
ABSTRACT
The extent of ageing in the musculoskeletal system during the life course affects the quality and length of life. Loss of bone, degraded articular cartilage, and degenerate, narrowed intervertebral discs are primary features of an ageing skeleton, and together they contribute to pain and loss of mobility. This review covers the cellular constituents that make up some key components of the musculoskeletal system and summarizes discussion from the 2015 Aarhus Regenerative Orthopaedic Symposium (AROS) (Regeneration in the Ageing Population) about how each particular cell type alters within the ageing skeletal microenvironment.
In the ageing skeleton, bone volume and mass declines in both sexes and in people of all ethnic backgrounds, often manifesting as osteoporosis and an increased risk of fracture. Loss of cartilage thickness in synovial joints with age contributes to osteoarthritis while decreasing structural integrity within the intervertebral discs (IVDs) leads to loss of disc height, and collapse and compression within the spine.
In these individual tissue compartments, there is a delicate cellular balance where the differentiation and activity of specific cell types are matched to the local requirements. Whereas some skeletal elements rely on individual cell types acting remotely and in isolation (e.g. osteocytes, chondrocytes), others depend on a diverse population of cells, derived from widely different stem cell lineages, to work together to reach the common goal of maintaining skeletal integrity throughout life.
Bone
Bone strength and volume is preserved as we age by the constant, balanced remodeling of the skeleton—a balance that is not maintained in the ageing bone microenvironment.
Stem cells and ageing bone
Bone-forming osteoblasts derive from mesenchymal stem cells (MSCs). MSCs can be stimulated to form a range of cell types including chondrocytes, myocytes, adipocytes, and fibroblasts depending on the local stimuli (Ferrari et al. Citation1998, Awad et al. Citation1999, Pittenger et al. Citation1999). Consequently, alterations in the number and activity of MSCs have the potential to affect a range of musculoskeletal tissues by altering progenitor cell numbers, not least osteoblasts. MSCs isolated from aged donors showed a reduced capacity to expand in vitro and appeared flat and widespread compared to rapidly proliferating, spindle-shaped cells from younger donors (Colter et al. Citation2001, Baxter et al. Citation2004). Increased granulation within the cytoplasm was also observed (Bonab et al. Citation2006), suggesting an accumulation of waste protein material that typically results from impaired autophagy, another age-related mechanism.
Aged MSCs also show increased levels of cellular senescence, a state of irreversible growth arrest, where increased expression of p53, p21, and senescence-associated β-galactosidase is seen in MSC cultures from old donors compared to young donors (Stenderup et al. Citation2003, Stolzing et al. Citation2008). A shift in differentiation potential is also clear, as aged MSCs favor adipocyte formation at the expense of osteoblast formation, leading to increased fat accumulation within the bone marrow (a feature common in ageing bone) and reduced bone formation (Moore and Dawson Citation1990, Sethe et al. Citation2006). This is probably driven by an age-related downregulation of osteogenic genes including Runx2 and osteocalcin (D’Ippolito et al. Citation1999, Lazarenko et al. Citation2007) coupled with an upregulation of adipocyte-specific genes such as PPAR-γ and the adipocyte fatty acid-binding protein (aP2) (Moerman et al. Citation2004, Jiang et al. Citation2008).
Sirtuins
The influence of ageing and longevity-related mechanisms on such crucial regulators of transcription within skeletal cells may also contribute strongly to the age-related change we see in normal cell biology. The sirtuin gene family is closely linked to the regulation of lifespan (Kaeberlein et al. Citation1999). Overexpression of the principal mammalian homolog SirT1 in early mesenchymal progenitors increased Runx2 and Osterix expression, while inhibition of SirT1 decreased Runx2 expression (Cohen-Kfir et al. Citation2011) and increased the activity of pro-adipocyte factor PPAR-γ (Picard et al. Citation2004). Importantly, SirT1 declines with ageing in a variety of tissues (including musculoskeletal sites) and may be a common mediator of both ageing and the onset of age-related disorders. Genetically modified mice lacking SirT1 had a bone loss phenotype similar to that seen in human age-related bone loss (Cohen-Kfir et al. Citation2011), while tissue-specific deletion of SirT1 in either osteoblast- or osteoclast-lineage cells decreased bone volume either through impaired bone formation or excessive bone resorption (Edwards et al. Citation2013, Iyer et al. Citation2014). The deacetylase activity of SirT1 offers many mechanisms through which SirT1 may mediate bone biology, including inhibition of NFkB (Yeung et al. Citation2004, Edwards et al. Citation2013) and PPAR-γ (Picard et al. Citation2004) transcription factors. Loss of SirT1 with increasing age may therefore enhance inflammatory responses, osteoclast formation, and adipocyte formation, common physiological features of ageing and crucial systems in the maintenance of musculoskeletal health. SirT1 also suppresses sclerostin (Cohen-Kfir et al. Citation2011), a potent inhibitor of the anabolic Wnt signaling pathway in bone. As SirT1 levels decline with age, sclerostin is released from inhibition to suppress bone formation. In support of this mechanism, circulating sclerostin levels are increased in elderly humans compared to young individuals (Modder et al. Citation2011), effectively acting as a brake on Wnt signaling in the ageing skeleton; it is also thought to be released by osteoclasts in aged mice—only to block coupled bone formation (Ota et al. Citation2013).
Wnt signaling activates β-catenin to control osteoblast differentiation and function. Signaling through LRP5/6 by Wnts prevents degradation of β-catenin, allowing its concentration to rise in the cytoplasm. After translocation to the nucleus, its association with the TCF/LEF transcription co-factors stimulates osteoblastogenesis and reduces osteoclastogenesis by activating genes for factors such as alkaline phosphatase and osteoprotegerin, to increase bone mass and reduce resorption and fat cell accumulation (Kolpakova and Olsen Citation2005, Rosen and MacDougald Citation2006).
Forkhead proteins
In a similar manner to sclerostin, the Forkhead transcription factor FoxO can also reduce Wnt signaling by sequestering of β-catenin. FoxO is implicated in ageing, where C. elegans and Drosophila studies have suggested a beneficial effect on longevity. Activation of FoxO proteins as a cellular response to oxidative stress appears to underscore many of their protective effects, and elevated levels of reactive oxygen species (ROS) such as superoxide anions are common in the ageing cellular environment. Indeed, posttranslational modifications induced by the presence of ROS are potent stimuli for FoxO1, -3, and -4 activity in mammals (Calnan and Brunet Citation2008). In opposition to the canonical Wnt pathway described above, activation of FoxO proteins under conditions of oxidative stress creates a competitive intracellular environment for the binding of available β-catenin, and where reduced numbers of osteoblasts are formed at the expense of cell survival mechanisms (Manolagas and Almeida Citation2007, Iyer et al. Citation2013). This may be suppressed in part by the action of SirT1 to deacetylate and inactivate FoxO (Iyer et al. Citation2014). However, the significance of FoxO proteins in skeletal homeostasis is highlighted by osteoblast-specific deletion of FoxO1, -3, and -4 where increased oxidative stress and osteoblast apoptosis are coupled to reduced osteoblast number, bone formation rate, and cortical and cancellous bone volume (Ambrogini et al. Citation2010), demonstrating clearly how changes in environmental stimuli—such as those experienced during ageing—alter normal biological processes.
mTOR signaling
Inhibition of the serine/threonine protein kinase mTOR is another ageing-associated mechanism that extends lifespan in model organisms and mammals (Vellai et al. Citation2003, Kapahi et al. Citation2004, Kaeberlein et al. Citation2005). mTOR expression influences cell growth, motility, survival, protein synthesis, and recycling. As such, changes in the regulation of this crucial protein have the potential to dramatically affect bone cells. In vitro inhibition of mTOR with rapamycin activates osteogenic genes in MSCs (e.g. Runx2, Osterix) while suppressing genes linked to stem-ness (e.g. Oct3/4, nanog). In mature osteoblasts, rapamycin promoted increased alkaline phosphatase production and mineralization (Lee et al. Citation2010), while disrupting the OPG/RANKL ratio to reduce osteoclastogenesis in bone marrow cells (Mogi and Kondo Citation2009). This finding is recapitulated in vivo, where use of the mTOR inhibitor everolimus protects against ovariectomy-induced bone loss by reducing osteoclast formation and release of the matrix-degrading enzyme cathepsin K (Kneissel et al. Citation2004).
Ageing-related hormonal effects
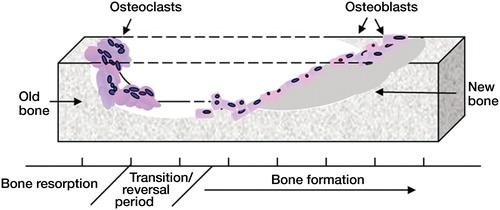
In the ageing skeleton, the effects of reduced osteoblast activity are compounded by increased osteoclast formation and function (). The resulting excessive bone destruction reduces skeletal volume and strength and predisposes to increased risk of fracture. Alongside changes in lifespan-regulating mechanisms, age-related changes in sex hormones also affect normal skeletal biology. In women, the decline in estrogen levels following menopause leads to a dramatic loss of bone in both cortical and trabecular compartments (Riggs et al. Citation1998, Manolagas Citation2000). This results from an elevated bone turnover rate where osteoclasts are released from estrogen inhibition to increase overall bone resorption. The coupled relationship between osteoclastic resorption and bone formation triggers an increase in osteoblasts (Han et al.Citation1997); the increase in bone formation rate is, however, insufficient to match the excessive destruction by overstimulated osteoclasts. In men, low levels of androgens also cause increased bone remodeling and net bone loss (Daniell Citation1997, Katznelson et al. Citation1996), attributed in part to reduced levels of estrogen, which is derived from aromatization of testosterone (Falahati-Nini et al. Citation2000).
Figure 1. Bone remodeling. Bone is continually remodeled throughout life. The coordinated activity of bone-resorbing osteoclasts and bone-forming osteoblasts ensures that adequate skeletal mass is maintained. As we age, the delicate balance between bone removal and bone production is disrupted, resulting in an overall loss of bone.
Cellular responses to the hormone insulin-like growth factor 1 (IGF1) are also altered with age, where proliferative osteoblast numbers are reduced with increasing age in both basal and IGF1-stimulated settings. In addition, increased age reduces the ability of IGF1 to form osteogenic progenitor cells (Cao et al. Citation2007). Pro-proliferative and anti-apoptotic effectors normally stimulated by IGF1 are also reduced in aged cells as compared to younger cells. Overall, this suggests that ageing leads to a reduction in cellular sensitivity to IGF1 signaling, thus contributing to bone loss.
The coupled, interdependent, local co-regulation of bone cells is also dysregulated in old skeletal microenvironments as compared to young ones. Stromal or osteoblastic cells co-cultured with osteoclast progenitors typically encourage osteoclastogenesis. However, osteoclastogenesis is dramatically increased when old stromal or osteoblastic cells are cultured (compared to cells from young donors), due to the increased expression with age of pro-osteoclastogenic factors such as RANKL and M-CSF (Cao et al. Citation2005). Interestingly, aged bone itself also stimulates higher levels of osteoclast formation and resorption (Henriksen et al. Citation2007), suggesting that the age-related change in matrix proteins within bone also affects the ageing cellular environment.
Furthermore, the ability of cells from many lineages to sense and respond to mechanical changes is altered with age, adversely affecting their capacity to resist tissue damage (Wu et al. Citation2011). Embedded within bone, the solitary isolated osteocyte is the principal mechanosensor, relaying signals to the surface of bone to trigger changes in bone and possibly also muscle (Bonewald Citation2015). Increased osteocyte apoptosis occurs with age, reducing the skeleton’s capacity to sense and respond to mechanical forces. Osteocytes respond to changes in loading in various ways, such as through alterations in calcium flux (Huang and Ogawa Citation2010) or interferon-γ concentrations, where older cells fail to transduce mechanical forces—such as fluid flow—into changes in cell function (Donahue et al. Citation2001).
Articular cartilage
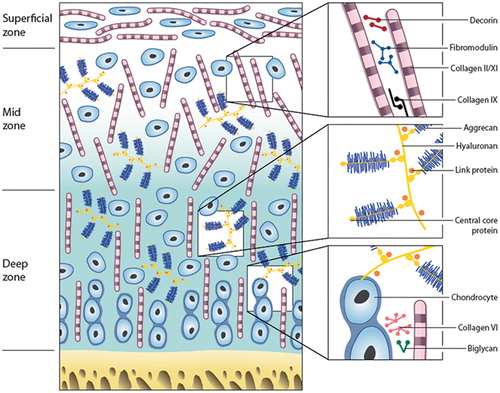
Articular cartilage shows distinct regional variations in the biochemistry and organization of the matrix, and of the cells within it. Similarly, the apparent simplicity of the avascular, aneural articular cartilage being populated with a single cell type—chondrocytes—is deceiving, as chondrocytes from different regions or zones of the cartilage have different capabilities (Aydelotte and Kuettner Citation1988), but how much of that is inherent and how much is dictated by the environment of the cells is unknown (Hayes et al. Citation2007). Those cells from the upper superficial zone were smaller and had higher activities of lysosomal enzymes, but synthesized less proteoglycans (PGs) and less keratan sulfate (KS) glycosaminoglycans (GAGs) than the larger cells in the deep zone. These in turn had more rough endoplasmic reticulum, had more glycogen and lipid deposits, proliferated faster in culture, and synthesized more GAGs (Aydelotte and Kuettner Citation1988). In addition to these zonal subpopulations, a further population of progenitor cells has been identified in cartilage, as in most other tissues (Dowthwaite et al. Citation2004, Candela et al. Citation2014). These cells may normally be quiescent but have the ability to respond to particular mechanical or biochemical stimuli ().
Figure 2. Zones and constituents of articular cartilage. A schematic representation of the different matrix components and their organization throughout articular cartilage.
During ageing, articular cartilage undergoes changes to its molecular structure with some, but not all, changes resembling those seen in degeneration associated with osteoarthritis (OA). The most notable quantitative changes are a decrease in water content and fragmentation of the core protein of the main PG, aggrecan, and also of the chondroitin sulfate (CS) GAGs, but there is an increase in KS content (Hardingham and Bayliss Citation1990). Hyaluronan, in contrast, increases 4-fold from birth to 90 years of age. In addition, there is increased proteolytic fragmentation of the collagen molecules by collagenase and cathepsin K with increasing age, resulting in reduced tensile strength (Dejica et al. Citation2012).
There are also many more subtle changes in the biochemistry of cartilage with increasing age, including altered sulfation patterns on the GAG chains, a switch in types of collagen produced (more type-I:II and increased type-IX:II), as well as an alteration in crosslinking between the collagen molecules. “Mature” hydroxypyridinium crosslinks, which are 5- to 10-fold higher in cartilage than in bone, increase from birth to ∼15 years of age in cartilage and then stay consistent to at least 80 years of age. In contrast, the reducible aldimine crosslinks, which are present during the development of cartilage, are virtually absent by the age of 15 years. In addition to these, there are of course a myriad of other age-related changes to other extracellular matrix components (ECM), including the small leucine-rich proteoglycans (SLRPs), cartilage oligomeric matrix protein (COMP), and the appropriately named advanced glycation end-products (AGEs) (described in reviews such as that by Heinegard and Saxne (Citation1991)).
Ageing chondrocytes
Of the various signatures of cellular ageing described (Lopez-Otin et al. Citation2013), cellular senescence has been shown to increase in human chondrocytes from the age of 1 to 87 years, with a concomitant decrease in telomere erosion and reduced mitochondria (Martin and Buckwalter Citation2001). This chondrocyte senescence has been linked to an age-related decrease in the chondrocyte’s responsiveness to anabolic growth factors such as IGF (Loeser et al. Citation2000) and to synthesis of smaller matrix molecules (e.g. aggrecan and link protein), with oxidative damage causing mitochondrial degeneration (Martin and Buckwalter Citation2002). Little is known about oxygen tensions within the avascular environment of articular cartilage. Reported measurements of partial pressure of oxygen in vitro are 1% to 2.5% in the mid-zone of cartilage plugs (Silver Citation1975). Chondrocytes, although suited to a low oxygen environment, do consume oxygen, albeit at low rates when compared to other cell types (Zhou et al. Citation2004). However, how chondrocytes use oxygen is unclear. They contain a very low number of mitochondria (Stockwell Citation1991) compared to most other cell types, and derive most of their energy from an oxygen-independent process via the Embden-Meyerhof-Parnas pathway of glycolysis (Lee and Urban Citation1997, Zhou et al. Citation2004).
Certainly, oxygen is required for proteoglycan synthesis, which is thought to decrease significantly with decreasing oxygen tension below 5% (Lee and Urban Citation1997, Grimshaw and Mason Citation2000). Although chondrocytes have been shown to survive for several days without oxygen (Grimshaw and Mason Citation2000, Zhou et al. Citation2004), it has been suggested that oxygen tensions as low as ∼1% are unlikely to exist in vivo, except under abnormal conditions. These abnormal conditions may relate to different disease pathologies, with the oxygen tension in synovial fluid varying depending on the pathology of the joint. For example, a pO2 of 0–6% has been reported in patients with rheumatoid arthritis, 3–9% in osteoarthritis, 6–11% in patients with traumatic exudates (Lund-Olesen Citation1970), and ∼1% in degenerate intervertebral disc (Rajpurohit et al. Citation2002) (discussed below). The level of oxygen in normal rabbit cartilage has been shown to be 3–10% (Ferrell and Najafipour Citation1992). The supply of oxygen, as well as of other nutrients, reaches the cartilage not only from the synovial fluid (which is in turn supplied by the vasculature of the joint capsule and the synovium) but also from the subchondral bone (Houard et al. Citation2013). Age-related changes in the subchondral bone, which often undergoes significant remodeling (Loeser Citation2010), would probably affect the oxygen tension in the joint, as would increasing calcification of the cartilage itself, which has also been associated with advancing age.
Like other tissues including bone, articular cartilage appears to be susceptible to damage caused by oxidative stress. Chondrocytes can produce both ROS such as superoxide anions and nitric oxide (NO), which is known to be upregulated by IL-1 and to react with tyrosine residues to form 3-nitrotyrosine (Loeser et al. Citation2002). This is found at increased levels both in aged and osteoarthritic articular cartilage in humans, and has been shown to cause damage in ageing-associated degeneration in other tissues such as brain and muscle. Since many cell signaling proteins use tyrosine phosphorylation for activation, the formation of nitrotyrosine residues will interfere with this, and may be responsible for the reduction in chondrocyte response to IGF, since NO can lead to a reduction in tyrosine phosphorylation of the IGF-1 receptor. van der Kraan et al. (Citation2012) have shown that there is a shift in SMAD 1/5/8 signaling with age. Since these SMAD pathways are involved in TGFβ signaling (via the activin receptor-like kinase (ALK) 1 and 5), the increased ratio of ALK 1 to ALK 5 found with age results in a correlated increase in MMP-13 expression, and possibly in the autolytic chondrocyte phenotype which is found in senescent chondrocytes. Another cytokine that has been shown to have elevated expression with increased age is IL-7 (Rubenhagen et al. Citation2012). The AGEs that accumulate in chondrocytes from older individuals appear to activate janus kinase 2 and 3 signal transducer and STAT 3 phosphorylation, and lead to increased MMP13 and ADAMTS-4 and -5 expression and activity (Huang et al. Citation2011).
Mutations in the mitochondrial DNA of chondrocytes (which may result from exposure to cytokines or ROS) may also contribute to the age-related changes in articular cartilage (Blanco et al. Citation2011). They can affect several pathways, including defective chondrocyte biosynthesis and increased apoptosis. Certainly, accumulation of ROS and AGEs can promote the senescent phenotype in chondrocytes (Leong and Sun Citation2011), conferring not only reduced anabolic activity and synthesis of proinflammatory cytokines and degradative enzymes, but also triggering an impaired response to the mechanical and inflammatory insults to the cartilage. This may be the reason for OA developing much more quickly in the destabilized medial meniscus (DMM) model of mechanical instability in 12-month-old mice than in 12-week-old mice (Loeser et al. Citation2012). There is differential expression of immune response genes in the old and young DMM mice, which may be a key factor in the association between the development of OA with increasing age, where an increasing degree of low-grade, chronic inflammation occurs (or “inflammaging” (Franceschi et al. Citation2007)). Greene and Loeser (Citation2015) suggested that this process is key to the increased likelihood of developing OA with advancing age, with overall adiposity playing an important part, where greater amounts of adipose tissue in older (or obese) individuals promote systemic inflammation and elevated levels of IL-6 and TNFα (Greene and Loeser Citation2015).
The genes controlling all the anabolic, catabolic, and signaling pathways can of course be affected by epigenetic influences, which add another complete level of complexity, and effector mechanisms. As described for bone cells above, SirT1 promotes chondrocyte survival and matrix gene expression also, and can be cleaved and inactivated by TNFα (Li et al. Citation2013). Similarly, some miRNAs have been identified that become upregulated with age (miR199a, miR193p, and miR193b) and which appear to be anti-anabolic, whereas miR320c expression is downregulated (reviewed by Li and colleagues (2013)).
If cell therapy is being used to repair or regenerate diseased joints, such as in OA, it is likely to be occurring in an older individual. If autologous therapy is being undertaken, the phenotype of the cells (in terms of senescence) to be implanted could affect their ability to effect repair. At our center (RJAH Orthopaedic Hospital NHS Trust), we have, however, seen no reduction in terms of clinical benefit from autologous chondrocyte implantation to date, which relates to the age of the patient (unpublished data).
Intervertebral discs
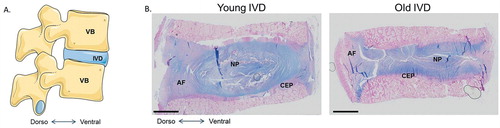
IVDs are composed of a central highly hydrated tissue named the nucleus pulposus (NP) surrounded by a fibrous ring, the annulus fibrosus (AF). Both tissues are enclosed between the vertebral endplates. In most mammals, the first signs of IVD degeneration start early in life and mainly affect the NP (Boos et al. Citation2002) (). During IVD growth, 2 cell types populate the NP: the nucleopulpocytes (NPCytes) and the notochordal cells (NTCs) (Hunter et al. Citation2004, Colombier et al. Citation2014). Nucleopulpocytes are the effector cells of the NP, which are responsible for the synthesis of extracellular matrix (ECM) components, while NTCs are considered to be the NP progenitors, maintaining homeostasis (Aguiar et al. Citation1999, Erwin et al. Citation2006, Citation2011, McCann et al. Citation2011). The progressive loss of NTCs during skeletal maturation may be one of the first events leading to the initiation of the degenerative cascade observed in ageing IVD. Some elements of the pathophysiology of the NP, the AF, and the cartilage endplates (CEPs) will be given below.
Figure 3. The human intervertebral disc. A. Schematic view of the human lumbar spine, composed of vertebral bodies (VB) and intervertebral discs (IVD). B. Histological appearance of young and old human intervertebral discs stained with Alcian blue.
Pathophysiology of the nucleus pulposus
The NP contains mainly NTCs and NPCytes embedded in a matrix that, like cartilage (described above), is rich in type-II collagen (COL2) and PGs (Clouet et al. Citation2009, Risbud et al. Citation2015). Aggrecan is the main PG found in the NP, containing ∼30 chains of sulfated GAGs, which contribute to the negative charge that gives the hyper-hydrated state of the NP. These aggrecan molecules are embedded in type-II collagen fibers to form a specific network that allows the NP to be elastic and deform under stress. A third cell type has recently been described, displaying progenitor properties similar to those of MSCs (Sakai et al. Citation2012). The expression of the notochordal markers brachyury and cytokeratin 8 by these progenitor cells has not been thoroughly described, however, making their origin unclear. NP cells are highly specialized, having adapted to survive in a more severe hypoxic environment than articular cartilage (1% O2). For this reason, the hypoxia-inducible transcription factors 1 and 2 (HIF-1 and HIF-2), which are key cellular regulators of the hypoxic response, were found to be constitutively active in NP cells (Rajpurohit et al. Citation2002). Interestingly, the promoter of the aggrecan gene has been found to be regulated by HIF-1 (Agrawal et al. Citation2007). The constitutive activity of HIF-1 might therefore be partly responsible for the large production of aggrecan by NP-resident cells.
Numerous studies have suggested the existence of a molecular dialog between NTCs and NPCytes responsible for the maturation of the NP, the synthesis of functional ECM, and the survival of NPCytes as we age (Nerlich et al. Citation2005, Erwin et al. Citation2006, Dahia et al. Citation2009, Citation2012, Erwin et al. Citation2011). NPCytes secrete TGF-β, which positively regulates the expression and secretion of the connective tissue growth factor (CTGF/CCN2) by NTC through a Smad-binding element in the CTGF/CCN2 promoter (Leask et al. Citation2001, Chen et al. Citation2002, Tran et al. Citation2010). The secretion of CTGF/CCN2 in turn ensures the proliferation of NPCytes and the synthesis of aggrecan (Erwin et al. Citation2006). In addition, some yet uncharacterized factors secreted by NTCs have the ability to inhibit the activation of the apoptotic caspases 3 and 9, thereby favoring the expression of aggrecan (Erwin et al. Citation2011). Finally, similar to the HIF-1 regulation in NP cells, it has been reported that increased oxygen tension negatively affects the expression of CTGF/CCN2 by NTCs (Tran et al. Citation2013a). Thus, this mechanism not only controls the hypoxic response in NP cells, but also the secretion of CTGF/CCN2—which controls PG production.
Taken together, these results highlight the fundamental role of NTCs in the survival and activity of NPCytes, which subsequently maintain NP homeostasis. The gradual disappearance of NTCs during skeletal maturation and ageing is therefore likely to disturb the molecular dialog between NP cells, and is the leading event in initiating degeneration of the NP.
Degenerative events
The complete disappearance of NTCs at skeletal maturity in humans is concomitant with the activation of ageing-related cellular responses including apoptosis, senescence, and autophagy. NP cell apoptosis could be initiated through several caspase-dependent pathways (Thornberry and Lazebnik Citation1998). This caspase activation during degeneration results either from the death receptor-mediated extrinsic pathway (Bachmeier et al. Citation2007) or the intrinsic pathway, which is linked to mitochondrial activity, especially during the most advanced stages of degeneration (Rannou et al. Citation2004, Wang et al. Citation2014). Resident NP cells have limited spontaneous repair properties. However, autophagy and senescence mechanisms are activated in NP cells during degeneration to protect from cell death.
Autophagy is a process that allows cells to generate their own energy under conditions of high stress, such as prolonged nutrient deficiency (Rubinsztein et al. Citation2011). During degeneration, both the number of autophagosomes and the autophagic flow were found to increase gradually (Ye et al. Citation2011). This upregulation of autophagy-associated events strongly suggests that NP cells have the potential to compensate for nutrient deprivation by generating their own energy when exposed to intense stress. The second mechanism of cell protection observed in NP degeneration relates to senescence. This cell mechanism involves the entry of cells into the non-proliferative G0 phase of the cell cycle. Upon entry into the G0 phase, cells no longer respond to environmental cues, which can allow evasion of programmed death (Herbig et al. Citation2006). Interestingly, it has been found that NP cells experience replicative senescence during ageing (Kim et al. Citation2009).
The cellular modifications observed in the NP are accompanied by ECM remodeling. Surprisingly, CTGF/CCN2, which is involved in the establishment of the NP-specific ECM during growth, has also been found to promote degeneration (Tran et al. Citation2010). This dual effect may be mainly explained by the loss of regulation of CTGF/CCN2 expression. Indeed, an abnormal accumulation of CTGF/CCN2 is observed in the NP while NTCs disappear during ageing. This growth factor presents several domains that interact with a large set of molecules including PGs, which may account for its accumulation throughout life (Tran et al. Citation2013a and Citationb). This accumulation may also be explained by the increase in TGF-β in the NP, which positively regulates the expression of CTGF/CCN2 (Lee et al. Citation2009). This increase in TGF-β and CTGF/CCN2 activates a fibrotic repair response of NP cells, characterized by the deposition of type-I collagen fibers.
A disequilibrium between the disappearance of NTC-secreted protective factors and the increase in factors associated with senescent cells (senescence-associated secretory phenotype) results in an increase in the expression of matrix metalloproteinases (including MMP3 and MMP7) and a disintegrin and metalloproteinase with thrombospondin repeats (ADAMTS) (Roberts et al. Citation2000, Freund et al. Citation2010). These enzymes are capable of degrading both collagen and PGs, leading to dehydration and progressive disorganization of ECM. Under these conditions, it has been observed that the ECM takes on a grainy appearance and shows cracks and tears. In addition, an increase in the secretion of interleukins (IL-6 and IL-8), vascular endothelial growth factor (VEGF), and nerve growth factor (NGF) has been reported following the increased ADAMTS and MMP expression seen in ageing, promoting vessel and neurite ingrowth in painful discs during degeneration (Burke et al. Citation2002, Ohba et al. Citation2009, Cornejo et al. Citation2015, Purmessur et al. Citation2015). The progressive change in the ECM of the NP ultimately leads to the loss of the biomechanical functions of IVDs that is associated with increasing age.
Pathophysiology of the annulus fibrosus
The AF can be divided into 2 distinct areas: the outer AF and the inner AF. The inner AF, which is also known as the transition zone, contains chondrogenic cells and a poorly organized ECM that is composed of type-II collagen, PGs, and water. In contrast, the outer AF is populated by fibroblasts and a highly organized ECM rich in type-I collagen, and type-II collagen and PGs are much less obvious in this region (Eyre and Muir Citation1976). AF cells have membranous mechanoreceptors and are very sensitive to mechanical stress (Dimitroulias et al. Citation2010). The secretion of sonic hedgehog (SHH) by NTCs in the NP is involved not only in the maintenance of the AF cell identity but also in the control of their secretory activity (type-I and type-II collagen) (Dahia et al. Citation2012). The ECM of the AF is organized into 15–25 concentric lamellae oriented at 65° relative to the vertical plane (Hayes et al. Citation2011). These lamellae are interconnected by PG aggregates, elastin and lubricin, and also type-VI collagen fibers (Melrose et al. Citation2008, Shine et al. Citation2009). Moreover, the outer AF has a higher resistance to tension than the inner AF. Collectively, the fibrous structure of the AF gives important mechanical properties that limit protrusion of the NP.
The interruption in SHH secretion by NTC during degeneration leads to a differentiation of AF cells into hypertrophic cells that are unable to secrete type-II collagen (Kondo et al. Citation2011, Haschtmann et al. Citation2012). This differentiation leads to modification of the tensile strength of the AF, and thus to a modification of cell response to overload. In this context, it has been described that AF cells undergo massive apoptotic cell death in response to overload (Rannou et al. Citation2004). Degradation of matrix components is later initiated in the AF, resulting in destabilization of its structure. During this process, the collagen fibers become thinner and more irregular, eventually leading to cracks.
Pathophysiology of the cartilage endplates
CEPs are composed of a thin layer of hyaline cartilage adjacent to the vertebral bony endplate. Like articular cartilage, these CEPs are composed of chondrocytes that synthesize a COL2- and PG-enriched ECM, with a ratio of PG to type-II collagen of approximately 2:1. These PGs efficiently allow hydration of the ECM, with a water content of around 50–60% (Roberts et al. Citation1989). The vertebral endplates are the site of a microscopic network of blood vessels that are responsible for nutritional intake during development and growth of IVDs (Taylor Citation1975, Benneker et al. Citation2005, Hee et al. Citation2011). Metabolites diffuse through the pores present in the CEP, based on their size and charge. Positive ions (e.g. sodium, calcium) or neutral molecules, such as glucose and oxygen, diffuse most readily (Grunhagen et al. Citation2006). Cartilage endplates also undergo degenerative events with increasing age, exacerbating NP degeneration. Indeed, the disappearance of NTCs in the NP is concomitant with the commitment of endplate chondrocytes to a hypertrophic phenotype. The activity of alkaline phosphatase increases and chondrocytes start to secrete type-X collagen, which ultimately leads to calcification of the ECM (Aigner et al. Citation1998, Sahlman et al. Citation2001, Paietta et al. Citation2013). Endplates become impermeable, which not only blocks the diffusion of nutrients but also the removal of metabolites in the NP. These events dramatically alter IVD homeostasis, mainly by inducing acidification and restricting the supply of oxygen and nutrients.
Musculoskeletal vasculature
For decades, the vasculature of the musculoskeletal system has been perceived merely as a means of supplying oxygen and nutrients, and for disposal of waste products. However, recent discoveries suggest a close crosstalk between the vascular and osteogenic niches and a bilateral influence of both. As in other tissues of the body, ageing-dependent changes occur within the vasculature also. Arteries become increasingly stiff and hard, due to changes in the vessel wall, where calcification and glycosylation are chiefly responsible. These processes take place in the inner 2 vessel layers, namely the intima and media. The outer layer, the adventitia, is also affected by ageing, with an increase in collagen fibers and fibroblasts (Kovacic et al. Citation2011). An intriguing “calcification paradox” has been suggested, where increased calcification of the vasculature occurs while the bone becomes less calcified with increasing age (Persy and D’Haese Citation2009). Deficiency of the protein osteoprotegerin in mice results in a severe decrease in bone density due to elevated osteoclastogenesis, but—interestingly—this is accompanied by elevated vascular calcification (Bucay et al. Citation1998), suggesting a link between the 2 processes. Also, mutation of the klotho gene leads to a condition resembling human ageing and involving vessel calcification and bone decalcification in the mouse (Kuro-o et al. Citation1997).
Pericytes are ubiquitously distributed around the endothelium throughout the human body. These cells are closely related to mesenchymal stromal cells and can dedifferentiate into osteoprogenitor cells with osteoblastic capabilities (Bostrom et al. Citation1993, Kovacic et al. Citation2011). Furthermore, phenotypic switching of endothelial cells to become osteoblasts and chondrocytes has been proposed (Medici et al. Citation2010). Recent discoveries in mice suggest a close relationship between a specific subset of endothelial cells and bone formation (Kusumbe et al. Citation2014, Ramasamy et al. Citation2014, Citation2015). These cells are known as type H endothelium; they are found in close proximity to osteoprogenitors and promote angiogenesis and bone formation via paracrine Dll4-Notch signaling (Ramasamy et al. Citation2014). Interestingly, the same pathway inhibits angiogenesis in many other tissues (Thomas et al. Citation2013). The impaired bone remodeling and bone-forming capacity in aged animals seems to be related to the observed age-dependent decrease in type H endothelium. Intriguingly, bone mass could be restored by increasing levels of HIF-1α, a transcription factor that is pivotal in regulation of cell metabolism and angiogenesis (Pugh and Ratcliffe Citation2003). HIF-1α was found to have a positive influence on type H endothelium, which upregulated Notch signaling and the release of Noggin downstream (Ramasamy et al. Citation2014).
Noggin is known as to be an antagonist of bone-morphogenic proteins and acts in a paracrine fashion on the cells of the osteogenic niche. Nevertheless, this pathway was able to restore bone mass and angiogenesis in aged mice. These findings have led some authors to name type H endothelium “vessels of rejuvenation” (le Noble and le Noble Citation2014). Type H endothelium expresses high levels of CD31, Pecam1, and endomucin and it produces significant levels of Pdgfa, Pdgfb, Tgfb1, Tgfb3, and Fgf1 transcripts, which attract osteogenic cell types. In line with this, bone-forming cells were predominantly found in close proximity to type H endothelium. This type of endothelium appears to be a central building block for bone formation and homeostasis (Kusumbe et al. Citation2014), and couples angiogenesis and osteogenesis, substantiating earlier observations that angiogenic growth factors also increase bone formation (Gerber et al. Citation1999, Rolfing Citation2014).
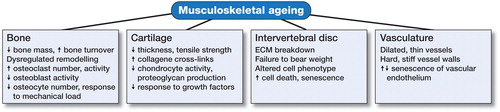
In summary, the musculoskeletal system is a dynamic environment consisting of a variety of tissues types made up of several different cellular components within complex arrangements of matrices. The interaction of each tissue type is essential for the adequate functioning of the skeleton as a whole, and as such, each region (bone, cartilage, tendon, disc, muscle vasculature) has the potential to significantly affect the others. As we age, the alterations in normal biological responses lead to impaired tissue function, manifest in features we recognize as ageing (). This is especially true in the musculoskeletal system, where the interdependent nature of skeletal components is particularly susceptible. Ongoing investigations identifying, characterizing, and targeting novel ageing-related mechanisms may serve to increase healthspan, lengthen lifespan, and prevent musculoskeletal deterioration as we age.
Figure 4. Musculoskeletal ageing. The various tissues that comprise the skeleton are each susceptible to ageing-related deterioration, and have a reduced capacity to function adequately with increasing lifespan.
JRE, SR, and CM are grateful for support from Arthritis Research UK (Fellowship 20631 (JRE) and Project Grant 19429 (SR, CM)) and from Orthopaedic Research UK (509) (JRE).
- Agrawal A, Guttapalli A, Narayan S, Albert T J, Shapiro I M, Risbud M V. Normoxic stabilization of HIF-1alpha drives glycolytic metabolism and regulates aggrecan gene expression in nucleus pulposus cells of the rat intervertebral disk. Am J Physiol Cell Physiol 2007; 293 (2): C621–31.
- Aguiar D J, Johnson S L, Oegema T R. Notochordal cells interact with nucleus pulposus cells: regulation of proteoglycan synthesis. Exp Cell Res 1999; 246 (1): 129–37.
- Aigner T, Gresk-otter K R, Fairbank J C, von der Mark K, Urban J P. Variation with age in the pattern of type X collagen expression in normal and scoliotic human intervertebral discs. Calcif Tissue Int1998; 63 (3): 263–8.
- Ambrogini E, Almeida M, Martin-Millan M, Paik J H, Depinho R A, Han L, et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab 2010; 11 (2): 136–46.
- Awad H A, Butler D L, Boivin G P, Smith F N, Malaviya P, Huibregtse B, et al. Autologous mesenchymal stem cell-mediated repair of tendon. Tissue Eng 1999; 5 (3): 267–77.
- Aydelotte M B, Kuettner K E. Differences between sub-populations of cultured bovine articular chondrocytes. I. Morphology and cartilage matrix production. Connect Tissue Res 1988; 18 (3): 205–22.
- Bachmeier B E, Nerlich A G, Weiler C, Paesold G, Jochum M, Boos N. Analysis of tissue distribution of TNF-alpha, TNF-alpha-receptors, and the activating TNF-alpha-converting enzyme suggests activation of the TNF-alpha system in the aging intervertebral disc. Ann N Y Acad Sci 2007; 1096: 44–54.
- Baxter M A, Wynn R F, Jowitt S N, Wraith J E, Fairbairn L J, Bellantuono I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004; 22 (5): 675–82.
- Benneker L M, Heini P F, Alini M, Anderson S E, Ito K. 2004 Young Investigator Award Winner: vertebral endplate marrow contact channel occlusions and intervertebral disc degeneration. Spine (Phila Pa 1976) 2005; 30 (2): 167–73.
- Blanco F J, Rego I, Ruiz-Romero C. The role of mitochondria in osteoarthritis. Nat Rev Rheumatol 2011; 7 (3): 161–9.
- Bonab M M, Alimoghaddam K, Talebian F, Ghaffari S H, Ghavamzadeh A, Nikbin B. Aging of mesenchymal stem cell in vitro. BMC Cell Biol 2006; 7: 14.
- Bonewald L F. Does defective bone lead to defective muscle? J Bone Miner Res 2015; 30 (4): 593–5.
- Boos N, Weissbach S, Rohrbach H, Weiler C, Spratt K F, Nerlich A G. Classification of age-related changes in lumbar intervertebral discs: 2002 Volvo Award in basic science. Spine 2002; 27 (23): 2631–44.
- Bostrom K, Watson K E, Horn S, Wortham C, Herman I M, Demer L L. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest 1993; 91 (4): 1800–9.
- Bucay N, Sarosi I, Dunstan C R, Morony S, Tarpley J, Capparelli C, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 1998; 12 (9): 1260–8.
- Burke J G, Watson R W, McCormack D, Dowling F E, Walsh M G, Fitzpatrick J M. Spontaneous production of monocyte chemoattractant protein-1 and interleukin-8 by the human lumbar intervertebral disc. Spine (Phila Pa 1976) 2002; 27 (13): 1402–7.
- Calnan D R, Brunet A. The FoxO code. Oncogene 2008; 27 (16): 2276–88.
- Candela M E, Yasuhara R, Iwamoto M, Enomoto-Iwamoto M. Resident mesenchymal progenitors of articular cartilage. Matrix Biol 2014; 39: 44–9.
- Cao J J, Wronski T J, Iwaniec U, Phleger L, Kurimoto P, Boudignon B, et al. Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. J Bone Miner Res 2005; 20 (9): 1659–68.
- Cao J J, Kurimoto P, Boudignon B, Rosen C, Lima F, Halloran B P. Aging impairs IGF-I receptor activation and induces skeletal resistance to IGF-I. J Bone Miner Res 2007; 22 (8): 1271–9.
- Chen Y, Blom I E, Sa S, Goldschmeding R, Abraham D J, Leask A. CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int 2002; 62 (4): 1149–59.
- Clouet J, Grimandi G l, Pot-Vaucel M, Masson M, Fellah H B, Guigand L, et al. Identification of phenotypic discriminating markers for intervertebral disc cells and articular chondrocytes. Rheumatology (Oxford) 2009; 48 (11): 1447–50.
- Cohen-Kfir E, Artsi H, Levin A, Abramowitz E, Bajayo A, Gurt I, et al. Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology 2011; 152 (12): 4514–24.
- Colombier P, Camus A, Lescaudron L, Clouet J, Guicheux J. Intervertebral disc regeneration: a great challenge for tissue engineers. Trends Biotechnol 2014; 32 (9): 433–5.
- Colter D C, Sekiya I, Prockop D J. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc Natl Acad Sci U S A 2001; 98 (14): 7841–5.
- Cornejo M C, Cho S K, Giannarelli C, Iatridis J C, Purmessur D. Soluble factors from the notochordal-rich intervertebral disc inhibit endothelial cell invasion and vessel formation in the presence and absence of pro-inflammatory cytokines. Osteoarthritis Cartilage 2015; 23 (3): 487–96.
- D’Ippolito G, Schiller P C, Ricordi C, Roos B A, Howard G A. Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res 1999; 14 (7): 1115–22.
- Dahia C L, Mahoney E J, Durrani A A, Wylie C. Intercellular signaling pathways active during intervertebral disc growth, differentiation, and aging. Spine (Phila Pa 1976) 2009; 34 (5): 456–62.
- Dahia C L, Mahoney E, Wylie C. Shh signaling from the nucleus pulposus is required for the postnatal growth and differentiation of the mouse intervertebral disc. PLoS One 2012; 7 (4): e35944.
- Daniell H W. Osteoporosis after orchiectomy for prostate cancer. J Urol 1997; 157 (2): 439–44.
- Dejica V M, Mort J S, Laverty S, Antoniou J, Zukor D J, Tanzer M, et al. Increased type II collagen cleavage by cathepsin K and collagenase activities with aging and osteoarthritis in human articular cartilage. Arthritis Res Ther 2012; 14 (3): R113.
- Dimitroulias A, Tsonidis C, Natsis K, Venizelos I, Djau S N, Tsitsopoulos P, et al. An immunohistochemical study of mechanoreceptors in lumbar spine intervertebral discs. J Clin Neurosci 2010; 17 (6): 742–5.
- Donahue S W, Jacobs C R, Donahue H J. Flow-induced calcium oscillations in rat osteoblasts are age, loading frequency, and shear stress dependent. Am J Physiol Cell Physiol 2001; 281 (5): C1635–41.
- Dowthwaite G P, Bishop J C, Redman S N, Khan I M, Rooney P, Evans D J, et al. The surface of articular cartilage contains a progenitor cell population. J Cell Sci 2004; 117 (Pt 6): 889–97.
- Edwards J R, Perrien D S, Fleming N, Nyman J S, Ono K, Connelly L, et al. Silent information regulator (Sir)T1 inhibits NF-kappaB signaling to maintain normal skeletal remodeling. J Bone Miner Res 2013; 28 (4): 960–9.
- Erwin W M, Ashman K, O’Donnel P, Inman R D. Nucleus pulposus notochord cells secrete connective tissue growth factor and up-regulate proteoglycan expression by intervertebral disc chondrocytes. Arthritis Rheum 2006; 54 (12): 3859–67.
- Erwin W M, Islam D, Inman R D, Fehlings M G, Tsui F W. Notochordal cells protect nucleus pulposus cells from degradation and apoptosis: implications for the mechanisms of intervertebral disc degeneration. Arthritis Res Ther 2011; 13 (6): R215.
- Eyre D R, Muir H. Types I and II collagens in intervertebral disc. Interchanging radial distributions in annulus fibrosus. Biochem J 1976; 157 (1): 267–70.
- Falahati-Nini A, Riggs B L, Atkinson E J, O’Fallon W M, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J Clinical Invest 2000; 106 (12): 1553–60.
- Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science 1998; 279 (5356): 1528–30.
- Ferrell W R, Najafipour H. Changes in synovial PO2 and blood flow in the rabbit knee joint due to stimulation of the posterior articular nerve. J Physiol 1992; 449: 607–17.
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 2007; 128 (1): 92–105.
- Freund A, Orjalo A V, Desprez P Y, Campisi J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 2010; 16 (5): 238–46.
- Gerber H P, Vu T H, Ryan A M, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 1999; 5 (6): 623–8.
- Greene M A, Loeser R F. Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage 2015; 23 (11): 1966–71.
- Grimshaw M J, Mason R M. Bovine articular chondrocyte function in vitro depends upon oxygen tension. Osteoarthritis Ccartilage 2000; 8 (5): 386–92.
- Grunhagen T, Wilde G, Soukane D M, Shirazi-Adl S A, Urban J P. Nutrient supply and intervertebral disc metabolism. J Bone Joint Surg Am 2006; 88Suppl2: 30–5.
- Han Z H, Palnitkar S, Rao D S, Nelson D, Parfitt A M. Effects of ethnicity and age or menopause on the remodeling and turnover of iliac bone: implications for mechanisms of bone loss. J Bone Miner Res 1997; 12 (4): 498–508.
- Hardingham T, Bayliss M. Proteoglycans of articular cartilage: changes in aging and in joint disease. Semin Arthritis Rheum1990; 20 (3 Suppl 1): 12–33.
- Haschtmann D, Ferguson S J, Stoyanov J V. BMP-2 and TGF-beta3 do not prevent spontaneous degeneration in rabbit disc explants but induce ossification of the annulus fibrosus. Eur Spine J 2012; 21 (9): 1724–33.
- Hayes A J, Hall A, Brown L, Tubo R, Caterson B. Macromolecular organization and in vitro growth characteristics of scaffold-free neocartilage grafts. J Histochem Cytochem 2007; 55 (8): 853–66.
- Hayes A J, Isaacs M D, Hughes C, Caterson B, Ralphs J R. Collagen fibrillogenesis in the development of the annulus fibrosus of the intervertebral disc. Eur Cell Mater 2011; 22: 226–41.
- Hee H T, Chuah Y J, Tan B H, Setiobudi T, Wong H K. Vascularization and morphological changes of the endplate after axial compression and distraction of the intervertebral disc. Spine (Phila Pa 1976) 2011; 36 (7): 505–11.
- Heinegard D, Saxne T. Macromolecular markers in joint disease. J Rheumatol Suppl 1991; 27: 27–9.
- Henriksen K, Leeming D J, Byrjalsen I, Nielsen R H, Sorensen M G, Dziegiel M H, et al. Osteoclasts prefer aged bone. Osteoporos Int 2007; 18 (6): 751–9.
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy J M. Cellular senescence in aging primates. Science 2006; 311 (5765): 1257.
- Houard X, Goldring M B, Berenbaum F. Homeostatic mechanisms in articular cartilage and role of inflammation in osteoarthritis. Curr Rheumatol Rep 2013; 15 (11): 375.
- Huang C, Ogawa R. Mechanotransduction in bone repair and regeneration. FASEB J 2010; 24 (10): 3625–32.
- Huang C Y, Lai K Y, Hung L F, Wu W L, Liu F C, Ho L J. Advanced glycation end products cause collagen II reduction by activating Janus kinase/signal transducer and activator of transcription 3 pathway in porcine chondrocytes. Rheumatology 2011; 50 (8): 1379–89.
- Hunter C J, Matyas J R, Duncan N A. Cytomorphology of notochordal and chondrocytic cells from the nucleus pulposus: a species comparison. J Anat 2004; 205 (5): 357–62.
- Iyer S, Ambrogini E, Bartell S M, Han L, Roberson P K, de Cabo R, et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest 2013; 123 (8): 3409–19.
- Iyer S, Han L, Bartell S M, Kim H N, Gubrij I, de Cabo R, et al. Sirtuin1 (Sirt1) promotes cortical bone formation by preventing beta-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J Biol Chem 2014; 289 (35): 24069–78.
- Jiang Y, Mishima H, Sakai S, Liu Y K, Ohyabu Y, Uemura T. Gene expression analysis of major lineage-defining factors in human bone marrow cells: effect of aging, gender, and age-related disorders. J Orthop Res 2008; 26 (7): 910–7.
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999; 13 (19): 2570–80.
- Kaeberlein M, Powers R W, 3rd, Steffen K K, Westman E A, Hu D, Dang N, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005; 310 (5751): 1193–6.
- Kapahi P, Zid B M, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 2004; 14 (10): 885–90.
- Katznelson L, Finkelstein J S, Schoenfeld D A, Rosenthal D I, Anderson E J, Klibanski A. Increase in bone density and lean body mass during testosterone administration in men with acquired hypogonadism. J Clin Endocrinol Metab 1996; 81 (12): 4358–65.
- Kim K W, Chung H N, Ha K Y, Lee J S, Kim Y Y. Senescence mechanisms of nucleus pulposus chondrocytes in human intervertebral discs. Spine J 2009; 9 (8): 658–66.
- Kneissel M, Luong-Nguyen N H, Baptist M, Cortesi R, Zumstein-Mecker S, Kossida S, et al. Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone 2004; 35 (5): 1144–56.
- Kolpakova E, Olsen B R. Wnt/beta-catenin–a canonical tale of cell-fate choice in the vertebrate skeleton. Dev Cell 2005; 8 (5): 626–7.
- Kondo N, Yuasa T, Shimono K, Tung W, Okabe T, Yasuhara R, et al. Intervertebral disc development is regulated by Wnt/beta-catenin signaling. Spine (Phila Pa 1976) 2011; 36 (8): E513–8.
- Kovacic J C, Moreno P, Hachinski V, Nabel E G, Fuster V. Cellular senescence, vascular disease, and aging: Part 1 of a 2-part review. Circulation 2011; 123 (15): 1650–60.
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997; 390 (6655): 45–51.
- Kusumbe A P, Ramasamy S K, Adams R H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014; 507 (7492): 323–8.
- Lazarenko O P, Rzonca S O, Hogue W R, Swain F L, Suva L J, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology 2007; 148 (6): 2669–80.
- le Noble F, le Noble J. Bone biology: Vessels of rejuvenation. Nature 2014; 507 (7492): 313–4.
- Leask A, Sa S, Holmes A, Shiwen X, Black C M, Abraham D J. The control of ccn2 (ctgf) gene expression in normal and scleroderma fibroblasts. Mol Pathol 2001; 54 (3): 180–3.
- Lee R B, Urban J P. Evidence for a negative Pasteur effect in articular cartilage. Biochem J 1997; 321 (Pt 1): 95–102.
- Lee S, Moon C S, Sul D, Lee J, Bae M, Hong Y, et al. Comparison of growth factor and cytokine expression in patients with degenerated disc disease and herniated nucleus pulposus. Clin Biochem 2009; 42 (15): 1504–11.
- Lee K W, Yook J Y, Son M Y, Kim M J, Koo D B, Han Y M, et al. Rapamycin promotes the osteoblastic differentiation of human embryonic stem cells by blocking the mTOR pathway and stimulating the BMP/Smad pathway. Stem Cells Dev 2010; 19 (4): 557–68.
- Leong D J, Sun H B. Events in articular chondrocytes with aging. Curr Osteoporosis Rep 2011; 9 (4): 196–201.
- Li Y, Wei X, Zhou J, Wei L. The age-related changes in cartilage and osteoarthritis. BioMed Res Int 2013; 2013: 916530.
- Loeser R F. Age-related changes in the musculoskeletal system and the development of osteoarthritis. Clin Geriatr Med 2010; 26 (3): 371–86.
- Loeser R F, Shanker G, Carlson C S, Gardin J F, Shelton B J, Sonntag W E. Reduction in the chondrocyte response to insulin-like growth factor 1 in aging and osteoarthritis: studies in a non-human primate model of naturally occurring disease. Arthritis Rheum 2000; 43 (9): 2110–20.
- Loeser R F, Carlson C S, Del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum 2002; 46 (9): 2349–57.
- Loeser R F, Olex A L, McNulty M A, Carlson C S, Callahan M F, Ferguson C M, et al. Microarray analysis reveals age-related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum 2012; 64 (3): 705–17.
- Lopez-Otin C, Blasco M A, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153 (6): 1194–217.
- Lund-Olesen K. Oxygen tension in synovial fluids. Arthritis Rheum 1970; 13 (6): 769–76.
- Manolagas S C. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 2000; 21 (2): 115–37.
- Manolagas S C, Almeida M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol 2007; 21 (11): 2605–14.
- Martin J A, Buckwalter J A. Telomere erosion and senescence in human articular cartilage chondrocytes. J Gerontol A Biol Sci Med Sci 2001; 56 (4): B172–9.
- Martin J A, Buckwalter J A. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology 2002; 3 (5): 257–64.
- McCann M R, Tamplin O J, Rossant J, Seguin C A. Tracing notochord-derived cells using a Noto-cre mouse: implications for intervertebral disc development. Dis Model Mech 2011; 5 (1): 73–82.
- Medici D, Shore E M, Lounev V Y, Kaplan F S, Kalluri R, Olsen B R. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 2010; 16 (12): 1400–6.
- Melrose J, Smith S M, Appleyard R C, Little C B. Aggrecan, versican and type VI collagen are components of annular translamellar crossbridges in the intervertebral disc. Eur Spine J 2008; 17 (2): 314–24.
- Modder U I, Clowes J A, Hoey K, Peterson J M, McCready L, Oursler M J, et al. Regulation of circulating sclerostin levels by sex steroids in women and in men. J Bone Miner Res 2011; 26 (1): 27–34.
- Moerman E J, Teng K, Lipschitz D A, Lecka-Czernik B. Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-gamma2 transcription factor and TGF-beta/BMP signaling pathways. Aging Cell 2004; 3 (6): 379–89.
- Mogi M, Kondo A. Down-regulation of mTOR leads to up-regulation of osteoprotegerin in bone marrow cells. Biochem Biophys Res Commun 2009; 384 (1): 82–6.
- Moore S G, Dawson K L. Red and yellow marrow in the femur: age-related changes in appearance at MR imaging. Radiology 1990; 175 (1): 219–23.
- Nerlich A G, Bachmeier B E, Boos N. Expression of fibronectin and TGF-beta1 mRNA and protein suggest altered regulation of extracellular matrix in degenerated disc tissue. Eur Spine J 2005; 14 (1): 17–26.
- Ohba T, Haro H, Ando T, Wako M, Suenaga F, Aso Y, et al. TNF-alpha-induced NF-kappaB signaling reverses age-related declines in VEGF induction and angiogenic activity in intervertebral disc tissues. J Orthop Res 2009; 27 (2): 229–35.
- Ota K, Quint P, Ruan M, Pederson L, Westendorf J J, Khosla S, et al. Sclerostin is expressed in osteoclasts from aged mice and reduces osteoclast-mediated stimulation of mineralization. J Cell Biochem 2013; 114 (8): 1901–7.
- Paietta R C, Burger E L, Ferguson V L. Mineralization and collagen orientation throughout aging at the vertebral endplate in the human lumbar spine. J Struct Biol 2013; 184 (2): 310–20.
- Persy V, D’Haese P. Vascular calcification and bone disease: the calcification paradox. Trends Mol Med 2009; 15 (9): 405–16.
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004; 429 (6993): 771–6.
- Pittenger M F, Mackay A M, Beck S C, Jaiswal R K, Douglas R, Mosca J D, et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999; 284 (5411): 143–7.
- Pugh C W, Ratcliffe P J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 2003; 9 (6): 677–84.
- Purmessur D, Cornejo M C, Cho S K, Roughley P J, Linhardt R J, Hecht A C, et al. Intact glycosaminoglycans from intervertebral disc-derived notochordal cell-conditioned media inhibit neurite growth while maintaining neuronal cell viability. Spine J 2015; 15 (5): 1060–9.
- Rajpurohit R, Risbud M V, Ducheyne P, Vresilovic E J, Shapiro I M. Phenotypic characteristics of the nucleus pulposus: expression of hypoxia inducing factor-1, glucose transporter-1 and MMP-2. Cell Tissue Res 2002; 308 (3): 401–7.
- Ramasamy S K, Kusumbe A P, Wang L, Adams R H. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014; 507 (7492): 376–80.
- Ramasamy S K, Kusumbe A P, Adams R H. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol 2015; 25 (3): 148–57.
- Rannou F, Lee T S, Zhou R H, Chin J, Lotz J C, Mayoux-Benhamou M A, et al. Intervertebral disc degeneration: the role of the mitochondrial pathway in annulus fibrosus cell apoptosis induced by overload. Am J Pathol 2004; 164 (3): 915–24.
- Riggs B L, Khosla S, Melton L J, 3rd. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res 1998; 13 (5): 763–73.
- Risbud M V, Schoepflin Z R, Mwale F, Kandel R A, Grad S, Iatridis J C, et al. Defining the phenotype of young healthy nucleus pulposus cells: Recommendations of the Spine Research Interest Group at the 2014 Annual ORS Meeting. J Orthop Res 2015; 33(3): 283–93.
- Roberts S, Menage J, Urban J P. Biochemical and structural properties of the cartilage end-plate and its relation to the intervertebral disc. Spine (Phila Pa 1976) 1989; 14 (2): 166–74.
- Roberts S, Caterson B, Menage J, Evans E H, Jaffray D C, Eisenstein S M. Matrix metalloproteinases and aggrecanase: their role in disorders of the human intervertebral disc. Spine (Phila Pa 1976) 2000; 25 (23): 3005–13.
- Rolfing J H. The effect of erythropoietin on bone. Acta Orthop 2014; 85 (Suppl 353): 1–27.
- Rosen E D, MacDougald O A. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 2006; 7 (12): 885–96.
- Rubenhagen R, Schuttrumpf J P, Sturmer K M, Frosch K H. Interleukin-7 levels in synovial fluid increase with age and MMP-1 levels decrease with progression of osteoarthritis. Acta Orthop 2012; 83 (1): 59–64.
- Rubinsztein D C, Marino G, Kroemer G. Autophagy and aging. Cell 2011; 146 (5): 682–95.
- Sahlman J, Inkinen R, Hirvonen T, Lammi M J, Lammi P E, Nieminen J, et al. Premature vertebral endplate ossification and mild disc degeneration in mice after inactivation of one allele belonging to the Col2a1 gene for Type II collagen. Spine 2001; 26 (23): 2558–65.
- Sakai D, Nakamura Y, Nakai T, Mishima T, Kato S, Grad S, et al. Exhaustion of nucleus pulposus progenitor cells with ageing and degeneration of the intervertebral disc. Nat Commun 2012; 3: 1264.
- Sethe S, Scutt A, Stolzing A. Aging of mesenchymal stem cells. Ageing Res Rev2006; 5 (1): 91–116.
- Shine K M, Simson J A, Spector M. Lubricin distribution in the human intervertebral disc. J Bone Joint Surg Am 2009; 91 (9): 2205–12.
- Silver I A. Measurement of pH and ionic composition of pericellular sites. Philos Trans R Soc Lond B Biol Sci 1975; 271 (912): 261–72.
- Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003; 33 (6): 919–26.
- Stockwell R A. Morphometry of cytoplasmic components of mammalian articular chondrocytes and corneal keratocytes: species and zonal variations of mitochondria in relation to nutrition. J Anat 1991; 175: 251–61.
- Stolzing A, Jones E, McGonagle D, Scutt A. Age-related changes in human bone marrow-derived mesenchymal stem cells: consequences for cell therapies. Mech Ageing Dev 2008; 129 (3): 163–73.
- Taylor J R. Growth of human intervertebral discs and vertebral bodies. J Anat 1975; 120 (Pt 1): 49–68.
- Thomas J L, Baker K, Han J, Calvo C, Nurmi H, Eichmann A C, et al. Interactions between VEGFR and Notch signaling pathways in endothelial and neural cells.Cell Mol Life Sci 2013; 70 (10): 1779–92.
- Thornberry N A, Lazebnik Y. Caspases: enemies within. Science 1998; 281 (5381): 1312–6.
- Tran C M, Markova D, Smith H E, Susarla B, Ponnappan R K, Anderson D G, et al. Regulation of CCN2/connective tissue growth factor expression in the nucleus pulposus of the intervertebral disc: role of Smad and activator protein 1 signaling. Arthritis Rheum 2010; 62 (7): 1983–92.
- Tran C M, Fujita N, Huang B L, Ong J R, Lyons K M, Shapiro I M, et al. HIF-1alpha and CCN2 form a regulatory circuit in hypoxic nucleus pulposus cells: CCN2 suppresses HIF-1alpha level and transcriptional activity. J Biol Chem 2013a; 288(18): 12654–66.
- Tran C M, Shapiro I M, Risbud M V. Molecular regulation of CCN2 in the intervertebral disc: Lessons learned from other connective tissues. Matrix Biol 2013b; 32 (6): 298–306.
- van der Kraan P M, Goumans M J, Blaney Davidson E, ten Dijke P. Age-dependent alteration of TGF-beta signalling in osteoarthritis. Cell Tissue Res 2012; 347 (1): 257–65.
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs A L, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 2003; 426 (6967): 620.
- Wang X, Wang H, Yang H, Li J, Cai Q, Shapiro I M, et al. Tumor necrosis factor-alpha- and interleukin-1beta-dependent matrix metalloproteinase-3 expression in nucleus pulposus cells requires cooperative signaling via syndecan 4 and mitogen-activated protein kinase-NF-kappaB axis: implications in inflammatory disc disease. Am J Pathol 2014; 184 (9): 2560–72.
- Wu M, Fannin J, Rice K M, Wang B, Blough E R. Effect of aging on cellular mechanotransduction. Ageing Res Rev 2011; 10 (1): 1–15.
- Ye W, Xu K, Huang D, Liang A, Peng Y, Zhu W, et al. Age-related increases of macroautophagy and chaperone-mediated autophagy in rat nucleus pulposus. Connect Tissue Res 2011; 52 (6): 472–8.
- Yeung F, Hoberg J E, Ramsey C S, Keller M D, Jones D R, Frye R A, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004; 23 (12): 2369–80.
- Zhou S, Cui Z, Urban J P. Factors influencing the oxygen concentration gradient from the synovial surface of articular cartilage to the cartilage-bone interface: a modeling study. Arthritis Rheum 2004; 50 (12): 3915–24.