX-478 was found to reduce anti-tumor responses by limiting CAR T-cell proliferation and cytotoxicity, highlighting a key role of HIF-1α in CAR T-cell function [Citation279]. In keeping with this, hypoxia pre-conditioned HER2-specific CAR T-cells upregulated HIF-1α which improved CAR T-cell expansion and anti-tumor responses against hypoxic SKOV3 tumors [Citation280]. Deletion of the HIF-1α negative regulator, VHL, in CAR T-cells also resulted in constitutive HIF-1α expression, leading to a more activated, cytotoxic and resident memory-like phenotype, enhanced CAR T-cell expansion and favorable transcriptomic expression [Citation281]. Similarly, when HER2-specific CAR T-cells were either treated with VHL inhibitors or transduced to express VHL-targeting miRNA, improved CAR T-cell expansion and survival was achieved in SKOV3 xenograft-bearing mice [Citation280].
The specificity of HIF-1α expression for hypoxia can be exploited to confine CAR T-cell activation to the tumor. Using a HRE to direct gene expression and/or fusion of an ODD to the CAR endodomain, selective CAR expression under conditions of hypoxia can be achieved [Citation89,Citation282–286]. By this means, risk of on-target, off-tumor toxicity can be mitigated [Citation89,Citation285]. Moreover, hypoxia-inducible CAR T-cells performed better than their non-engineered counterparts in hypoxia and demonstrated hypoxia-specific tumor lysis and cytokine production with greatest stringency of hypoxia-dependent expression achieved when combining both HRE and ODD elements [Citation89,Citation282,Citation283,Citation285,Citation286]. In addition to tumor-specific CAR T-cell activation, hypoxia-responsive CAR T-cells can also be engineered to specifically secrete typically toxic pro-inflammatory cytokines such as IL-12 during hypoxia, while maintaining safety. Co-expression of IL-12 boosts CAR T-cell efficacy [Citation287], but results in substantial toxicity. To localize IL-12 to the tumor site, CD19-directed CAR T-cells were co-expressed with an IL-12-ODD fusion protein [Citation287]. Although minimal differences were observed in anti-tumor responses when infused into OCI-Ly3-bearing hamsters, toxicity was reduced when IL-12 was fused to an ODD domain [Citation287].
Oxygen depletion within the TME causes an increase in reactive oxygen species (ROS) provoking oxidative stress due to an imbalance in ROS generation and anti-oxidant defense mechanisms. ROS is secreted during oxidative phosphorylation as part of aerobic respiration. Due to the increased proliferation and metabolism observed in cancer, ROS accumulate within the TME. ROS is also secreted by tumor-infiltrating myeloid cells and neutrophils. However, excessive ROS are inhibitory to lymphocytes. Among all TME-derived ROS molecules, H2O2 is the most stable and highly produced and reduces both lymphocyte viability and CAR T-cell/tumor immunological synapse formation [Citation276,Citation288]. H2O2 can be neutralized by anti-oxidant molecules. However, T-cells express minimal anti-oxidants and this renders them susceptible to the detrimental effects of oxidative stress [Citation288]. It is therefore of interest to armor CAR T-cells with anti-oxidant defense mechanisms.
Nrf2 is a master regulator of the anti-oxidant response and can be stimulated by auranofin. As expected, auranofin reduces ROS and, in combination with CD19-specific CAR T-cells, improves CAR T-cell-mediated tumor killing of Raji and N6/ADR cells in the presence of H2O2 [Citation278]. Catalase is the predominant enzyme responsible for H2O2 breakdown and thus can help shield T-cells from H2O2-induced oxidative stress. Indeed, overexpression of catalase by HER2-specific CAR T-cells reduced ROS production and improved CAR T-cell survival, proliferation, and cytotoxicity in the presence of H2O2 when co-cultured with SKOV-3 tumor cells [Citation289]. Additionally, CAR T-cell expression of catalase rescued bystander immune cells from the immunosuppressive effects of oxidative stress [Citation289]. Likewise, CAR T-cells and CAR-NK cells demonstrated enhanced immune cell expansion, cytokine production, and anti-tumor cytotoxicity in the presence of H2O2 or glucose oxidase when supplemented with other anti-oxidants including TRX1, PRDX1, and manganese dioxide [Citation276,Citation288,Citation290].
Finally, hypoxia also causes acidification of the TME which limits CAR T-cell responses and cytokine production [Citation291]. Countermeasures that may help to reverse this include the inhibition or deletion of anion exchanger Ae2, which was found to enhance CAR T-cell function at low pH [Citation291]. Conversely, co-expression of proton extruder Hvcn1 in GPC3-specific CAR T-cells improved anti-tumor responses when engrafted into mice bearing PM299L-GPC3 tumors [Citation291].
3. Conclusion
In this review, we have considered a range of interactions between CAR T-cells and the TME and how engineering/armoring strategies may be used to perturb these. Engineering CAR T-cells to express specific chemokine receptors improves tumor recruitment. Furthermore, CAR T-cell secretion of immune cell recruiting chemokines can reinstate the host immune response and contribute to epitope spreading. Targeting of neo-angiogenesis or the tumor vasculature directly can not only starve the tumor of oxygen and nutrients but also improve CAR T-cell access to the tumor. CAR T-cell targeting of CAFs can both alleviate immunosuppression and reduce the physical barrier generated by the ECM. A combination of CAR T-cells and immune checkpoint blockers can also reduce CAR T-cell inhibition and has been extensively explored clinically, although the best way to deploy this strategy remains uncertain. Inhibition of immune checkpoint signaling can also be achieved through CAR T-cell secretion of neutralizing antibodies, immune checkpoint gene knockout, or expression of dominant negative and switch receptors. Additionally, transgenic expression of TGF-β and adenosine dominant negative receptors, switch receptors, or genetic ablation may also attenuate immunosuppression. To counteract the suppressive activity of oxidative stress, CAR T-cells can be engineered to overexpress components of the anti-oxidant pathway. Hypoxia-dependent CAR expression can limit on-target, off-tumor toxicity and can also be utilized to confine expression of typically toxic pro-inflammatory cytokines to the hypoxic tumor site. Implementation of one or more of the strategies described here may enable the development of more effective CAR T-cell solutions for patients with solid tumors ().
Figure 3. CAR T-cell armouring strategies to help combat obstacles associated with the tumor microenvironment.
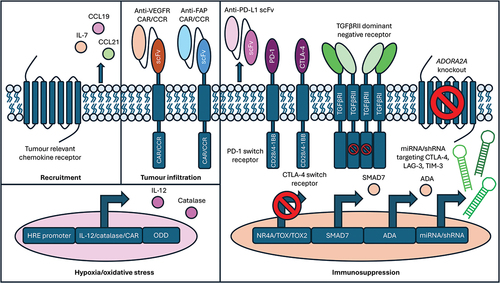
4. Expert opinion
CAR T-cell targeting of solid tumors remains insufficiently effective. Key challenges include the array of physical, chemical, and biological barriers that operate within the TME battleground where these cells must operate. This has been considered in detail in this review together with armoring strategies that may be utilized to favorably influence therapeutic impact. Indeed, many of the approaches presented here are now undergoing clinical investigation in cancer patients (). Since these concepts have generally been derived from imperfect pre-clinical model systems, these emerging clinical data will provide invaluable insights into next steps required to bridge the gap toward successful solid tumor CAR T-cell immunotherapy.
Although most of the approaches described here are directed against a single molecular target or a specific TME feature, it is evident that tumors exhibit substantial inter- and intra-tumoral heterogeneity. Consequently, strategies will probably need to be individualized for specific cancer types. Furthermore, not all tumors are encumbered by a profound stromal component or hypoxia and thus not all cancers may benefit from enhancements in these areas.
CAR T-cells also demonstrate significant intra- and inter-donor heterogeneity. While limiting CAR T-cell immunosuppression using a single targeted intervention may improve their activity, expression of alternative inhibitory molecules may facilitate tumor counter-evasion and restore CAR T-cell inhibition. Simultaneous targeting of multiple immunosuppressive pathways may consequently be required to avoid resistance and sustain CAR T-cell responses.
It should also be appreciated that despite the breakthrough made by second-generation CAR T-cells in the treatment of hematological malignancies, this 25-year-old technology may not be sufficiently robust for the treatment of solid tumors. Rather than focussing on armoring alone, there may also be a need to develop better CAR systems that achieve greater functional persistence of CAR T-cells in vivo. Recently, this has been exemplified through the delivery of synergistic dual co-stimulation using a parallel CAR format [Citation292,Citation293].
Although CAR T-cells have demonstrated a significant ability to specifically kill tumors and to persist long term, over time tumors can become refractory through target downregulation or mutation and hence previously responding patients may relapse. To circumvent this, CAR T-cells can be targeted against multiple target antigens simultaneously or engineered to stimulate the host immune response in concert. CAR T-cell secretion of dendritic cell and T-cell recruiting chemokines, production of immunosuppressant blockers and expression of anti-oxidant enzymes can reinvigorate local host immune cells. Armoring strategies may also be focussed on the stimulation of epitope spreading to harness endogenous tumor-reactive T-cells. Such strategies include the co-expression of pro-inflammatory cytokines such as IL-12 [Citation294,Citation295] or IL-18 [Citation296–299]. Alternatively, Flt3 ligand armoring may be employed to stimulate cross-presenting dendritic cells [Citation300]. Together, these approaches may result in enhanced host anti-tumor immunity and contribute to epitope spreading, thereby removing any reliance upon a single tumor antigen.
Finally, although improvements can be achieved in tumor recruitment, infiltration, and CAR T-cell activity independently, a mixture of approaches may be required to ensure adequate CAR T-cell efficacy for solid tumors. Therefore, improvements should be made in tumor recruitment, infiltration, and resistance to suppression concurrently. Personalization to the specific cancer type and stimulation of host immunity are also desirable endpoints that may rescue CAR T-cell hyporesponsiveness when targeting solid tumors.
Article highlights
Armouring strategies can augment CAR T-cell responses against solid tumors.
Engineering CAR T-cells to co-express chemokine receptors improves tumor recruitment and infiltration.
CAR T-cell secretion of dendritic cell and T-cell chemokines improves immune cell recruitment to the tumor and subsequent tumor surveillance.
Inhibition of tumor-associated neo-angiogenesis can simultaneously induce tumor starvation while improving CAR T-cell tumor infiltration.
CAR T-cells can be redirected to kill cancer-associated fibroblasts (CAFs) or degrade extracellular matrix (ECM) components to improve tumor infiltration.
Concomitant treatment with immune checkpoint inhibitors, such as those neutralizing PD-1, or direct secretion of derived scFvs by CAR T-cells can reduce immunosuppression.
Reduction of CAR T-cell PD-1 expression either through gene knockdown or knockout can limit sensitivity to the inhibitory effects of PD-L1.
Switch receptors such as PD-1/CD28 fusions offer the opportunity to harness PD-L1 engagement to deliver a co-stimulatory signal.
The transforming growth factor β receptor II (TGFβRII) dominant negative receptor abrogates inhibitory TGF-β signaling and can also be converted into a switch receptor.
Expression of the A2AR dominant negative receptor, knockout of ADORA2A, or overexpression of adenosine deaminase can limit sensitivity to adenosine-mediated immunosuppression.
Hypoxia-dependent CAR expression can be stringently achieved through the use of a hypoxia-responsive promoter and/or fused oxygen-dependent degradation domain. By this means, CAR expression can be restricted to hypoxic tumor areas, limiting on-target, off-tumor toxicity.
Co-expression of anti-oxidant enzymes by CAR T-cells can improve responses in the pro-oxidative tumor milieu.
A combination of different approaches will likely be required to improve CAR T-cell tumor recruitment, infiltration, and resistance to immunosuppressive mechanisms.
Declaration of interest
C Taylor and M Glover are employees of Leucid Bio. J Maher is Chief Scientific Officer, paid consultant and shareholder in Leucid Bio. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosure
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Finney HM, Lawson AD, Bebbington CR, et al. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998 Sep 15;161(6):2791–2797. doi: 10.4049/jimmunol.161.6.2791
- Bouchkouj N, Lin X, Wang X, et al. FDA approval summary: brexucabtagene autoleucel for treatment of adults with relapsed or refractory B-Cell precursor acute lymphoblastic leukemia. Oncologist. 2022 Oct 1;27(10):892–899. doi: 10.1093/oncolo/oyac163
- Bouchkouj N, Zimmerman M, Kasamon YL, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory follicular lymphoma. Oncologist. 2022 Jul 5;27(7):587–594. doi: 10.1093/oncolo/oyac054
- Elmacken M, Peredo-Pinto H, Wang C, et al. FDA approval summary: Lisocabtagene Maraleucel for second-line treatment of large B-Cell lymphoma. Clin Cancer Res. 2024 Feb 7;30(11):2309–2316. doi: 10.1158/1078-0432.CCR-23-2967
- King AC, Orozco JS. Axicabtagene Ciloleucel: the first FDA-Approved CAR T-Cell therapy for Relapsed/Refractory large B-Cell lymphoma. J Adv Pract Oncol. 2019 Nov;10(8):878–882. doi: 10.6004/jadpro.2019.10.8.9
- O’Leary MC, Lu X, Huang Y, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin Cancer Res. 2019 Feb 15;25(4):1142–1146. doi: 10.1158/1078-0432.CCR-18-2035
- Sharma P, Kasamon YL, Lin X, et al. FDA approval summary: Axicabtagene Ciloleucel for second-line treatment of large B-Cell lymphoma. Clin Cancer Res. 2023 Nov 1;29(21):4331–4337. doi: 10.1158/1078-0432.CCR-23-0568
- Letter TM. Ciltacabtagene autoleucel (Carvykti) for multiple myeloma. Med Lett Drugs Ther. 2022 Nov 14;64(1663):e188–e189.
- Mullard A. FDA approves second BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discov. 2022 Apr;21(4):249. doi: 10.1038/d41573-022-00048-8
- Sharma P, Kanapuru B, George B, et al. FDA approval summary: idecabtagene vicleucel for relapsed or refractory multiple myeloma. Clin Cancer Res. 2022 May 2;28(9):1759–1764. doi: 10.1158/1078-0432.CCR-21-3803
- Maher J, Davies DM. CAR-Based immunotherapy of solid tumours–A survey of the emerging targets. Cancers (Basel). 2023 Feb 11;15(4):1171. doi: 10.3390/cancers15041171
- Maher J, Davies DM. CAR based immunotherapy of solid tumours–A clinically based review of target antigens. Biology (Basel). 2023;12(2):287. doi: 10.3390/biology12020287
- Glover M, Avraamides S, Maher J. How can we engineer CAR T cells to overcome resistance? Biologics. 2021;15:175–198. doi: 10.2147/BTT.S252568
- Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987 Dec 31;149(3):960–968. doi: 10.1016/0006-291X(87)90502-X
- Vonderheide RH, Bear AS. Tumor-derived myeloid cell Chemoattractants and T cell exclusion in pancreatic cancer. Front Immunol. 2020;11:605619. doi: 10.3389/fimmu.2020.605619
- Fujimura T, Aiba S. Significance of immunosuppressive cells as a target for immunotherapies in melanoma and non-melanoma skin cancers. Biomolecules. 2020 Jul 22;10(8):1087. doi: 10.3390/biom10081087
- Hege KM, Bergsland EK, Fisher GA, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5(1):22. doi: 10.1186/s40425-017-0222-9
- Tchou J, Zhao Y, Levine BL, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. 2017 Dec;5(12):1152–1161. doi: 10.1158/2326-6066.CIR-17-0189
- Papa S, Adami A, Metoudi M, et al. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC). J Clin Oncol. 2018;36(15_suppl):3046–3046. doi: 10.1200/JCO.2018.36.15_suppl.3046
- Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-Cell therapy. N Engl J Med. 2016 Dec 29;375(26):2561–2569. doi: 10.1056/NEJMoa1610497
- Whilding LM, Halim L, Draper B, et al. CAR T-Cells targeting the Integrin αvβ6 and co-expressing the Chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel). 2019 May 14;11(5):674. doi: 10.3390/cancers11050674
- Korbecki J, Kupnicka P, Chlubek M, et al. CXCR2 receptor: regulation of expression, signal transduction, and involvement in cancer. Int J Mol Sci. 2022 Feb 16;23(4):2168. doi: 10.3390/ijms23042168
- Kershaw MH, Wang G, Westwood JA, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002 Nov 1;13(16):1971–1980. doi: 10.1089/10430340260355374
- Jin L, Tao H, Karachi A, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019 Sep 5;10(1):4016. doi: 10.1038/s41467-019-11869-4
- Liu G, Rui W, Zheng H, et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. 2020 May;50(5):712–724. doi: 10.1002/eji.201948457
- Craddock JA, Lu A, Bear A, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010 Oct;33(8):780–788. doi: 10.1097/CJI.0b013e3181ee6675
- Moon EK, Carpenito C, Sun J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011 Jul 15;17(14):4719–4730. doi: 10.1158/1078-0432.CCR-11-0351
- Siddiqui I, Erreni M, van Brakel M, et al. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: importance of the chemokine gradient. J Immunother Cancer. 2016;4(1):21. doi: 10.1186/s40425-016-0125-1
- Cadilha BL, Benmebarek MR, Dorman K, et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci Adv. 2021 Jun;7(24). doi: 10.1126/sciadv.abi5781
- Jin L, Cao L, Zhu Y, et al. Enhance anti-lung tumor efficacy of chimeric antigen receptor-T cells by ectopic expression of C-C motif chemokine receptor 6. Sci Bull (Beijing). 2021 Apr 30;66(8):803–812. doi: 10.1016/j.scib.2020.12.027
- Lesch S, Blumenberg V, Stoiber S, et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat Biomed Eng. 2021 Nov;5(11):1246–1260. doi: 10.1038/s41551-021-00737-6
- Sun R, Sun Y, Wu C, et al. CXCR4-modified CAR-T cells suppresses MDSCs recruitment via STAT3/NF-κB/SDF-1α axis to enhance efficacy against pancreatic cancer. Mol Ther. 2023 Nov 1;31(11):3193–3209. doi: 10.1016/j.ymthe.2023.09.010
- Dai Z, Lin X, Wang X, et al. Ectopic CXCR2 expression cells improve the anti-tumor efficiency of CAR-T cells and remodel the immune microenvironment of pancreatic ductal adenocarcinoma. Cancer Immunol Immunother. 2024 Mar 2;73(4):61. doi: 10.1007/s00262-024-03648-y
- Adachi K, Kano Y, Nagai T, et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018 Apr;36(4):346–351. doi: 10.1038/nbt.4086
- Pang N, Shi J, Qin L, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol. 2021 Jul 29;14(1):118. doi: 10.1186/s13045-021-01128-9
- Luo H, Su J, Sun R, et al. Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clin Cancer Res. 2020 Oct 15;26(20):5494–5505. doi: 10.1158/1078-0432.CCR-20-0777
- Moon EK, Wang LS, Bekdache K, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018;7(3):e1395997. doi: 10.1080/2162402X.2017.1395997
- Newick K, O’Brien S, Sun J, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase a localization. Cancer Immunol Res. 2016 Jun;4(6):541–551. doi: 10.1158/2326-6066.CIR-15-0263
- Ribatti D. Aberrant tumor vasculature. Facts and pitfalls. Front Pharmacol. 2024;15:1384721. doi: 10.3389/fphar.2024.1384721
- Deng C, Zhao J, Zhou S, et al. The vascular disrupting agent CA4P improves the antitumor efficacy of CAR-T cells in preclinical models of solid human tumors. Mol Ther. 2020 Jan 8;28(1):75–88. doi: 10.1016/j.ymthe.2019.10.010
- Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993 Apr 29;362(6423):841–844. doi: 10.1038/362841a0
- Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA. 1995 Nov 7;92(23):10457–10461. doi: 10.1073/pnas.92.23.10457
- Bocca P, Di Carlo E, Caruana I, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology. 2017;7(1):e1378843. doi: 10.1080/2162402X.2017.1378843
- Dong X, Ren J, Amoozgar Z, et al. Anti-VEGF therapy improves EGFR-vIII-CAR-T cell delivery and efficacy in syngeneic glioblastoma models in mice. J Immunother Cancer. 2023 Mar;11(3):e005583. doi: 10.1136/jitc-2022-005583
- Wu X, Luo H, Shi B, et al. Combined antitumor effects of sorafenib and GPC3-CAR T cells in mouse models of hepatocellular carcinoma. Mol Ther. 2019 Aug 7;27(8):1483–1494. doi: 10.1016/j.ymthe.2019.04.020
- Li H, Ding J, Lu M, et al. CAIX-specific CAR-T cells and sunitinib show synergistic effects against metastatic renal cancer models. J Immunother. 2020 Jan;43(1):16–28. doi: 10.1097/CJI.0000000000000301
- Kershaw MH, Westwood JA, Zhu Z, et al. Generation of gene-modified T cells reactive against the angiogenic kinase insert domain-containing receptor (KDR) found on tumor vasculature. Hum Gene Ther. 2000 Dec 10;11(18):2445–2452. doi: 10.1089/10430340050207939
- Niederman TM, Ghogawala Z, Carter BS, et al. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci USA. 2002 May 14;99(10):7009–7014. doi: 10.1073/pnas.092562399
- Chinnasamy D, Yu Z, Theoret MR, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest. 2010 Nov;120(11):3953–3968. doi: 10.1172/JCI43490
- Chinnasamy D, Yu Z, Kerkar SP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012 Mar 15;18(6):1672–1683. doi: 10.1158/1078-0432.CCR-11-3050
- Chinnasamy D, Tran E, Yu Z, et al. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013 Jun 1;73(11):3371–3380. doi: 10.1158/0008-5472.CAN-12-3913
- Kanagawa N, Yanagawa T, Nakagawa T, et al. Tumor vessel-injuring ability improves antitumor effect of cytotoxic T lymphocytes in adoptive immunotherapy. Cancer Gene Ther. 2013 Jan;20(1):57–64. doi: 10.1038/cgt.2012.85
- Inoo K, Inagaki R, Fujiwara K, et al. Immunological quality and performance of tumor vessel-targeting CAR-T cells prepared by mRNA-EP for clinical research. Mol Ther Oncolytics. 2016;3:16024. doi: 10.1038/mto.2016.24
- Kulemzin SV, Gorchakov AA, Chikaev AN, et al. VEGFR2-specific FnCAR effectively redirects the cytotoxic activity of T cells and YT NK cells. Oncotarget. 2018 Feb 6;9(10):9021–9029. doi: 10.18632/oncotarget.24078
- Hajari Taheri F, Hassani M, Sharifzadeh Z, et al. T cell engineered with a novel nanobody-based chimeric antigen receptor against VEGFR2 as a candidate for tumor immunotherapy. IUBMB Life. 2019 Sep;71(9):1259–1267. doi: 10.1002/iub.2019
- Lanitis E, Kosti P, Ronet C, et al. VEGFR-2 redirected CAR-T cells are functionally impaired by soluble VEGF-A competition for receptor binding. J Immunother Cancer. 2021 Aug;9(8):e002151. doi: 10.1136/jitc-2020-002151
- Lanitis E, Rota G, Kosti P, et al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med. 2021 Feb 1;218(2). doi: 10.1084/jem.20192203
- Duan Z, Li Z, Wang Z, et al. Chimeric antigen receptor macrophages activated through TLR4 or IFN-γ receptors suppress breast cancer growth by targeting VEGFR2. Cancer Immunol Immunother. 2023 Oct;72(10):3243–3257. doi: 10.1007/s00262-023-03490-8
- Zhong M, Chalbatani GM, Deng M, et al. Functional characterization and development of novel human kinase insert domain receptor chimeric antigen receptor T-cells for immunotherapy of non-small cell lung cancer. Eur J Pharm Sci. 2023 Jan 1;180:106331. doi: 10.1016/j.ejps.2022.106331
- Englisch A, Altvater B, Kailayangiri S, et al. VEGFR2 as a target for CAR T cell therapy of Ewing sarcoma. Pediatr Blood Cancer. 2020 Oct;67(10):e28313. doi: 10.1002/pbc.28313
- Rosenberg SA. CAR T cell receptor immunotherapy targeting VEGFR2 for patients with metastatic cancer: ClinicalTrials.Gov. 2015. Available from: https://clinicaltrials.gov/study/NCT01218867
- Wang W, Ma Y, Li J, et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. 2013 Oct;20(10):970–978. doi: 10.1038/gt.2013.19
- Xing H, Yang X, Xu Y, et al. Anti-tumor effects of vascular endothelial growth factor/vascular endothelial growth factor receptor binding domain-modified chimeric antigen receptor T cells. Cytotherapy. 2021 Sep;23(9):810–819. doi: 10.1016/j.jcyt.2021.05.008
- Demircioglu F, Hodivala-Dilke KA. Integrin and tumour blood vessels-learning from the past to shape the future. Curr Opin Cell Biol. 2016 Oct;42:121–127. doi: 10.1016/j.ceb.2016.07.008
- Fu X, Rivera A, Tao L, et al. Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int J Cancer. 2013 Nov 15;133(10):2483–2492. doi: 10.1002/ijc.28269
- Wallstabe L, Mades A, Frenz S, et al. CAR T cells targeting α(v)β(3) integrin are effective against advanced cancer in preclinical models. Adv Cell Gene Ther. 2018 Sep;1(2):e11. doi: 10.1002/acg2.11
- Zhang E, Gu J, Xue J, et al. Accurate control of dual-receptor-engineered T cell activity through a bifunctional anti-angiogenic peptide. J Hematol Oncol. 2018 Mar 20;11(1):44. doi: 10.1186/s13045-018-0591-7
- Cobb DA, de Rossi J, Liu L, et al. Targeting of the alpha v beta 3 integrin complex by CAR-T cells leads to rapid regression of diffuse intrinsic pontine glioma and glioblastoma. J Immunother Cancer. 2022 Feb;10(2):e003816. doi: 10.1136/jitc-2021-003816
- Xie YJ, Dougan M, Jailkhani N, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci USA. 2019 Apr 16;116(16):7624–7631. doi: 10.1073/pnas.1817147116
- Wagner J, Wickman E, Shaw TI, et al. Antitumor effects of CAR T cells redirected to the EDB splice variant of fibronectin. Cancer Immunol Res. 2021 Mar;9(3):279–290. doi: 10.1158/2326-6066.CIR-20-0280
- Martin-Otal C, Lasarte-Cia A, Serrano D, et al. Targeting the extra domain a of fibronectin for cancer therapy with CAR-T cells. J Immunother Cancer. 2022 Aug;10(8):e004479. doi: 10.1136/jitc-2021-004479
- Zhang Z, Liu C, Wang M, et al. Treating solid tumors with TCR-based chimeric antigen receptor targeting extra domain B-containing fibronectin. J Immunother Cancer. 2023 Aug;11(8):e007199. doi: 10.1136/jitc-2023-007199
- Altvater B, Kailayangiri S, Spurny C, et al. CAR T cells as micropharmacies against solid cancers: combining effector T-cell mediated cell death with vascular targeting in a one-step engineering process. Cancer Gene Ther. 2023 Oct;30(10):1355–1368. doi: 10.1038/s41417-023-00642-x
- Conway RE, Petrovic N, Li Z, et al. Prostate-specific membrane antigen regulates angiogenesis by modulating integrin signal transduction. Mol Cell Biol. 2006 Jul;26(14):5310–5324. doi: 10.1128/MCB.00084-06
- Santoro SP, Kim S, Motz GT, et al. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res. 2015 Jan;3(1):68–84. doi: 10.1158/2326-6066.CIR-14-0192
- Robinson J, Whitworth K, Jinks E, et al. An evaluation of the tumour endothelial marker CLEC14A as a therapeutic target in solid tumours. J Pathol Clin Res. 2020 Oct;6(4):308–319. doi: 10.1002/cjp2.176
- Zhuang X, Maione F, Robinson J, et al. CAR T cells targeting tumor endothelial marker CLEC14A inhibit tumor growth. JCI Insight. 2020 Oct 2;5(19). doi: 10.1172/jci.insight.138808
- St Croix B, Rago C, Velculescu V, et al. Genes expressed in human tumor endothelium. Science. 2000 Aug 18;289(5482):1197–1202. doi: 10.1126/science.289.5482.1197
- MacFadyen JR, Haworth O, Roberston D, et al. Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett. 2005 May 9;579(12):2569–2575. doi: 10.1016/j.febslet.2005.03.071
- Huber MA, Kraut N, Schweifer N, et al. Expression of stromal cell markers in distinct compartments of human skin cancers. J Cutan Pathol. 2006 Feb;33(2):145–155. doi: 10.1111/j.0303-6987.2006.00446.x
- Bagley RG, Honma N, Weber W, et al. Endosialin/TEM 1/CD248 is a pericyte marker of embryonic and tumor neovascularization. Microvasc Res. 2008 Nov;76(3):180–188. doi: 10.1016/j.mvr.2008.07.008
- Christian S, Winkler R, Helfrich I, et al. Endosialin (Tem1) is a marker of tumor-associated myofibroblasts and tumor vessel-associated mural cells. Am J Pathol. 2008 Feb;172(2):486–494. doi: 10.2353/ajpath.2008.070623
- Simonavicius N, Robertson D, Bax DA, et al. Endosialin (CD248) is a marker of tumor-associated pericytes in high-grade glioma. Mod Pathol. 2008 Mar;21(3):308–315. doi: 10.1038/modpathol.3801006
- Ebert LM, Yu W, Gargett T, et al. Endothelial, pericyte and tumor cell expression in glioblastoma identifies fibroblast activation protein (FAP) as an excellent target for immunotherapy. Clin Transl Immunol. 2020;9(10):e1191. doi: 10.1002/cti2.1191
- Ash SL, Orha R, Mole H, et al. Targeting the activated microenvironment with endosialin (CD248)-directed CAR-T cells ablates perivascular cells to impair tumor growth and metastasis. J Immunother Cancer. 2024 Feb 27;12(2):e008608. doi: 10.1136/jitc-2023-008608
- Byrd TT, Fousek K, Pignata A, et al. TEM8/ANTXR1-specific CAR T cells as a targeted therapy for triple-negative breast cancer. Cancer Res. 2018 Jan 15;78(2):489–500. doi: 10.1158/0008-5472.CAN-16-1911
- Petrovic K, Robinson J, Whitworth K, et al. TEM8/ANTXR1-specific CAR T cells mediate toxicity in vivo. PLoS One. 2019;14(10):e0224015. doi: 10.1371/journal.pone.0224015
- Legler DF, Johnson-Léger C, Wiedle G, et al. The alpha v beta 3 integrin as a tumor homing ligand for lymphocytes. Eur J Immunol. 2004 Jun;34(6):1608–1616. doi: 10.1002/eji.200424938
- Kosti P, Opzoomer JW, Larios-Martinez KI, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med. 2021 Apr 20;2(4):100227. doi: 10.1016/j.xcrm.2021.100227
- Glabman RA, Choyke PL, Sato N. Cancer-associated fibroblasts: tumorigenicity and targeting for cancer therapy. Cancers (Basel). 2022 Aug 12;14(16). doi: 10.3390/cancers14163906
- Wright K, Ly T, Kriet M, et al. Cancer-associated fibroblasts: master tumor microenvironment modifiers. Cancers (Basel). 2023 Mar 22;15(6):1899. doi: 10.3390/cancers15061899
- Belle JI, Sen D, Baer JM, et al. Senescence defines a distinct subset of myofibroblasts that orchestrates immunosuppression in pancreatic cancer. Cancer Discov. 2024 Apr 29;14(7):1324–1355.
- Sakemura R, Hefazi M, Siegler EL, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood. 2022 Jun 30;139(26):3708–3721. doi: 10.1182/blood.2021012811
- Chuangchot N, Jamjuntra P, Yangngam S, et al. Enhancement of PD-L1-attenuated CAR-T cell function through breast cancer-associated fibroblasts-derived IL-6 signaling via STAT3/AKT pathways. Breast Cancer Res. 2023 Jul 21;25(1):86. doi: 10.1186/s13058-023-01684-7
- Baldari S, Di Modugno F, Nistico P, et al. Strategies for efficient targeting of tumor collagen for cancer therapy. Cancers (Basel). 2022 Sep 27;14(19):4706. doi: 10.3390/cancers14194706
- Kuczek DE, Larsen AMH, Thorseth ML, et al. Collagen density regulates the activity of tumor-infiltrating T cells. J Immunother Cancer. 2019 Mar 12;7(1):68. doi: 10.1186/s40425-019-0556-6
- Vijver SV, Singh A, Mommers-Elshof E, et al. Collagen fragments produced in cancer mediate T cell suppression through leukocyte-associated immunoglobulin-like receptor 1. Front Immunol. 2021;12:733561. doi: 10.3389/fimmu.2021.733561
- Zhao R, Cui Y, Zheng Y, et al. Human hyaluronidase PH20 potentiates the antitumor activities of mesothelin-specific CAR-T cells against gastric cancer. Front Immunol. 2021;12:660488. doi: 10.3389/fimmu.2021.660488
- Xiong X, Xi J, Liu Q, et al. Co-expression of IL-7 and PH20 promote anti-GPC3 CAR-T tumour suppressor activity in vivo and in vitro. Liver Int. 2021 May;41(5):1033–1043. doi: 10.1111/liv.14771
- Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015 May;21(5):524–529. doi: 10.1038/nm.3833
- Zhang W, Liu L, Su H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer. 2019 Nov;121(10):837–845. doi: 10.1038/s41416-019-0578-3
- Lee YE, Go GY, Koh EY, et al. Synergistic therapeutic combination with a CAF inhibitor enhances CAR-NK-mediated cytotoxicity via reduction of CAF-released IL-6. J Immunother Cancer. 2023 Feb;11(2):e006130. doi: 10.1136/jitc-2022-006130
- Garin-Chesa P, Old LJ, Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci USA. 1990 Sep;87(18):7235–7239. doi: 10.1073/pnas.87.18.7235
- Hofheinz RD, Al-Batran SE, Hartmann F, et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003 Feb;26(1):44–48. doi: 10.1159/000069863
- Scott AM, Wiseman G, Welt S, et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003 May;9(5):1639–1647.
- Welt S, Divgi CR, Scott AM, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol. 1994 Jun;12(6):1193–1203. doi: 10.1200/JCO.1994.12.6.1193
- Kakarla S, Chow KK, Mata M, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013 Aug;21(8):1611–1620. doi: 10.1038/mt.2013.110
- Schuberth PC, Hagedorn C, Jensen SM, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. 2013 Aug 12;11(1):187. doi: 10.1186/1479-5876-11-187
- Tran E, Chinnasamy D, Yu Z, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013 Jun 3;210(6):1125–1135. doi: 10.1084/jem.20130110
- Wang LC, Lo A, Scholler J, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014 Feb;2(2):154–166. doi: 10.1158/2326-6066.CIR-13-0027
- Lo A, Wang LS, Scholler J, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-Expressing stromal cells. Cancer Res. 2015 Jul 15;75(14):2800–2810. doi: 10.1158/0008-5472.CAN-14-3041
- Li F, Zhao S, Wei C, et al. Development of Nectin4/FAP-targeted CAR-T cells secreting IL-7, CCL19, and IL-12 for malignant solid tumors. Front Immunol. 2022;13:958082. doi: 10.3389/fimmu.2022.958082
- Das S, Valton J, Duchateau P, et al. Stromal depletion by TALEN-edited universal hypoimmunogenic FAP-CAR T cells enables infiltration and anti-tumor cytotoxicity of tumor antigen-targeted CAR-T immunotherapy. Front Immunol. 2023;14:1172681. doi: 10.3389/fimmu.2023.1172681
- Liu Y, Sun Y, Wang P, et al. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18.2-targeted CAR-T against pancreatic cancer. J Transl Med. 2023 Apr 12;21(1):255. doi: 10.1186/s12967-023-04080-z
- Xiao Z, Todd L, Huang L, et al. Desmoplastic stroma restricts T cell extravasation and mediates immune exclusion and immunosuppression in solid tumors. Nat Commun. 2023 Aug 22;14(1):5110. doi: 10.1038/s41467-023-40850-5
- Gulati P, Ruhl J, Kannan A, et al. Aberrant lck signal via CD28 costimulation augments antigen-specific functionality and tumor control by redirected T cells with PD-1 blockade in humanized mice. Clin Cancer Res. 2018 Aug 15;24(16):3981–3993. doi: 10.1158/1078-0432.CCR-17-1788
- Moon EK, Wang LC, Dolfi DV, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014 Aug 15;20(16):4262–4273. doi: 10.1158/1078-0432.CCR-13-2627
- Wang E, Wang LC, Tsai CY, et al. Generation of potent T-cell immunotherapy for cancer using DAP12-based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res. 2015 Jul;3(7):815–826. doi: 10.1158/2326-6066.CIR-15-0054
- Roberts EW, Deonarine A, Jones JO, et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013 Jun 3;210(6):1137–1151. doi: 10.1084/jem.20122344
- Hiltbrunner S, Britschgi C, Schuberth P, et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann Oncol. 2021 Jan;32(1):120–121. doi: 10.1016/j.annonc.2020.10.474
- Petrausch U, Schuberth PC, Hagedorn C, et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer. 2012;12(1):615. doi: 10.1186/1471-2407-12-615
- Millul J, Bassi G, Mock J, et al. An ultra-high-affinity small organic ligand of fibroblast activation protein for tumor-targeting applications. Proc Natl Acad Sci USA. 2021 Apr 20;118(16). doi: 10.1073/pnas.2101852118
- Loureiro LR, Hoffmann L, Neuber C, et al. Immunotheranostic target modules for imaging and navigation of UniCAR T-cells to strike FAP-expressing cells and the tumor microenvironment. J Exp Clin Cancer Res. 2023 Dec 15;42(1):341. doi: 10.1186/s13046-023-02912-w
- Wehrli M, Guinn S, Birocchi F, et al. Mesothelin CAR T-cells secreting anti-FAP/anti-CD3 molecules efficiently target pancreatic adenocarcinoma and its stroma. Clinical Cancer Research. 2024 Feb 23;30(9):1859–1877. doi: 10.1158/1078-0432.CCR-23-3841
- Van den Eynde A, Gehrcken L, Verhezen T, et al. IL-15-secreting CAR natural killer cells directed toward the pan-cancer target CD70 eliminate both cancer cells and cancer-associated fibroblasts. J Hematol Oncol. 2024 Feb 9;17(1):8. doi: 10.1186/s13045-024-01525-w
- Pal SK, Tran B, Haanen J, et al. CD70-targeted allogeneic CAR T-Cell therapy for advanced clear cell renal cell carcinoma. Cancer Discov. 2024 Apr 5;14(7):1176–1189.
- Im SJ, Hashimoto M, Gerner MY, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016 Sep 15;537(7620):417–421. doi: 10.1038/nature19330
- Scott-Browne JP, López-Moyado IF, Trifari S, et al. Dynamic changes in chromatin accessibility occur in CD8(+) T cells responding to viral infection. Immunity. 2016 Dec 20;45(6):1327–1340. doi: 10.1016/j.immuni.2016.10.028
- Mognol GP, Spreafico R, Wong V, et al. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci USA. 2017 Mar 28;114(13):E2776–e2785. doi: 10.1073/pnas.1620498114
- Snell LM, MacLeod BL, Law JC, et al. CD8(+) T cell priming in established chronic viral infection preferentially directs differentiation of memory-like cells for sustained immunity. Immunity. 2018 Oct 16;49(4):678–694.e5. doi: 10.1016/j.immuni.2018.08.002
- Alfei F, Kanev K, Hofmann M, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature. 2019 Jul;571(7764):265–269. doi: 10.1038/s41586-019-1326-9
- Chen J, Lopez-Moyado IF, Seo H, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019 Mar;567(7749):530–534. doi: 10.1038/s41586-019-0985-x
- Khan O, Giles JR, McDonald S, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019 Jul;571(7764):211–218. doi: 10.1038/s41586-019-1325-x
- Scott AC, Dündar F, Zumbo P, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019 Jul;571(7764):270–274. doi: 10.1038/s41586-019-1324-y
- Seo H, Chen J, Gonzalez-Avalos E, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci USA. 2019 Jun 18;116(25):12410–12415. doi: 10.1073/pnas.1905675116
- Li P, Yang L, Li T, et al. The third generation anti-HER2 chimeric antigen receptor mouse T cells alone or together with anti-PD1 antibody inhibits the growth of mouse breast tumor cells expressing HER2 in vitro and in immune competent mice. Front Oncol. 2020;10:1143. doi: 10.3389/fonc.2020.01143
- Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017 Apr 7;7(1):737. doi: 10.1038/s41598-017-00462-8
- Li F, Zhang Z, Xuan Y, et al. PD-1 abrogates the prolonged persistence of CD8(+) CAR-T cells with 4-1BB co-stimulation. Signal Transduct Target Ther. 2020 Aug 25;5(1):164. doi: 10.1038/s41392-020-00277-6
- Jacobson CA, Hunter B, Armand P, et al. Axicabtagene Ciloleucel in the real world: outcomes and predictors of response, resistance and toxicity. Blood. 2018;132(Supplement 1):92. doi: 10.1182/blood-2018-99-117199
- Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016 Aug 1;126(8):3130–3144. doi: 10.1172/JCI83092
- Hirayama AV, Kimble EL, Wright JH, et al. Timing of anti-PD-L1 antibody initiation affects efficacy/toxicity of CD19 CAR T-cell therapy for large B-cell lymphoma. Blood Adv. 2024 Jan 23;8(2):453–467. doi: 10.1182/bloodadvances.2023011287
- Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B-Cell lymphomas. N Engl J Med. 2017 Dec 28;377(26):2545–2554. doi: 10.1056/NEJMoa1708566
- Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018 May;24(5):563–571. doi: 10.1038/s41591-018-0010-1
- Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell lymphoma. N Engl J Med. 2019 Jan 3;380(1):45–56. doi: 10.1056/NEJMoa1804980
- John LB, Devaud C, Duong CP, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013 Oct 15;19(20):5636–5646. doi: 10.1158/1078-0432.CCR-13-0458
- Beavis PA, Henderson MA, Giuffrida L, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017 Mar 1;127(3):929–941. doi: 10.1172/JCI89455
- Giuffrida L, Sek K, Henderson MA, et al. IL-15 preconditioning augments CAR T cell responses to checkpoint blockade for improved treatment of solid tumors. Mol Ther. 2020 Nov 4;28(11):2379–2393. doi: 10.1016/j.ymthe.2020.07.018
- Strecker MI, Wlotzka K, Strassheimer F, et al. AAV-mediated gene transfer of a checkpoint inhibitor in combination with HER2-targeted CAR-NK cells as experimental therapy for glioblastoma. Oncoimmunology. 2022;11(1):2127508. doi: 10.1080/2162402X.2022.2127508
- Gargett T, Yu W, Dotti G, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016 Jun;24(6):1135–1149. doi: 10.1038/mt.2016.63
- Serganova I, Moroz E, Cohen I, et al. Enhancement of PSMA-Directed CAR adoptive immunotherapy by PD-1/PD-L1 blockade. Mol Ther Oncolytics. 2017 Mar 17;4:41–54. doi: 10.1016/j.omto.2016.11.005
- Yin Y, Boesteanu AC, Binder ZA, et al. Checkpoint blockade reverses anergy in IL-13Rα2 humanized scFv-based CAR T cells to treat murine and canine gliomas. Mol Ther Oncolytics. 2018 Dec 21;11:20–38. doi: 10.1016/j.omto.2018.08.002
- Harrasser M, Gohil SH, Lau H, et al. Inducible localized delivery of an anti-PD-1 scFv enhances anti-tumor activity of ROR1 CAR-T cells in TNBC. Breast Cancer Res. 2022 Jun 3;24(1):39. doi: 10.1186/s13058-022-01531-1
- Grover NS, Ivanova A, Moore DT, et al. CD30-directed CAR-T cells Co-expressing CCR4 in Relapsed/Refractory Hodgkin lymphoma and CD30+ cutaneous T cell lymphoma. Blood. 2021;138(Supplement 1):742–2. doi: 10.1182/blood-2021-148102
- Duan D, Wang K, Wei C, et al. The BCMA-Targeted fourth-generation CAR-T cells secreting IL-7 and CCL19 for therapy of Refractory/Recurrent multiple myeloma. Front Immunol. 2021;12:609421. doi: 10.3389/fimmu.2021.609421
- Lei W, Zhao A, Liu H, et al. Safety and feasibility of anti-CD19 CAR T cells expressing inducible IL-7 and CCL19 in patients with relapsed or refractory large B-cell lymphoma. Cell Discov. 2024 Jan 9;10(1):5. doi: 10.1038/s41421-023-00625-0
- Heczey A, Louis CU, Savoldo B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017 Sep 6;25(9):2214–2224. doi: 10.1016/j.ymthe.2017.05.012
- Adusumilli PS, Zauderer MG, Riviere I, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discov. 2021 Nov;11(11):2748–2763. doi: 10.1158/2159-8290.CD-21-0407
- Chong EA, Alanio C, Svoboda J, et al. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood. 2022 Feb 17;139(7):1026–1038. doi: 10.1182/blood.2021012634
- Jaeger U, Worel N, McGuirk JP, et al. Safety and efficacy of tisagenlecleucel plus pembrolizumab in patients with r/r DLBCL: phase 1b PORTIA study results. Blood Adv. 2023 Jun 13;7(11):2283–2286. doi: 10.1182/bloodadvances.2022007779
- Bagley SJ, Binder ZA, Lamrani L, et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer. 2024 Mar;5(3):517–531. doi: 10.1038/s43018-023-00709-6
- Osborne W, Marzolini M, Tholouli E, et al. Phase I alexander study of AUTO3, the first CD19/22 dual targeting CAR T cell therapy, with pembrolizumab in patients with relapsed/refractory (r/r) DLBCL. J Clin Oncol. 2020;38(15_suppl):8001–8001. doi: 10.1200/JCO.2020.38.15_suppl.8001
- Zhang Y, Geng H, Zeng L, et al. Tislelizumab augment the efficacy of CD19/22 dual-targeted chimeric antigen receptor T cell in advanced stage relapsed or refractory B-cell non-Hodgkin lymphoma. Hematol Oncol. 2024 Jan;42(1):e3227. doi: 10.1002/hon.3227
- Zhang J, Hu Y, Yang J, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. 2022 Sep;609(7926):369–374. doi: 10.1038/s41586-022-05140-y
- Hu Y, Zu C, Zhang M, et al. Safety and efficacy of CRISPR-based non-viral PD1 locus specifically integrated anti-CD19 CAR-T cells in patients with relapsed or refractory non-Hodgkin’s lymphoma: a first-in-human phase I study. EClinicalMedicine. 2023 Jun;60:102010. doi: 10.1016/j.eclinm.2023.102010
- Liu H, Lei W, Zhang C, et al. CD19-specific CAR T cells that express a PD-1/CD28 chimeric switch-receptor are effective in patients with PD-L1-positive B-Cell lymphoma. Clin Cancer Res. 2021 Jan 15;27(2):473–484. doi: 10.1158/1078-0432.CCR-20-1457
- Ma Q, He X, Zhang B, et al. A PD-L1-targeting chimeric switch receptor enhances efficacy of CAR-T cell for pleural and peritoneal metastasis. Signal Transduct Target Ther. 2022 Nov 19;7(1):380. doi: 10.1038/s41392-022-01198-2
- Jacobson CA, Locke FL, Miklos DB, et al. End of phase 1 results from zuma-6: Axicabtagene Ciloleucel (axi-cel) in combination with atezolizumab for the treatment of patients with refractory diffuse large B cell lymphoma. Blood. 2018;132(Supplement 1):4192. doi: 10.1182/blood-2018-99-111523
- Liu H, Ma Y, Yang C, et al. Severe delayed pulmonary toxicity following PD-L1-specific CAR-T cell therapy for non-small cell lung cancer. Clin Transl Immunology. 2020;9(10):e1154. doi: 10.1002/cti2.1154
- Narayan V, Barber-Rotenberg JS, Jung IY, et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. 2022 Apr;28(4):724–734. doi: 10.1038/s41591-022-01726-1
- Chong EA, Melenhorst JJ, Lacey SF, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017 Feb 23;129(8):1039–1041. doi: 10.1182/blood-2016-09-738245
- Chong EA, Svoboda J, Dwivedy Nasta S, et al. Sequential anti-CD19 directed chimeric antigen receptor modified T-Cell therapy (CART19) and PD-1 blockade with pembrolizumab in patients with relapsed or refractory B-Cell non-Hodgkin Lymphomas. Blood. 2018;132(Supplement 1):4198. doi: 10.1182/blood-2018-99-119502
- Hill BT, Roberts ZJ, Xue A, et al. Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone Marrow Transplant. 2020 Jun;55(6):1184–1187. doi: 10.1038/s41409-019-0657-3
- Wang D, Zeng C, Xu B, et al. Anti-CD30 chimeric antigen receptor T cell therapy for relapsed/refractory CD30(+) lymphoma patients. Blood Cancer J. 2020 Jan 23;10(1):8. doi: 10.1038/s41408-020-0274-9
- Sang W, Wang X, Geng H, et al. Anti-PD-1 therapy enhances the efficacy of CD30-directed chimeric antigen receptor T cell therapy in patients with Relapsed/Refractory CD30+ lymphoma. Front Immunol. 2022;13:858021. doi: 10.3389/fimmu.2022.858021
- Andreu-Saumell I, Rodriguez-Garcia A, Muhlgrabner V, et al. CAR affinity modulates the sensitivity of CAR-T cells to PD-1/PD-L1-mediated inhibition. Nat Commun. 2024 Apr 26;15(1):3552. doi: 10.1038/s41467-024-47799-z
- Yamaguchi Y, Gibson J, Ou K, et al. PD-L1 blockade restores CAR T cell activity through IFN-γ-regulation of CD163+ M2 macrophages. J Immunother Cancer. 2022 Jun;10(6):e004400. doi: 10.1136/jitc-2021-004400
- Rosewell Shaw A, Porter CE, Watanabe N, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. 2017 Nov 1;25(11):2440–2451. doi: 10.1016/j.ymthe.2017.09.010
- Li X, Zhu T, Wang R, et al. Genetically programmable vesicles for enhancing CAR-T therapy against solid tumors. Adv Mater. 2023 May;35(19):e2211138. doi: 10.1002/adma.202211138
- Li S, Siriwon N, Zhang X, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017 Nov 15;23(22):6982–6992. doi: 10.1158/1078-0432.CCR-17-0867
- Rafiq S, Yeku OO, Jackson HJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018 Oct;36(9):847–856. doi: 10.1038/nbt.4195
- Yuti P, Wutti-In Y, Sawasdee N, et al. Anti-CD19 chimeric antigen receptor T cells secreting anti-PD-L1 single-chain variable fragment attenuate PD-L1 mediated T cell inhibition. Int Immunopharmacol. 2022 Dec;113(Pt B):109442. doi: 10.1016/j.intimp.2022.109442
- Nakajima M, Sakoda Y, Adachi K, et al. Improved survival of chimeric antigen receptor-engineered T (CAR-T) and tumor-specific T cells caused by anti-programmed cell death protein 1 single-chain variable fragment-producing CAR-T cells. Cancer Sci. 2019 Oct;110(10):3079–3088. doi: 10.1111/cas.14169
- Ping Y, Li F, Nan S, et al. Augmenting the effectiveness of CAR-T cells by enhanced self-delivery of PD-1-Neutralizing scFv. Front Cell Dev Biol. 2020;8:803. doi: 10.3389/fcell.2020.00803
- Zhou JT, Liu JH, Song TT, et al. EGLIF-CAR-T cells secreting PD-1 blocking antibodies significantly mediate the elimination of gastric cancer. Cancer Manag Res. 2020;12:8893–8902. doi: 10.2147/CMAR.S260915
- Chen J, Zhu T, Jiang G, et al. Target delivery of a PD-1-TREM2 scFv by CAR-T cells enhances anti-tumor efficacy in colorectal cancer. Mol Cancer. 2023 Aug 10;22(1):131. doi: 10.1186/s12943-023-01830-x
- Suarez ER, Chang de K, Sun J, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016 Jun 7;7(23):34341–34355. doi: 10.18632/oncotarget.9114
- de Campos NSP, de Oliveira Beserra A, Pereira PHB, et al. Immune checkpoint blockade via PD-L1 potentiates more CD28-based than 4-1BB-Based anti-carbonic anhydrase IX chimeric antigen receptor T cells. Int J Mol Sci. 2022 May 13;23(10):5448. doi: 10.3390/ijms23105448
- Raftery MJ, Franzén AS, Radecke C, et al. Next generation CD44v6-specific CAR-NK cells effective against triple negative breast cancer. Int J Mol Sci. 2023 May 20;24(10):9038. doi: 10.3390/ijms24109038
- Simon B, Harrer DC, Schuler-Thurner B, et al. The siRNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in vitro CAR-T-cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp Dermatol. 2018 Jul;27(7):769–778. doi: 10.1111/exd.13678
- Zou F, Lu L, Liu J, et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat Commun. 2019 Sep 11;10(1):4109. doi: 10.1038/s41467-019-11893-4
- Liu G, Zhang Q, Li D, et al. PD-1 silencing improves anti-tumor activities of human mesothelin-targeted CAR T cells. Hum Immunol. 2021 Feb;82(2):130–138. doi: 10.1016/j.humimm.2020.12.002
- Hamilton AG, Swingle KL, Joseph RA, et al. Ionizable lipid nanoparticles with integrated immune checkpoint inhibition for mRNA CAR T cell engineering. Adv Healthc Mater. 2023 Dec;12(30):e2301515. doi: 10.1002/adhm.202301515
- Ren J, Liu X, Fang C, et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017 May 1;23(9):2255–2266. doi: 10.1158/1078-0432.CCR-16-1300
- Choi BD, Yu X, Castano AP, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. 2019 Nov 14;7(1):304. doi: 10.1186/s40425-019-0806-7
- Hu W, Zi Z, Jin Y, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019 Mar;68(3):365–377. doi: 10.1007/s00262-018-2281-2
- Liu X, Zhang Y, Cheng C, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017 Jan;27(1):154–157. doi: 10.1038/cr.2016.142
- Agarwal S, Aznar MA, Rech AJ, et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity. 2023 Oct 10;56(10):2388–2407.e9. doi: 10.1016/j.immuni.2023.09.001
- Ren J, Zhang X, Liu X, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017 Mar 7;8(10):17002–17011. doi: 10.18632/oncotarget.15218
- Wei J, Luo C, Wang Y, et al. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J Immunother Cancer. 2019 Aug 7;7(1):209. doi: 10.1186/s40425-019-0685-y
- Liu X, Ranganathan R, Jiang S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016 Mar 15;76(6):1578–1590. doi: 10.1158/0008-5472.CAN-15-2524
- Chen C, Gu YM, Zhang F, et al. Construction of PD1/CD28 chimeric-switch receptor enhances anti-tumor ability of c-met CAR-T in gastric cancer. Oncoimmunology. 2021 Mar 31;10(1):1901434. doi: 10.1080/2162402X.2021.1901434
- Lorenzini T, Cadilha BL, Obeck H, et al. Rational design of PD-1-CD28 immunostimulatory fusion proteins for CAR T cell therapy. Br J Cancer. 2023 Sep;129(4):696–705. doi: 10.1038/s41416-023-02332-9
- Qin L, Cui Y, Yuan T, et al. Co-expression of a PD-L1-specific chimeric switch receptor augments the efficacy and persistence of CAR T cells via the CD70-CD27 axis. Nat Commun. 2022 Oct 13;13(1):6051. doi: 10.1038/s41467-022-33793-w
- Lynch A, Hawk W, Nylen E, et al. Adoptive transfer of murine T cells expressing a chimeric-PD1-Dap10 receptor as an immunotherapy for lymphoma. Immunology. 2017 Nov;152(3):472–483. doi: 10.1111/imm.12784
- Parriott G, Deal K, Crean S, et al. T-cells expressing a chimeric-PD1-Dap10-CD3zeta receptor reduce tumour burden in multiple murine syngeneic models of solid cancer. Immunology. 2020 Jul;160(3):280–294. doi: 10.1111/imm.13187
- Yang CY, Fan MH, Miao CH, et al. Engineering chimeric antigen receptor T cells against immune checkpoint inhibitors PD-1/PD-L1 for treating pancreatic cancer. Mol Ther Oncolytics. 2020 Jun 26;17:571–585. doi: 10.1016/j.omto.2020.05.009
- Li D, English H, Hong J, et al. A novel PD-L1-targeted shark V(NAR) single-domain-based CAR-T cell strategy for treating breast cancer and liver cancer. Mol Ther Oncolytics. 2022 Mar 17;24:849–863. doi: 10.1016/j.omto.2022.02.015
- Liu WN, So WY, Harden SL, et al. Successful targeting of PD-1/PD-L1 with chimeric antigen receptor-natural killer cells and nivolumab in a humanized mouse cancer model. Sci Adv. 2022 Nov 25;8(47):eadd1187. doi: 10.1126/sciadv.add1187
- Li D, Qin J, Zhou T, et al. Bispecific GPC3/PD‑1 CAR‑T cells for the treatment of HCC. Int J Oncol. 2023 Apr;62(4). doi: 10.3892/ijo.2023.5501
- Prinz LF, Riet T, Neureuther DF, et al. An anti-CD19/CTLA-4 switch improves efficacy and selectivity of CAR T cells targeting CD80/86-upregulated DLBCL. Cell Rep Med. 2024 Feb 20;5(2):101421. doi: 10.1016/j.xcrm.2024.101421
- Zhang Y, Zhang X, Cheng C, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. 2017 Dec;11(4):554–562. doi: 10.1007/s11684-017-0543-6
- Condomines M, Arnason J, Benjamin R, et al. Tumor-targeted human T cells expressing CD28-based chimeric antigen receptors circumvent CTLA-4 inhibition. PLoS One. 2015;10(6):e0130518. doi: 10.1371/journal.pone.0130518
- Tang L, Shao H, Wu Y, et al. Dominant negative TGFβ receptor II and truncated TIM3 enhance the antitumor efficacy of CAR-T-cell therapy in prostate cancer. Int Immunopharmacol. 2023 Nov;124(Pt A):110807. doi: 10.1016/j.intimp.2023.110807
- Alabanza LM, Xiong Y, Vu B, et al. Armored BCMA CAR T cells eliminate multiple myeloma and are resistant to the suppressive effects of TGF-β. Front Immunol. 2022;13:832645. doi: 10.3389/fimmu.2022.832645
- Alishah K, Birtel M, Masoumi E, et al. CRISPR/Cas9-mediated TGFβRII disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells in vitro. J Transl Med. 2021 Nov 27;19(1):482. doi: 10.1186/s12967-021-03146-0
- Golumba-Nagy V, Kuehle J, Hombach AA, et al. CD28-ζ CAR T cells resist TGF-β repression through IL-2 signaling, which can Be mimicked by an engineered IL-7 Autocrine loop. Mol Ther. 2018 Sep 5;26(9):2218–2230. doi: 10.1016/j.ymthe.2018.07.005
- Li Y, Wu H, Chen G, et al. Arming anti-EGFRvIII CAR-T with TGFβ trap improves antitumor efficacy in glioma mouse models. Front Oncol. 2020;10:1117. doi: 10.3389/fonc.2020.01117
- Li G, Liao G, Xie J, et al. Overexpression of SMAD7 improves the function of EGFR-targeted human CAR-T cells against non-small-cell lung cancer. Respirology. 2023 Sep;28(9):869–880. doi: 10.1111/resp.14541
- Liang S, Zheng R, Zuo B, et al. SMAD7 expression in CAR-T cells improves persistence and safety for solid tumors. Cell Mol Immunol. 2024 Jan 4;21(3):213–226. doi: 10.1038/s41423-023-01120-y
- Noh KE, Lee JH, Choi SY, et al. TGF-β/IL-7 chimeric switch receptor-expressing CAR-T cells inhibit recurrence of CD19-positive B cell lymphoma. Int J Mol Sci. 2021 Aug 13;22(16):8706. doi: 10.3390/ijms22168706
- Stüber T, Monjezi R, Wallstabe L, et al. Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J Immunother Cancer. 2020 Apr;8(1):e000676. doi: 10.1136/jitc-2020-000676
- Tang N, Cheng C, Zhang X, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020 Feb 27;5(4). doi: 10.1172/jci.insight.133977
- Zhang PF, Huang Y, Liang X, et al. Enhancement of the antitumor effect of HER2-directed CAR-T cells through blocking epithelial-mesenchymal transition in tumor cells. FASEB J. 2020 Aug;34(8):11185–11199. doi: 10.1096/fj.202000080RR
- Hao Y, Chen P, Guo S, et al. Tumor-derived exosomes induce initial activation by exosomal CD19 antigen but impair the function of CD19-specific CAR T-cells via TGF-β signaling. Front Med. 2023 Oct 23;18(1):128–146. doi: 10.1007/s11684-023-1010-1
- Wang Z, Liu Q, Risu N, et al. Galunisertib enhances chimeric antigen receptor-modified T cell function. Eur J Histochem. 2020 Jun 19;64(s2). doi: 10.4081/ejh.2020.3122
- Zhang L, Yu Z, Muranski P, et al. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013 May;20(5):575–580. doi: 10.1038/gt.2012.75
- Chen X, Yang S, Li S, et al. Secretion of bispecific protein of anti-PD-1 fused with TGF-β trap enhances antitumor efficacy of CAR-T cell therapy. Mol Ther Oncolytics. 2021 Jun 25;21:144–157. doi: 10.1016/j.omto.2021.03.014
- Li Y, Xiao F, Zhang A, et al. Oncolytic adenovirus targeting TGF-β enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell Immunol. 2020 Feb;348:104041. doi: 10.1016/j.cellimm.2020.104041
- Chang ZL, Lorenzini MH, Chen X, et al. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018 Mar;14(3):317–324. doi: 10.1038/nchembio.2565
- Hou AJ, Chang ZL, Lorenzini MH, et al. TGF-β-responsive CAR-T cells promote anti-tumor immune function. Bioeng Transl Med. 2018 May;3(2):75–86. doi: 10.1002/btm2.10097
- Werchau N, Kotter B, Criado-Moronati E, et al. Combined targeting of soluble latent TGF-ß and a solid tumor-associated antigen with adapter CAR T cells. Oncoimmunology. 2022;11(1):2140534. doi: 10.1080/2162402X.2022.2140534
- Wieser R, Attisano L, Wrana JL, et al. Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol Cell Biol. 1993 Dec;13(12):7239–7247. doi: 10.1128/MCB.13.12.7239
- Ebner R, Chen RH, Shum L, et al. Cloning of a type I TGF-beta receptor and its effect on TGF-beta binding to the type II receptor. Science. 1993 May 28;260(5112):1344–1348. doi: 10.1126/science.8388127
- Bollard CM, Rossig C, Calonge MJ, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002 May 1;99(9):3179–3187. doi: 10.1182/blood.V99.9.3179
- Foster AE, Dotti G, Lu A, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother. 2008 Jun;31(5):500–505. doi: 10.1097/CJI.0b013e318177092b
- Kloss CC, Lee J, Zhang A, et al. Dominant-negative TGF-β receptor enhances PSMA-Targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018 Jul 5;26(7):1855–1866. doi: 10.1016/j.ymthe.2018.05.003
- Webster B, Xiong Y, Hu P, et al. Self-driving armored CAR-T cells overcome a suppressive milieu and eradicate CD19(+) Raji lymphoma in preclinical models. Mol Ther. 2021 Sep 1;29(9):2691–2706. doi: 10.1016/j.ymthe.2021.05.006
- Chaudhry K, Geiger A, Dowlati E, et al. Co-transducing B7H3 CAR-NK cells with the DNR preserves their cytolytic function against GBM in the presence of exogenous TGF-β. Mol ther methods Clin Dev. Mol Ther – Methods & Clin Devel. 2022 Dec 8;27:415–430. doi: 10.1016/j.omtm.2022.10.010
- Zanvit P, van Dyk D, Fazenbaker C, et al. Antitumor activity of AZD0754, a dnTGFβRII-armored, STEAP2-targeted CAR-T cell therapy, in prostate cancer. J Clin Invest. 2023 Nov 15;133(22). doi: 10.1172/JCI169655
- Editorial. CAR T-cell therapy for solid tumours. Lancet Oncol. 2021;22(7):893. doi: 10.1016/S1470-2045(21)00353-3
- Koehler H, Kofler D, Hombach A, et al. CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. 2007 Mar 1;67(5):2265–2273. doi: 10.1158/0008-5472.CAN-06-2098
- Weimin S, Abula A, Qianghong D, et al. Chimeric cytokine receptor enhancing PSMA-CAR-T cell-mediated prostate cancer regression. Cancer Biol Ther. 2020 Jun 2;21(6):570–580. doi: 10.1080/15384047.2020.1739952
- Sukumaran S, Watanabe N, Bajgain P, et al. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. 2018 Aug;8(8):972–987. doi: 10.1158/2159-8290.CD-17-1298
- Beatson RE, Parente-Pereira AC, Halim L, et al. TGF-beta1 potentiates Vgamma9Vdelta2 T cell adoptive immunotherapy of cancer. Cell Rep Med. 2021 Dec 21;2(12):100473. doi: 10.1016/j.xcrm.2021.100473
- Synnestvedt K, Furuta GT, Comerford KM, et al. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002 Oct;110(7):993–1002. doi: 10.1172/JCI0215337
- Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007 Aug 15;110(4):1225–1232. doi: 10.1182/blood-2006-12-064527
- Dwyer KM, Deaglio S, Gao W, et al. CD39 and control of cellular immune responses. Purinergic Signal. 2007 Mar;3(1–2):171–180. doi: 10.1007/s11302-006-9050-y
- Kobie JJ, Shah PR, Yang L, et al. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J Immunol. 2006 Nov 15;177(10):6780–6786. doi: 10.4049/jimmunol.177.10.6780
- Fallah-Mehrjardi K, Mirzaei HR, Masoumi E, et al. Pharmacological targeting of immune checkpoint A2aR improves function of anti-CD19 CAR T cells in vitro. Immunol Lett. 2020 Jul;223:44–52. doi: 10.1016/j.imlet.2020.04.005
- Giuffrida L, Sek K, Henderson MA, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. 2021 May 28;12(1):3236. doi: 10.1038/s41467-021-23331-5
- Li N, Tang N, Cheng C, et al. Improving the anti-solid tumor efficacy of CAR-T cells by inhibiting adenosine signaling pathway. Oncoimmunology. 2020 Sep 24;9(1):1824643. doi: 10.1080/2162402X.2020.1824643
- Liu G, Zhang Q, Liu G, et al. Disruption of adenosine 2A receptor improves the anti-tumor function of anti-mesothelin CAR T cells both in vitro and in vivo. Exp Cell Res. 2021 Dec 1;409(1):112886. doi: 10.1016/j.yexcr.2021.112886
- Masoumi E, Jafarzadeh L, Mirzaei HR, et al. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J Exp Clin Cancer Res. 2020 Mar 10;39(1):49. doi: 10.1186/s13046-020-01546-6
- Seifert M, Benmebarek MR, Briukhovetska D, et al. Impact of the selective A2(A)R and A2(B)R dual antagonist AB928/etrumadenant on CAR T cell function. Br J Cancer. 2022 Dec;127(12):2175–2185. doi: 10.1038/s41416-022-02013-z
- Siriwon N, Kim YJ, Siegler E, et al. CAR-T cells surface-engineered with drug-encapsulated nanoparticles can ameliorate intratumoral T-cell hypofunction. Cancer Immunol Res. 2018 Jul;6(7):812–824. doi: 10.1158/2326-6066.CIR-17-0502
- Renauer P, Park JJ, Bai M, et al. Immunogenetic metabolomics reveals key enzymes that modulate CAR T-cell metabolism and function. Cancer Immunol Res. 2023 Aug 3;11(8):1068–1084. doi: 10.1158/2326-6066.CIR-22-0565
- Klysz DD, Fowler C, Malipatlolla M, et al. Inosine induces stemness features in CAR-T cells and enhances potency. Cancer Cell. 2024 Feb 12;42(2):266–282.e8. doi: 10.1016/j.ccell.2024.01.002
- Qu Y, Dunn ZS, Chen X, et al. Adenosine deaminase 1 overexpression enhances the antitumor efficacy of chimeric antigen receptor-engineered T cells. Hum Gene Ther. 2022 Mar;33(5–6):223–236. doi: 10.1089/hum.2021.050
- Cox JR, Jennings M, Lenahan C, et al. Rational engineering of an improved adenosine deaminase 2 enzyme for weaponizing T-cell therapies. Immunooncol Technol. 2023 Sep;19:100394. doi: 10.1016/j.iotech.2023.100394
- Akbari B, Soltantoyeh T, Shahosseini Z, et al. PGE2-EP2/EP4 signaling elicits mesoCAR T cell immunosuppression in pancreatic cancer. Front Immunol. 2023;14:1209572. doi: 10.3389/fimmu.2023.1209572
- Sullivan KM, Jiang X, Guha P, et al. Blockade of interleukin 10 potentiates antitumour immune function in human colorectal cancer liver metastases. Gut. 2023 Feb;72(2):325–337. doi: 10.1136/gutjnl-2021-325808
- Hombach AA, Heiders J, Foppe M, et al. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology. 2012 Jul 1;1(4):458–466. doi: 10.4161/onci.19855
- Gorby C, Sotolongo Bellón J, Wilmes S, et al. Engineered IL-10 variants elicit potent immunomodulatory effects at low ligand doses. Sci Signal. 2020 Sep 15;13(649). doi: 10.1126/scisignal.abc0653
- Zhao Y, Chen J, Andreatta M, et al. IL-10-expressing CAR T cells resist dysfunction and mediate durable clearance of solid tumors and metastases. Nat Biotechnol. 2024 Jan 2. doi: 10.1038/s41587-023-02060-8
- Huang Q, Xia J, Wang L, et al. miR-153 suppresses IDO1 expression and enhances CAR T cell immunotherapy. J Hematol Oncol. 2018 Apr 23;11(1):58. doi: 10.1186/s13045-018-0600-x
- Ninomiya S, Narala N, Huye L, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015 Jun 18;125(25):3905–3916. doi: 10.1182/blood-2015-01-621474
- Towers R, Trombello L, Fusenig M, et al. Bone marrow-derived mesenchymal stromal cells obstruct AML-targeting CD8(+) clonal effector and CAR T-cell function while promoting a senescence-associated phenotype. Cancer Immunol Immunother. 2024 Jan 17;73(1):8. doi: 10.1007/s00262-023-03594-1
- Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc Trans. 2016 Apr 15;44(2):333–341. doi: 10.1042/BST20150245
- Lo AS, Taylor JR, Farzaneh F, et al. Harnessing the tumour-derived cytokine, CSF-1, to co-stimulate T-cell growth and activation. Mol Immunol. 2008 Mar;45(5):1276–1287. doi: 10.1016/j.molimm.2007.09.010
- LaMarche NM, Hegde S, Park MD, et al. An IL-4 signalling axis in bone marrow drives pro-tumorigenic myelopoiesis. Nature. 2024 Jan;625(7993):166–174. doi: 10.1038/s41586-023-06797-9
- Wilkie S, Burbridge SE, Chiapero-Stanke L, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010 Aug 13;285(33):25538–25544. doi: 10.1074/jbc.M110.127951
- Zebley CC, Brown C, Mi T, et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. 2021 Nov 30;37(9):110079. doi: 10.1016/j.celrep.2021.110079
- Collins SM, Alexander KA, Lundh S, et al. TOX2 coordinates with TET2 to positively regulate central memory differentiation in human CAR T cells. Sci Adv. 2023 Jul 21;9(29):eadh2605. doi: 10.1126/sciadv.adh2605
- Guo T, Ma D, Lu TK. Sense-and-respond payload delivery using a novel antigen-inducible promoter improves suboptimal CAR-T activation. ACS Synth Biol. 2022 Apr 15;11(4):1440–1453. doi: 10.1021/acssynbio.1c00236
- Berahovich R, Liu X, Zhou H, et al. Hypoxia selectively impairs CAR-T cells in vitro. Cancers (Basel). 2019 Apr 30;11(5):602. doi: 10.3390/cancers11050602
- Balta E, Janzen N, Kirchgessner H, et al. Expression of TRX1 optimizes the antitumor functions of human CAR T cells and confers resistance to a pro-oxidative tumor microenvironment. Front Immunol. 2022;13:1063313. doi: 10.3389/fimmu.2022.1063313
- Mane MM, Cohen IJ, Ackerstaff E, et al. Lactate dehydrogenase a depletion alters MyC-CaP tumor metabolism, microenvironment, and CAR T cell therapy. Mol Ther Oncolytics. 2020 Sep 25;18:382–395. doi: 10.1016/j.omto.2020.07.006
- Renken S, Nakajima T, Magalhaes I, et al. Targeting of Nrf2 improves antitumoral responses by human NK cells, TIL and CAR T cells during oxidative stress. J Immunother Cancer. 2022 Jun;10(6):e004458. doi: 10.1136/jitc-2021-004458
- Panahi Meymandi AR, Akbari B, Soltantoyeh T, et al. PX-478, an HIF-1α inhibitor, impairs mesoCAR T cell antitumor function in cervical cancer. Front Oncol. 2024;14:1357801. doi: 10.3389/fonc.2024.1357801
- Cunha PP, Minogue E, Krause LCM, et al. Oxygen levels at the time of activation determine T cell persistence and immunotherapeutic efficacy. Elife. 2023 May 11;12. doi: 10.7554/eLife.84280
- Liikanen I, Lauhan C, Quon S, et al. Hypoxia-inducible factor activity promotes antitumor effector function and tissue residency by CD8+ T cells. J Clin Invest. 2021 Apr 1;131(7). doi: 10.1172/JCI143729
- Juillerat A, Marechal A, Filhol JM, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017 Jan 20;7(1):39833. doi: 10.1038/srep39833
- Liao Q, He H, Mao Y, et al. Engineering T cells with hypoxia-inducible chimeric antigen receptor (HiCAR) for selective tumor killing. Biomark Res. 2020 Oct 30;8(1):56. doi: 10.1186/s40364-020-00238-9
- Greenshpan Y, Sharabi O, Ottolenghi A, et al. Synthetic promoters to induce immune-effectors into the tumor microenvironment. Commun Biol. 2021 Jan 29;4(1):143. doi: 10.1038/s42003-021-01664-7
- He H, Liao Q, Zhao C, et al. Conditioned CAR-T cells by hypoxia-inducible transcription amplification (HiTA) system significantly enhances systemic safety and retains antitumor efficacy. J Immunother Cancer. 2021 Oct;9(10):e002755. doi: 10.1136/jitc-2021-002755
- Zhu X, Chen J, Li W, et al. Hypoxia-responsive CAR-T cells exhibit reduced exhaustion and enhanced efficacy in solid tumors. Cancer Res. 2024 Jan 2;84(1):84–100. doi: 10.1158/0008-5472.CAN-23-1038
- Zhou W, Miao J, Cheng Z, et al. Hypoxia-regulated secretion of IL-12 enhances antitumor activity and safety of CD19 CAR-T cells in the treatment of DLBCL. Mol Ther Oncolytics. 2023 Sep 21;30:216–226. doi: 10.1016/j.omto.2023.08.009
- Klopotowska M, Bajor M, Graczyk-Jarzynka A, et al. PRDX-1 supports the survival and antitumor activity of primary and CAR-Modified NK cells under oxidative stress. Cancer Immunol Res. 2022 Feb;10(2):228–244. doi: 10.1158/2326-6066.CIR-20-1023
- Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, et al. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol. 2016 Jan 15;196(2):759–766. doi: 10.4049/jimmunol.1401710
- Duan J, Zhao S, Duan Y, et al. Mno x nanoenzyme armed CAR-NK cells enhance solid tumor immunotherapy by alleviating the immunosuppressive microenvironment. Adv Healthcare Materials. 2024 Jan 31;13(11):e2303963. doi: 10.1002/adhm.202303963
- Navarro F, Casares N, Martín-Otal C, et al. Overcoming T cell dysfunction in acidic pH to enhance adoptive T cell transfer immunotherapy. Oncoimmunology. 2022;11(1):2070337. doi: 10.1080/2162402X.2022.2070337
- Muliaditan T, Halim L, Whilding LM, et al. Synergistic T cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep Med. 2021 Dec 21;2(12):100457. doi: 10.1016/j.xcrm.2021.100457
- Halim L, Das KK, Larcombe-Young D, et al. Engineering of an avidity-optimized CD19-specific parallel chimeric antigen receptor that delivers dual CD28 and 4-1BB Co-stimulation. Front Immunol. 2022;13:836549. doi: 10.3389/fimmu.2022.836549
- Chmielewski M, Kopecky C, Hombach AA, et al. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011 Sep 1;71(17):5697–5706. doi: 10.1158/0008-5472.CAN-11-0103
- Pegram HJ, Lee JC, Hayman EG, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012 May 3;119(18):4133–4141. doi: 10.1182/blood-2011-12-400044
- Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-Bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017 Dec 12;21(11):3205–3219. doi: 10.1016/j.celrep.2017.11.063
- Avanzi MP, Yeku O, Li X, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018 May 15;23(7):2130–2141. doi: 10.1016/j.celrep.2018.04.051
- Hu B, Ren J, Luo Y, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017 Sep 26;20(13):3025–3033. doi: 10.1016/j.celrep.2017.09.002
- Hull CM, Larcombe-Young D, Mazza R, et al. Granzyme B-activated IL18 potentiates αβ and γδ CAR T cell immunotherapy in a tumor-dependent manner. Mol Ther. 2024 May 14;32(7):2373–2392. doi: 10.1016/j.ymthe.2024.05.013
- Lai J, Mardiana S, House IG, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. 2020 Aug;21(8):914–926. doi: 10.1038/s41590-020-0676-7